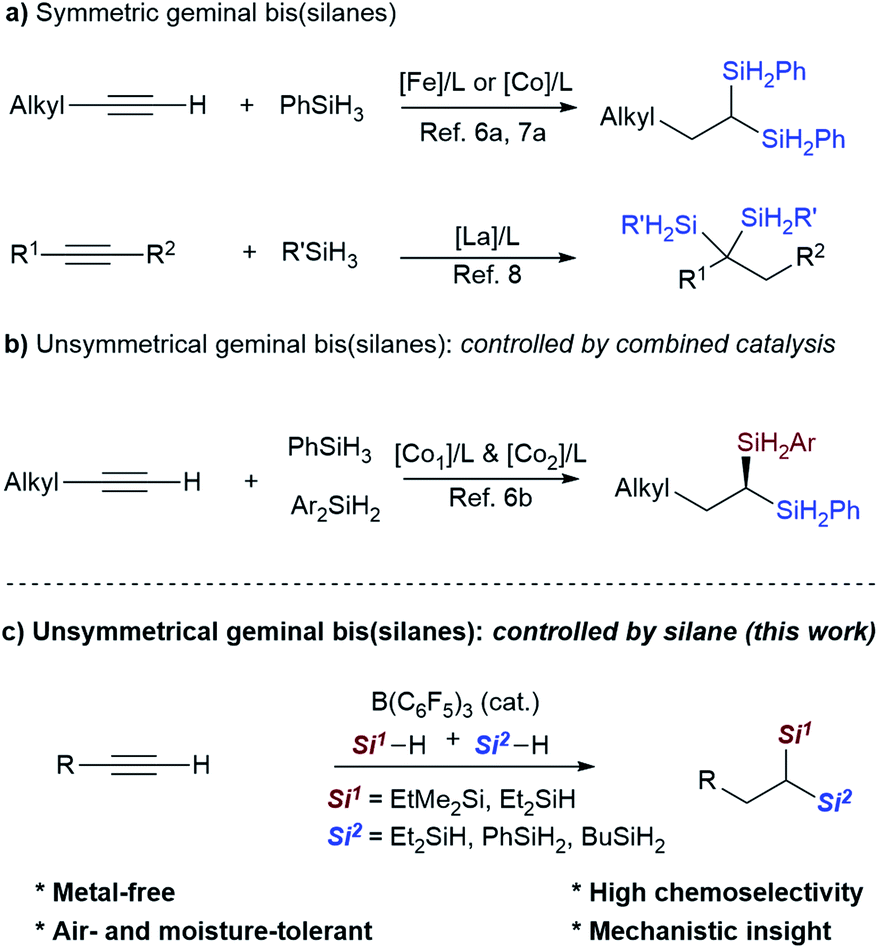DOI:
10.1039/D1SC02769G
(Edge Article)
Chem. Sci., 2021,
12, 10883-10892
Borane-catalyzed selective dihydrosilylation of terminal alkynes: reaction development and mechanistic insight†
Received
21st May 2021
, Accepted 13th July 2021
First published on 21st July 2021
Abstract
Here, we describe simple B(C6F5)3-catalyzed mono- and dihydrosilylation reactions of terminal alkynes by using a silane-tuned chemoselectivity strategy, affording vinylsilanes and unsymmetrical geminal bis(silanes). This strategy is applicable to the dihydrosilylation of both aliphatic and aryl terminal alkynes with different silane combinations. Gram-scale synthesis and conducting the reaction without the exclusion of air and moisture demonstrate the practicality of this methodology. The synthetic utility of the resulting products was further highlighted by the structural diversification of geminal bis(silanes) through transforming the secondary silane into other silyl groups. Comprehensive theoretical calculations combined with kinetical isotope labeling studies have shown that a prominent kinetic differentiation between the hydrosilylation of alkynes and vinylsilane is responsible for the chemoselective construction of unsymmetrical 1,1-bis(silanes).
Introduction
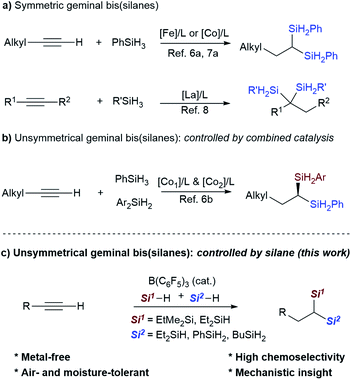
Organosilanes have a multitude of applications in a variety of fields, ranging from organic synthesis to functional materials and pharmaceutical sciences.1 Geminal bis(silanes) are particularly attractive due to their versatile synthetic utilities, and valuable structural and conformational properties.2,3 However, the scarcity of practical protocols for their synthesis limits their structural diversity and their further synthetic utilities. Previous strategies for the synthesis of geminal bis(silanes) are mainly based on prefunctionalized precursors, such as retro-[1,4]-Brook rearrangement of 3-silyl allyloxy-silanes,3a,b,d insertion of benzylic carbenes into disilanes,4 and double C–Si coupling of geminal dibromides.5
Synthesis of geminal bis(silanes) through dihydrosilylation of alkynes is an ideal way due to the high step- and atom-economy. Recently, several research groups have reported dihydrosilylation reaction of alkynes through transition-metal catalysis. For example, Lu's group and Zhu's group reported the catalytic 1,1-dihydrosilylation of terminal aliphatic alkynes with Co- and Fe-catalysts, affording symmetrical 1,1-bis(silanes), respectively (Scheme 1a).6a,7 Besides, Cui's group also reported the dihydrosilylation of internal alkynes with rare-earth ate complexes, providing access to 1,1-di- or 1,1,1-trisilylated products.8 More importantly, Lu and co-workers elegantly developed sequential catalysis with a combination of two Co-catalysts (CoBr2, Xantphos and CoBr2, OIP (OIP = oxazoline-iminopyridine)) to access unsymmetrical 1,1-bis(silanes) with aliphatic terminal alkynes (Scheme 1b).6b Although significant advances have been achieved, currently available synthetic protocols are mainly applicable to terminal aliphatic alkynes or internal alkynes. The synthesis of 1,1-bis(silanes) with arylacetylenes is less reported. Scope investigation on Fe-catalyzed dihydrosilylation of terminal alkynes has shown that the reaction of 1-arylacetylene with hydrosilane only affords the monohydrosilylated product due to the steric effect.7a,9
 |
| | Scheme 1 Catalytic dihydrosilylation of alkynes. R1 = alkyl; R2 = aryl, alkyl or silyl group; R = alkyl or aryl. | |
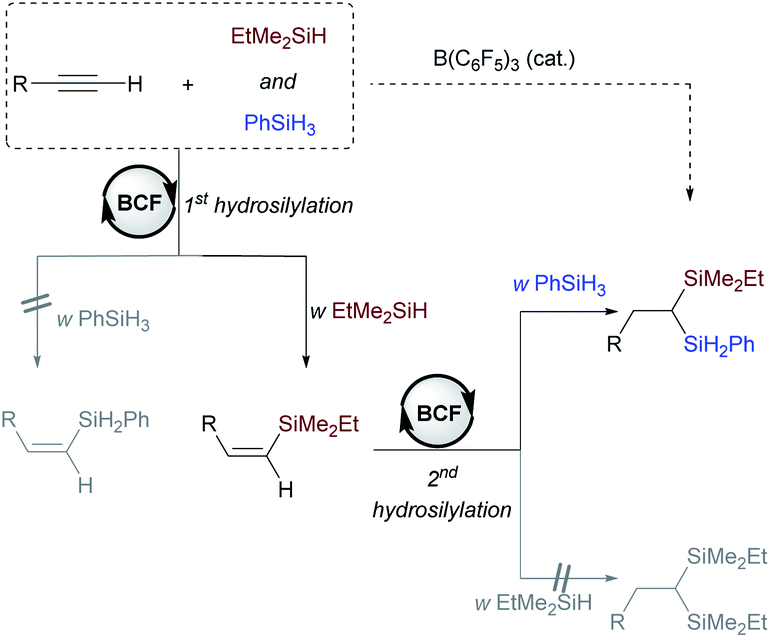
Recently, the combination of commercially available tris(pentafluoro) borane (B(C6F5)3)10,11 and hydrosilanes (Si–H) has emerged as a highly versatile catalytic system for the hydrosilylation, silylation and selective defunctionalization of organic molecules to valuable intermediates,12–15 providing a powerful alternative to the classical transition-metal catalysis. It should be noted that Gevorgyan's group has previously disclosed B(C6F3)3-catalyzed hydrosilylation of alkenes about twenty years ago,13c but the catalytic hydro- or dihydrosilylation of terminal alkynes with the versatile B(C6F5)3/Si–H system remains unknown. We herein report the development of a metal-free strategy for the selective dihydrosilylation of aliphatic or aryl-terminal alkynes with B(C6F5)3 as the catalyst (Scheme 1c). This method allows the direct construction of unsymmetrical geminal bis(silanes) with a quaternary and a secondary silyl group, in which the Si–H bonds can be readily converted to other silyl groups. Mechanistic studies reveal that tertiary silanes (like EtMe2SiH) favor the hydrosilylation of alkynes (1st hydrosilylation) while primary hydrosilanes (such as PhSiH3 and BuSiH3) favor the hydrosilylation of vinylsilane (2nd hydrosilylation).
Results and discussion
Reaction development
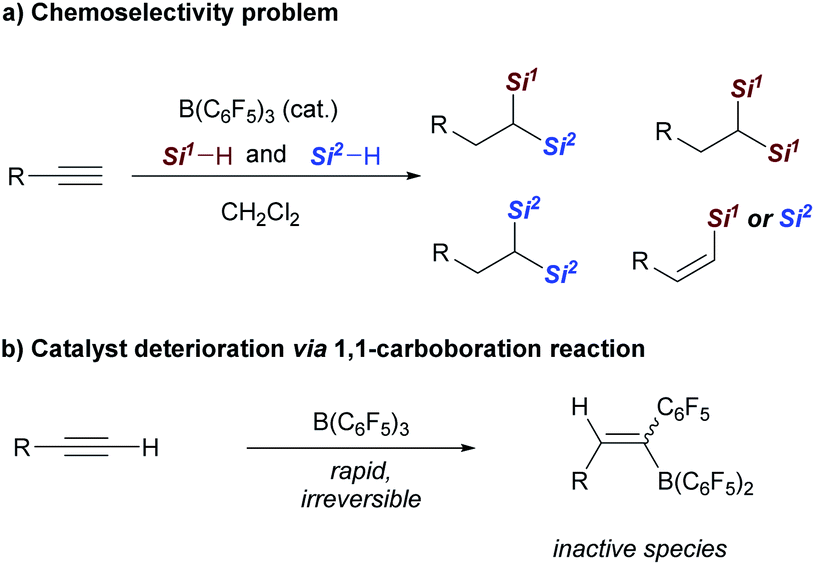
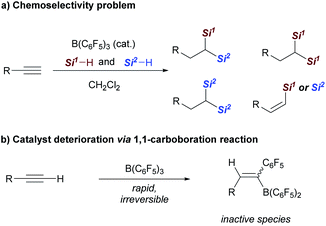
Although the realization of this strategy could provide a conceptually different approach for the synthesis of 1,1-bis(silanes), several possible problems should be overcome: first, hydrosilylation of alkynes generally yields alkenyl silanes, which are difficult for further hydrosilylation owing to the steric effect.7a,9 Second, the control of chemoselectivity in the presence of two different hydrosilanes is also problematic (Scheme 2a). Third, the rapid and irreversible 1,1-carboboration reaction between a terminal alkyne and B(C6F5)3 may lead to catalyst deterioration (Scheme 2b).16,17 As a result, despite the broad application of B(C6F5)3 in hydrosilylation reactions, its capability in the dihydrosilylation of alkynes has, to our knowledge, not been demonstrated.
 |
| | Scheme 2 Challenges in catalytic 1,1-dihydrosilylation of terminal alkynes with B(C6F5)3. | |
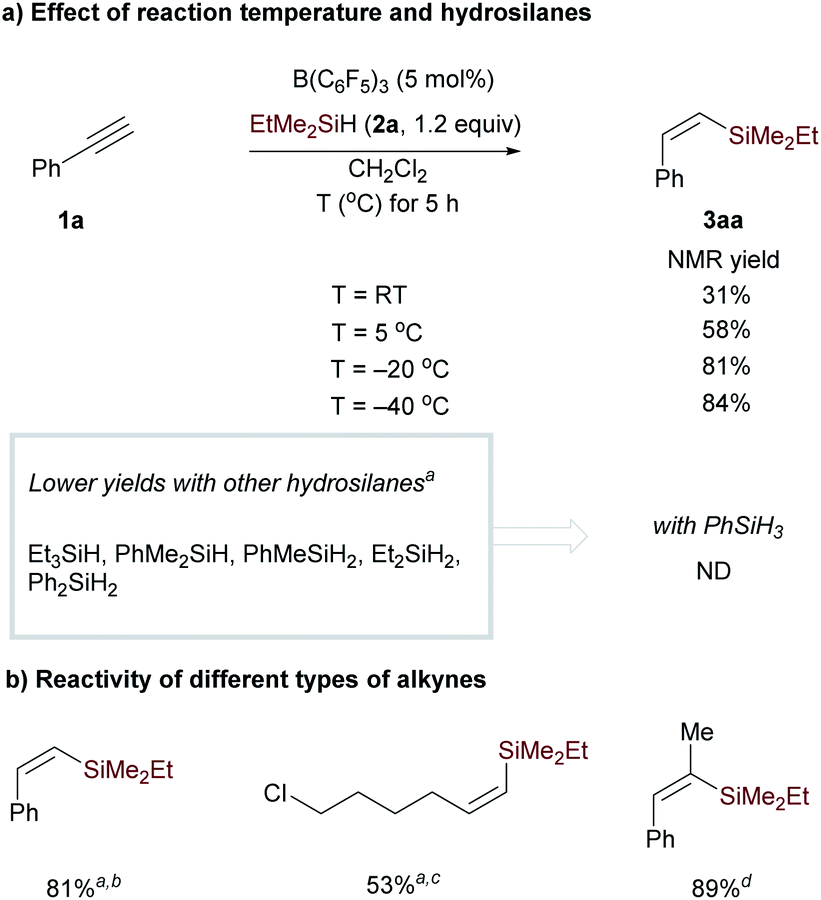
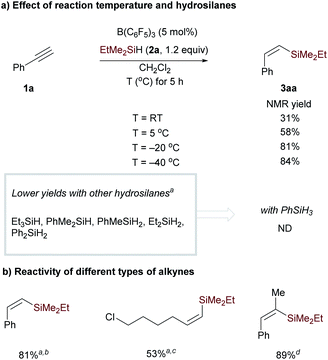
We first examined the reaction conditions for catalytic monohydrosilylation of a terminal alkyne (Scheme 3a). At room temperature, the hydrosilylation of phenylacetylene 1a using B(C6F5)3/EtMe2SiH combination only affords Z-vinylsilane 3aa in 31% yield.18 This may probably be attributed to the occurrence of 1,1-carboboration reaction between phenylacetylene and B(C6F5)3.16,17 From a mechanistic point of view, the hydrosilylation pathway, involving a concerted three-molecular Si-centered displacement of a boron-coordinated hydride by the alkyne transition state (SN2@Si),13,19 requires more entropy-cost than the two-molecular transition state toward the carboboration pathway. Therefore, we hypothesized that low temperatures might facilitate the entropy-disfavored hydrosilylation pathway. We were delighted to find that the yield was significantly improved from 58% to 84% when lowering the temperature from 5 to −40 °C. Then, the effect of hydrosilylation reagent was examined in the presence of 5 mol% of B(C6F5)3 at −40 °C. Other hydrosilanes, including tertiary, secondary, and primary silanes, were not as effective as EtMe2SiH for monohydrosilylation of phenylacetylene (see Table S1† in the ESI for optimization details). It is worth noting that with secondary hydrosilane Et2SiH2, Z-vinyl silane was formed in 55% yield but accompanied by 25% yield of dihydrosilylated product. These results indicate that low temperature and suitable hydrosilanes are necessary to synthesize vinylsilane from terminal alkynes using B(C6F5)3 as the catalyst. In addition to phenylacetylene, the reactivity of aliphatic terminal alkynes and internal alkynes was also investigated for the B(C6F5)3-catalyzed monohydrosilylation reaction. As shown in Scheme 3b, this method is applicable to both terminal and internal alkynes. It should be noted that the hydrosilylation of 1-phenylpropyne exhibits excellent regioselectivity and efficiency, the corresponding Z-vinylsilane could be prepared in 89% yield (9 mmol scale) with only 0.25 mol% of B(C6F5)3.
 |
| | Scheme 3 Initial investigation on the monohydrosilylation of terminal alkynes. aPerformed at −40 °C. bWith 5 mol% B(C6F5)3. cWith 10 mol% B(C6F5)3. dIn 9 mmol scale, performed at room temperature with 0.25 mol% B(C6F5)3. ND = not detected. | |
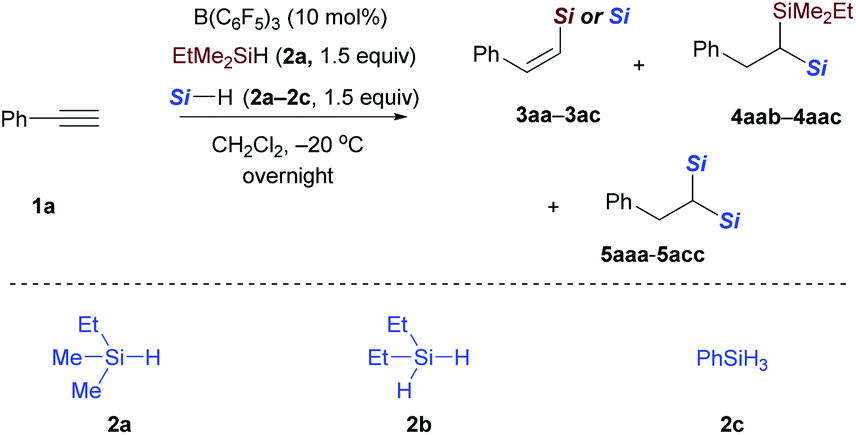
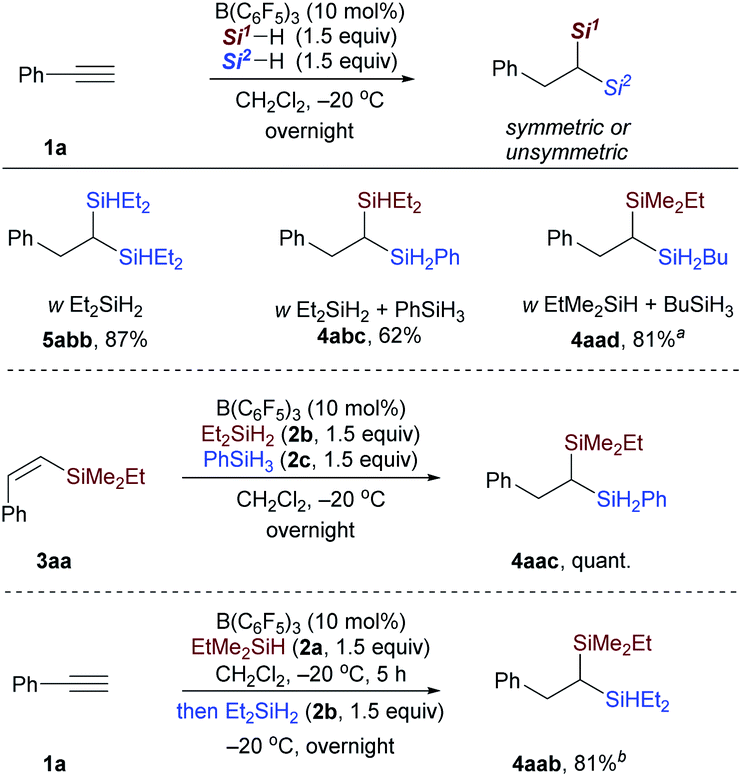
After the monohydrosilylation reaction conditions were established, we then explored the dihydrosilylation of alkynes (Table 1). Using EtMe2SiH as the sole hydrosilane, only traces of dihydrosilylated product 5aaa were observed, and most of the monohydrosilylated product 3aa (76% NMR yield) was left unreacted at −20 °C in the presence of 5 mol% of B(C6F5)3 (entry 1). Further increasing the catalyst loading from 5 mol% to 10 mol% did not significantly improve the yield of 5aaa (entry 2). Replacing EtMe2SiH by an equimolar mixture of EtMe2SiH and Et2SiH2, unsymmetrical geminal bis(silane) 4aab was formed in 66% yield (entry 3), but accompanied by 34% yield of symmetrical 1,1-bis(silane) 5abb (dihydrosilylated by Et2SiH2). When a combination of EtMe2SiH and PhSiH3 was used, phenylacetylene underwent two sequential hydrosilylation reactions to give the corresponding unsymmetrical 1,1-bis(silane) 4aac in 92% isolated yield (entry 4).20 Reaction performed overnight at a lower temperature (−40 °C) did not decrease the yield of the desired product (entry 5).
Table 1 Optimization of the dihydrosilylation reaction of alkynesa
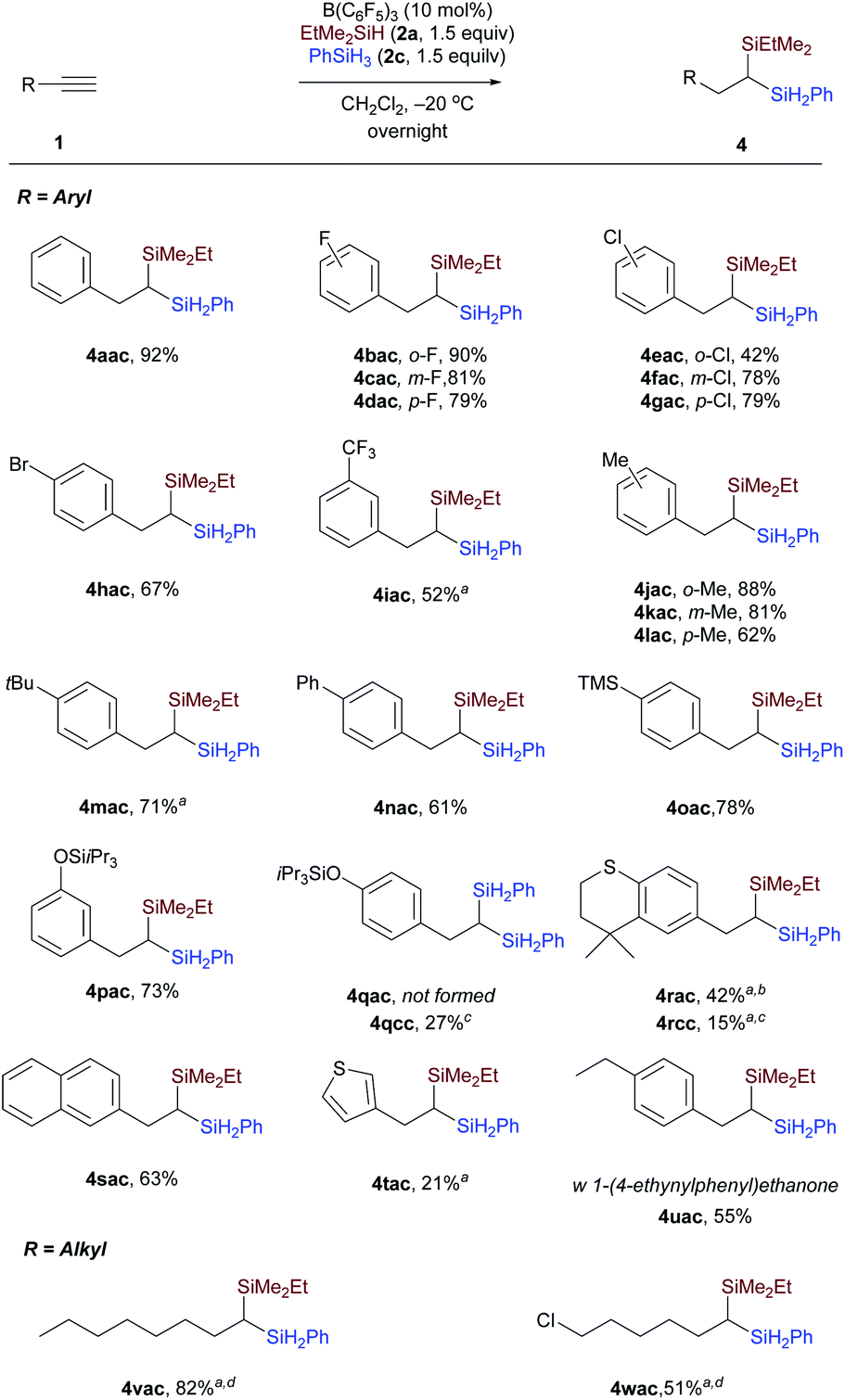
Examination of substrate scope
We then evaluated a series of terminal alkynes to probe the substrate scope (Scheme 4). Substrates with electron-withdrawing (1b–1i) or electron-donating (1j–1n) substituents underwent dihydrosilylation with good efficiency, transforming the terminal alkyne into the corresponding 1,1-bis(silanes) in reaction with an equimolar mixture of EtMe2SiH and PhSiH3. Functional groups, including halides (F, Cl, Br, 1b–1h), trifluoromethyl (CF3, 1i), and trimethylsilyl (TMS, 1o) were tolerated. 3-Triisopropylsilyl-O-substituted phenylacetylene was selectively dihydrosilylated to yield unsymmetrical 1,1-bis(silane) 4pac. The reaction of 1-ethynyl-4-[[tris(1-methylethyl)silyl]oxy]benzene with EtMe2SiH and PhSiH3 under standard conditions only affords the symmetrical geminal silane 4qcc in 27% (dihydrosilylated by PhSiH3). This is probably due to the fact that the kinetics or reaction mechanism of the substrate with strong electron-donating substituents is different from phenylacetylenes with weak electron-donating substituents. Similarly, alkyne 1r with a cyclic thioether also did not afford the corresponding unsymmetrical bis(silane) 4rac according to the general procedure, but afforded 4rcc in 15% yield (dihydrosilylated by PhSiH3). The unsymmetrical 1,1-bis(silane) 4rac could be obtained in 42% yield by slightly changing the reaction conditions. Aside from phenylacetylene derivatives, a β-naphthyl and a thiophene-3-yl as in 1s and 1t were also compatible with the reaction conditions, leading to products 4sac and 4tac, respectively, with moderate and slightly lower yields. However, functional groups including ether, ester, aldehyde, ketone, carboxylic acid, and amide were not tolerated because of the high reduction or defunctionalization ability of B(C6F5)3/hydrosilane combinations. For example, when 1-(4-ethynylphenyl)ethanone was subjected to the mixture of B(C6F5)3 and hydrosilane at −20 °C, we could only isolate the decarbonylated 1,1-bis(silane) 4uac. In addition to phenylacetylenes, terminal alkynes with aliphatic side chains were also viable (Scheme 4, bottom). Oct-1-yne 1v and 6-chlorohex-1-yne 1w could be dihydrosilylated to give the related 1,1-bis(silanes) 4vac and 4wac by simply stirring the reaction mixture at −40 °C for 5 hours and then at room temperature for an additional 24 hours. The reactivity difference between aryl and aliphatic terminal alkynes can be rationalized by the stability of different types of carbon cations involved. However, the reaction of the internal alkyne, 1-phenyl-1-propyne, only affords the monohydrosilylated compound as the major product. Different from Fe-catalyzed dihydrosilylation of terminal alkynes, in which only aliphatic terminal alkynes can be transformed into the corresponding symmetrical geminal bis(silanes) with primary silanes,7a our approach is applicable to both aryl and alkyl-substituted terminal alkynes.
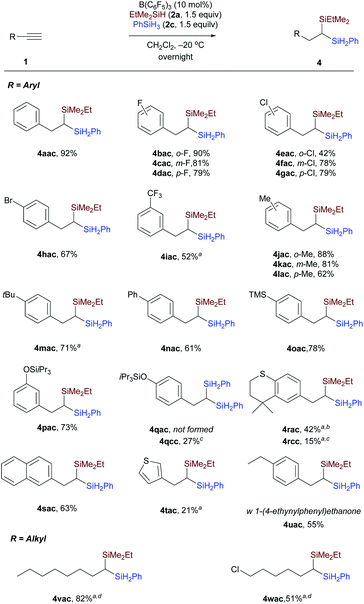 |
| | Scheme 4 Substrate scope. Isolated yields after flash chromatography. a20 mol% B(C6F5)3 was used. bPhSiH3 was added in 5 hours delay after the initial EtMe2SiH addition. cDihydrosilylated by PhSiH3. dStirred at −40 °C for 5 h, and then 24 h at room temperature. | |
Then, the reactivity observed in Table 1 intrigued us to construct geminal bis(silanes) with other hydrosilane combinations. As shown in Scheme 5, using Et2SiH2 as the sole silane, a symmetrical geminal bis(silane) 5abb with two tertiary silane moieties was obtained in excellent yield. Moreover, the reaction of Et2SiH2/PhSiH3 and EtMe2SiH2/BuSiH3 combination with phenylacetylene also worked effectively and selectively to give the unsymmetrical 1,1-bis(silane) 4abc and 4aad in 62% and 81% yield, respectively. To further identify the silane effect in the second hydrosilylation step, we subjected 3aa to the standard procedures with an equimolar mixture of Et2SiH2 and PhSiH3, and detected the exclusive formation of 4aac (Scheme 5, middle; selectively hydrosilylated by PhSiH3). These results confirmed that the primary silane is more reactive than secondary and tertiary hydrosilanes in the second hydrosilylation step (vinylsilane hydrosilylation). This trend is consistent with the size of the hydrosilanes. Although the use of EtMe2SiH and Et2SiH2 combination gives a mixture of two different dihydrosilylation products (cf.Table 1, entry 3), the unsymmetrical 1,1-bis(silane) 4aab can be obtained in 81% yield by simply adding Et2SiH2 in 5 hours delay after the initial EtMe2SiH addition (Scheme 5, bottom).
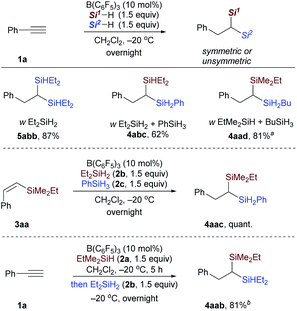 |
| | Scheme 5 Reactivity of other hydrosilane combinations. Isolated yields after flash chromatography. aA slight amount of symmetric geminal bis(silane) was detected (unsymmetric![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :symmetric > 10:1). bEt2SiH2 was added in 5 hours delay after the initial EtMe2SiH addition. :symmetric > 10:1). bEt2SiH2 was added in 5 hours delay after the initial EtMe2SiH addition. | |
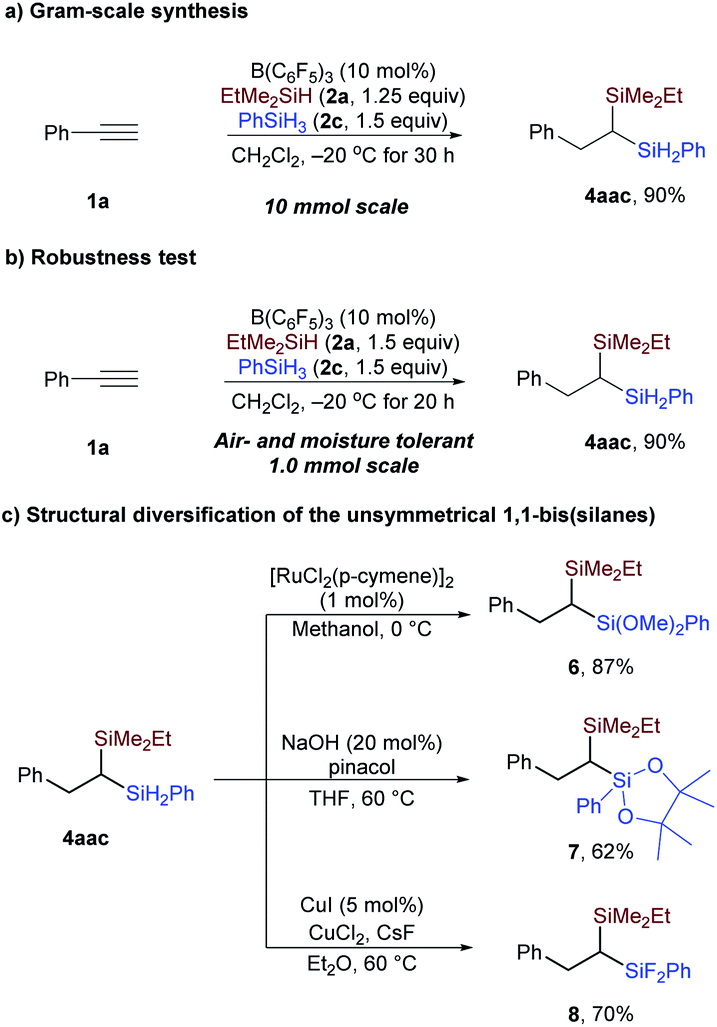
To demonstrate the practicality of this protocol in preparative organic synthesis, a gram-scale reaction was conducted. As shown in Scheme 6a, in the presence of 10 mol% B(C6F5)3, 2.7 grams of 4aac could be prepared with phenylacetylene in 90% yield. Moreover, the reaction could be conducted with all reagents handled in the open atmosphere without exclusion of air and moisture; the yield was 90% in 1 mmol scale (Scheme 6b).21 The two Si–H bonds in the secondary silane of 4aac can be readily converted to Si–O (6 and 7) and Si–F bonds (8) (Scheme 6c), enriching the structural diversity of the geminal bis(silanes).
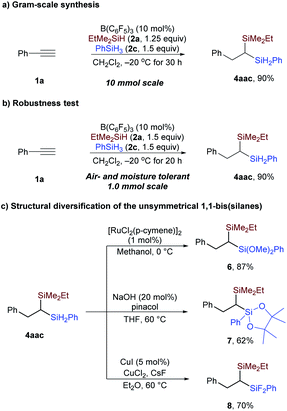 |
| | Scheme 6 Reaction practicality and product transformation. | |
Mechanism of borane-catalyzed dihydrosilylation of terminal alkynes
Comparison of the influence of hydrosilanes on alkyne hydrosilylation and vinylsilane hydrosilylation.
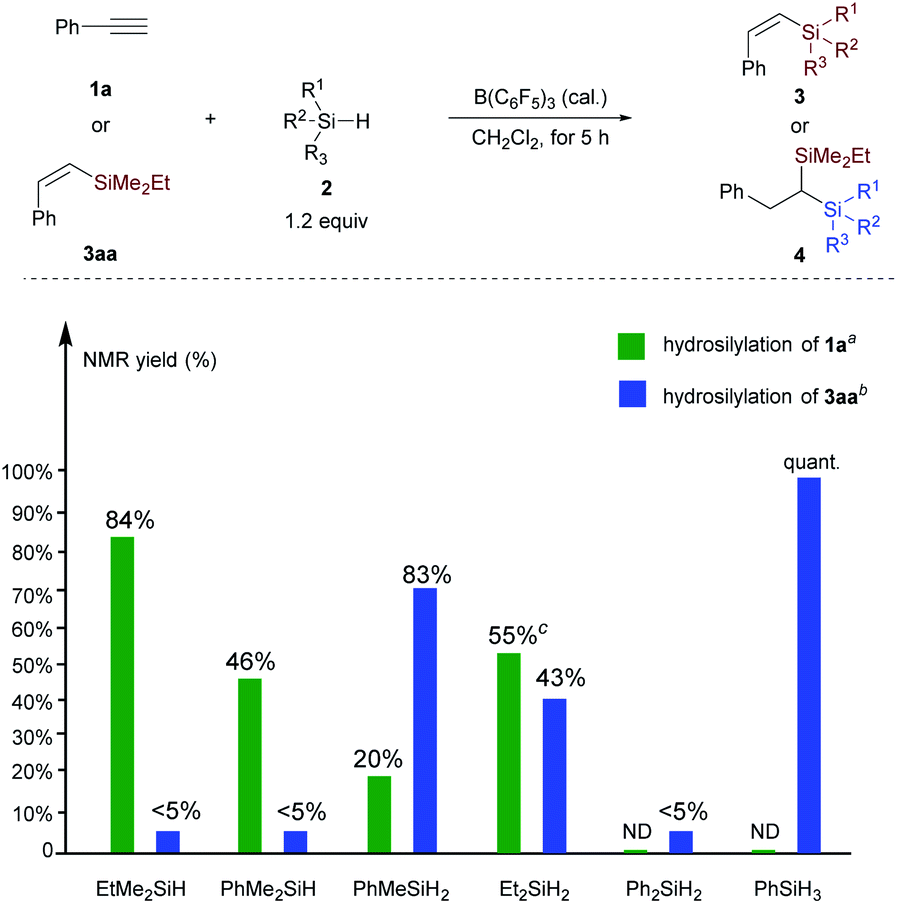
The experimental results collected in Scheme 7 show that for the hydrosilylation of phenylacetylene, replacement of the ethyl group in EtMe2SiH by phenyl (PhMe2SiH) leads to a reduced yield of vinylsilane (84% versus 46%). Hydrosilylation of 1a with Et2SiH2 exhibits higher reactivity than PhMeSiH2, albeit accompanied by the formation of symmetric geminal bis(silane); both Ph2SiH2 and PhSiH3 are inactive for the hydrosilylation of phenylacetylene. These results suggest that hydrosilylation of phenylacetylene is controlled by the electronic effect, and the observed reactivity order is consistent with the electron-donating ability (Me > Ph > H) of hydrosilanes as measured by Mayr et al.22 However, the reactivity of vinylsilane hydrosilylation is significantly different from alkyne hydrosilylation. Tertiary silane is less reactive than secondary and primary hydrosilane. Replacement of the two ethyl groups in Et2SiH2 by phenyl groups leads to little reactivity. Besides, we also performed the hydrosilylation reaction of 3aa in the presence of two secondary hydrosilanes Et2SiH2 (2b) and PhMeSiH2 (2e). The reaction rate of 3aa with PhMeSiH2 is faster than that of Et2SiH2 (4aab/4aae = 1:3.5, see Scheme S2 and Fig. S3† for details). This result confirms the importance of the steric effect in the hydrosilylation of vinylsilane. However, when performing the hydrosilylation reaction of 3aa in the presence of two primary silanes PhSiH3 and BuSiH3, alkylsilane BuSiH3 shows higher reactivity than PhSiH3; and the corresponding geminal bis(silanes) were obtained in 2.4:1 ratio (see Scheme S2 and Fig. S4† for details). To conclude from these control experiments, we can get the reactivity order for the hydrosilylation of vinylation: EtMe2SiH < Et2SiH2 < PhMeSiH2 < PhSiH3 < BuSiH3. These observations suggest that hydrosilylation of vinylsilane mainly depends on the steric effect, but electronic effects also play a role in the hydrosilylation of vinylsilane with primary hydrosilanes.
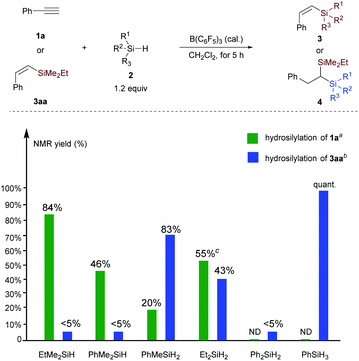 |
| | Scheme 7 Comparison of the influence of hydrosilanes on alkyne hydrosilylation and vinylsilane hydrosilylation. aHydrosilylation of phenylacetylene 1a: performed at −40 °C with 10 mol% B(C6F5)3 in 0.4 mmol scale. bHydrosilylation of vinylsilane 3aa: performed at −20 °C with 5 mol% B(C6F5)3 in 0.2 mmol scale. cDihydrosilylated product formed with Et2SiH2, see Table S1† for details. | |
DFT calculations on the full catalytic cycle.
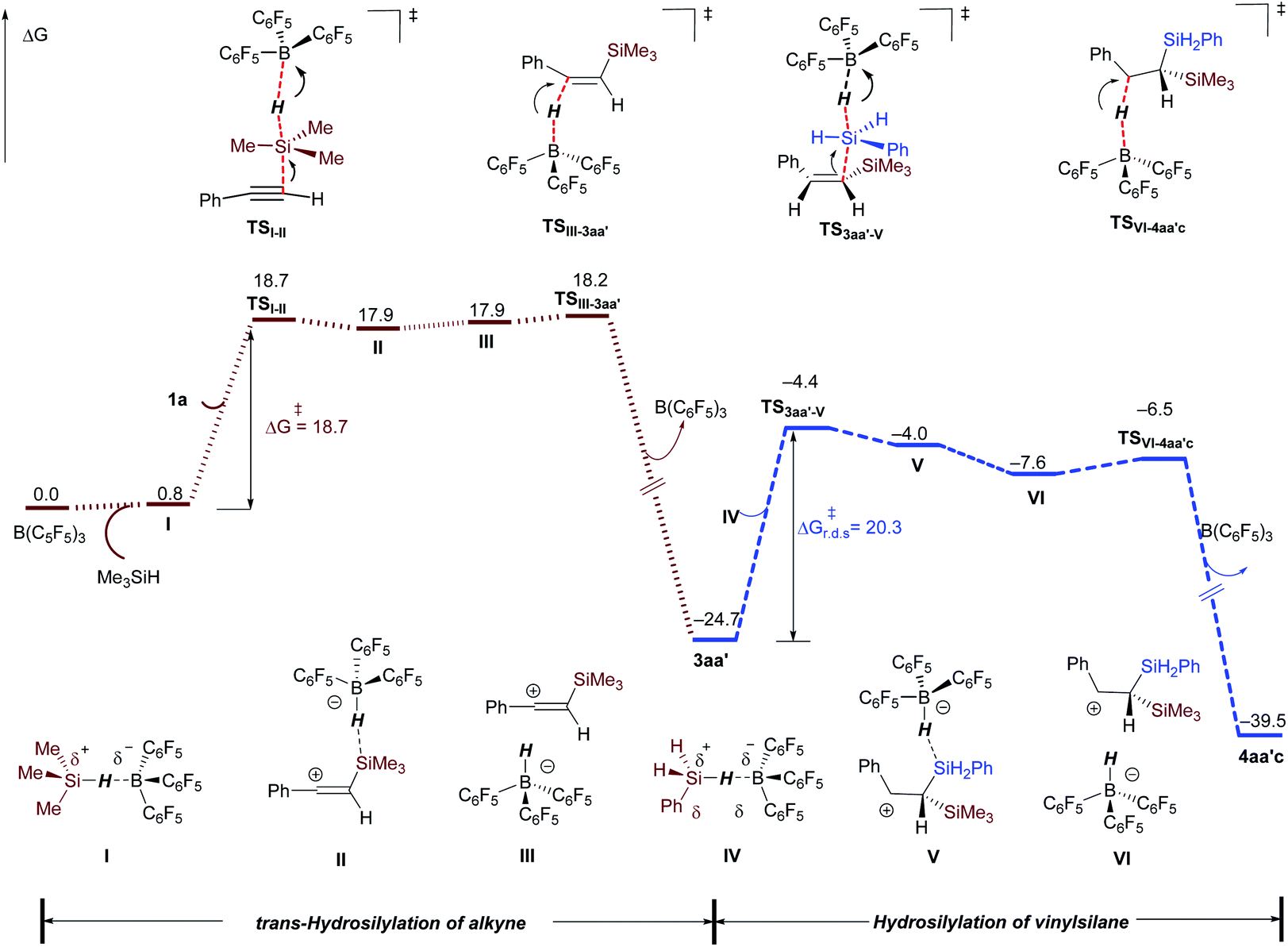
Computational studies with the M06-2X functional23 were undertaken to gain more mechanistic insight. The solvent effect was treated with the PCM24 solvation model and with dichloromethane as the solvent in structural optimization and single-point energy calculations (see ESI† for computational details).25 The dihydrosilylation of phenylacetylene 1a with Me3SiH/PhSiH3 combination was selected as the model reaction. The complete free energy profile is displayed in Fig. 1 (see Fig. S6† for optimized structures). The sequential dihydrosilylation of alkyne proceeds through a classical Piers–Oestreich-type mechanism,13,19 wherein substrate 1a (alkyne hydrosilylation, in brown line) or in situ formed vinylsilane 3aa′ (vinylsilane hydrosilylation, in blue line) attacks the Si center in the B(C6F5)3-hydrosilane η1-adduct I or IV (viaTSI-II or TS3aa′-V) to form the ion-pair species II or V. The barrier for Si–H bond cleavage in adduct I with alkyne (viaTSI-II) is computed to be 18.7 kcal mol−1, and the Si–H bond cleavage in adduct IV with vinylsilane as a nucleophile (TS3aa′-V) requires a slightly higher activation barrier of 20.3 kcal mol−1. Both ion-pair species II and IV are readily reorganized to their isomers III and VI,19 in which the anion B–H is oriented towards the carbon cation center (vinyl cation or β-silylcarbenium ion). Then, hydride transfer from the borohydride to the electrophilic carbon atom in the ion-pair III (viaTSIII-3aa′, ΔG‡ = 18.2 kcal mol−1) or VI (viaTSVI-4aa′c, ΔG‡ = 18.2 kcal mol−1) generates the trans-hydrosilylation product 3aa′ or dihydrosilylated product 4aa′c, respectively. In addition, the cis-hydrosilylation pathway involving hydride transfer from borohydride to the vinyl cation at the same side of the silyl group (leading to E-3aa′) was also considered. This process is less kinetically favorable than the trans-hydrosilylation pathway (ΔG‡ = 24.1 versus 18.2 kcal mol−1, see Fig. S7†). Along the whole dihydrosilylation pathway, nucleophilic attack of vinylsilane 3aa′ at the Si center of the B(C6F5)3-PhSiH3 adduct IV (TS3aa′-V) with an activation barrier of 20.3 kcal mol−1 is the rate-limiting step, which is consistent with the mild reaction conditions.
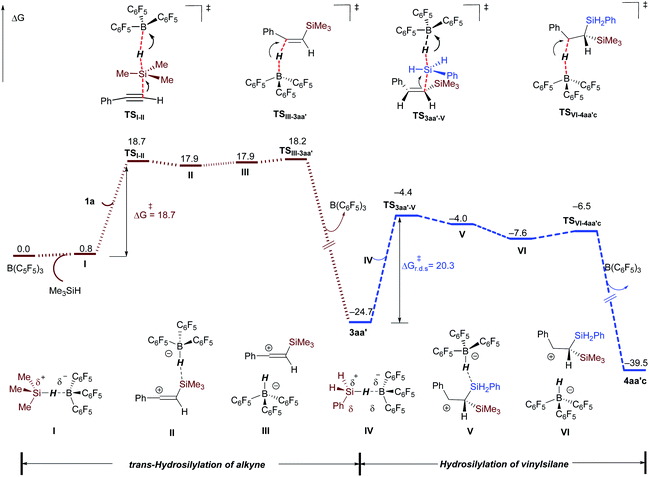 |
| | Fig. 1 Gibbs free energy profile (298.15 K) for the B(C6F5)3-catalyzed dihydrosilylation of alkyne (in kcal mol−1). Performed at the M06-2X/cc-pVTZ/(PCM, CH2Cl2)//M06-2X/6-311G(d,p)/(PCM, CH2Cl2) level of theory. r.d.s = rate-determining step. | |
Rationalization of the reactivity difference of hydrosilanes
Based on the reactivity difference observed in dihydrosilylation of terminal alkynes that depend on hydrosilanes, we then focused on gaining insights into the origin of chemoselectivity in this catalytic system.
Hydrosilylation of terminal alkynes (1st hydrosilylation).
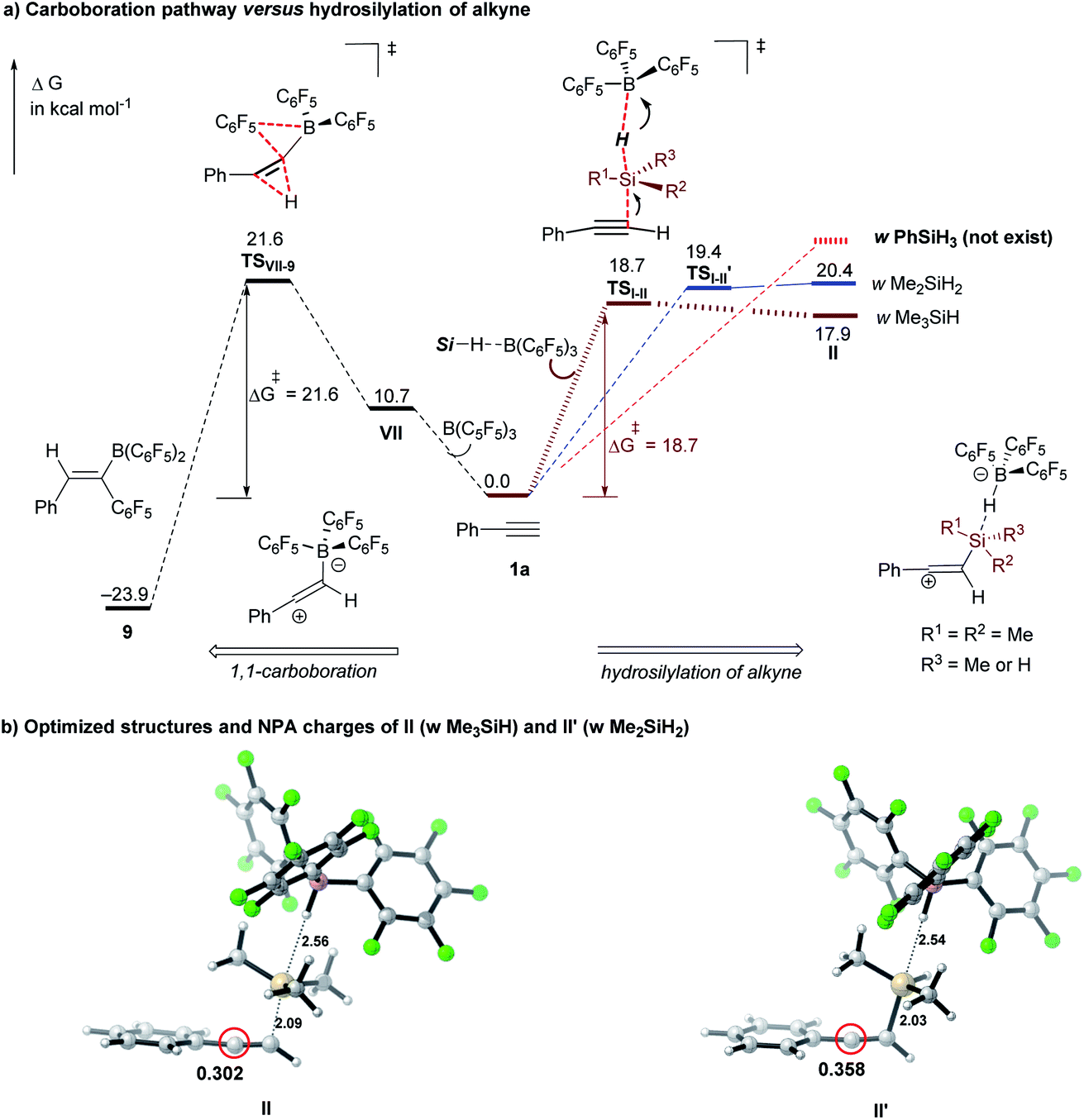
To compare the reactivity of silanes on the hydrosilylation of alkynes, we computed the energy barriers of the 1st hydrosilylation step (I → II) with representative hydrosilanes (Me3SiH, Me2SiH2, and PhSiH3). As shown in Fig. 2, the activation barrier for the secondary hydrosilane Me2SiH2 here is slightly higher than that with Me3SiH (19.4 versus 18.7 kcal mol−1); but both of which are lower than the barrier of the carboboration reaction between B(C6F5)3 and phenylacetylene 1a (21.6 kcal mol−1, see Fig. S8† in the ESI). With PhSiH3, however, the related SN2@Si transition state could not be located after extensive endeavors (Fig. S10†). Our further calculations show that the resulting ion-pair intermediate doesn't exist as a minimum structure on the potential energy surface (Fig. 2, red dashed line). These results suggest that both Me3SiH and Me2SiH2 are suitable for the hydrosilylation of terminal alkynes, but the primary silane PhSiH3 is not effective. NPA charge calculations reveal that the benzylic carbon of the ion-pair intermediate II′ for Me2SiH2 (Fig. 2b, II′: 0.358e) is more positively charged than that in II (for Me3SiH, II: 0.302e). These computational results also support that the hydrosilylation of the terminal alkyne is probably controlled by the electronic effect, being consistent with these experimental results.
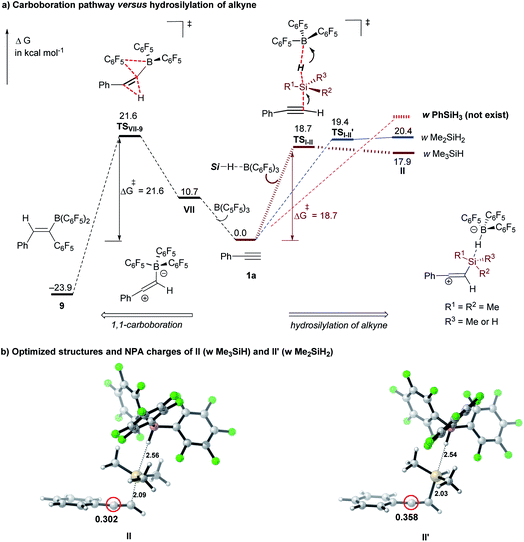 |
| | Fig. 2 (a) Comparison of the carboboration pathway and the B(C6F5)3-catalyzed hydrosilylation of alkyne. Performed at the M06-2X/cc-pVTZ/(PCM, CH2Cl2)//M06-2X/6-311G(d,p)/(PCM, CH2Cl2) level of theory (energies are in kcal mol−1); (b) optimized structures and NPA charges for ion-pair species II (with Me3SiH) and II′ (with Me2SiH2). | |
To understand the origin of reactivity difference, we undertook NMR analysis on the hydrosilylation reactions of 1a with EtMe2SiH or Et2SiH and PhSiH3, respectively. The crude 1H NMR spectra clearly show the formation of the hydrosilylation or dihydrosilylation product with EtMe2SiH or Et2SiH2 in the presence of 20 mol% B(C6F5)3 (Fig. S1 in the ESI†). We were able to detect the characteristic peaks of B(C6F5)3 (i.e., 128.1, 144.0, 160.9 ppm) from the 19F NMR spectra (Fig. S2 in the ESI†). However, only a trace amount of dihydrosilylated product was detected with PhSiH3 as the silane source. The 19F NMR spectrum of the reaction mixture of PhSiH3, 1a and 20 mol% of B(C6F5)3 confirms the deterioration of B(C6F5)3 in the case of PhSiH3. We attribute this to the occurrence of the carboboration reaction of 1a with B(C6F5)3 or other unknown oligomerization or polymerization reactions (see Scheme S1† for Computational analysis). These results are in line with our theoretical calculations that both EtMe2SiH and Et2SiH2 are suitable silanes for the first hydrosilylation step.
Hydrosilylation of vinylation (2nd hydrosilylation).
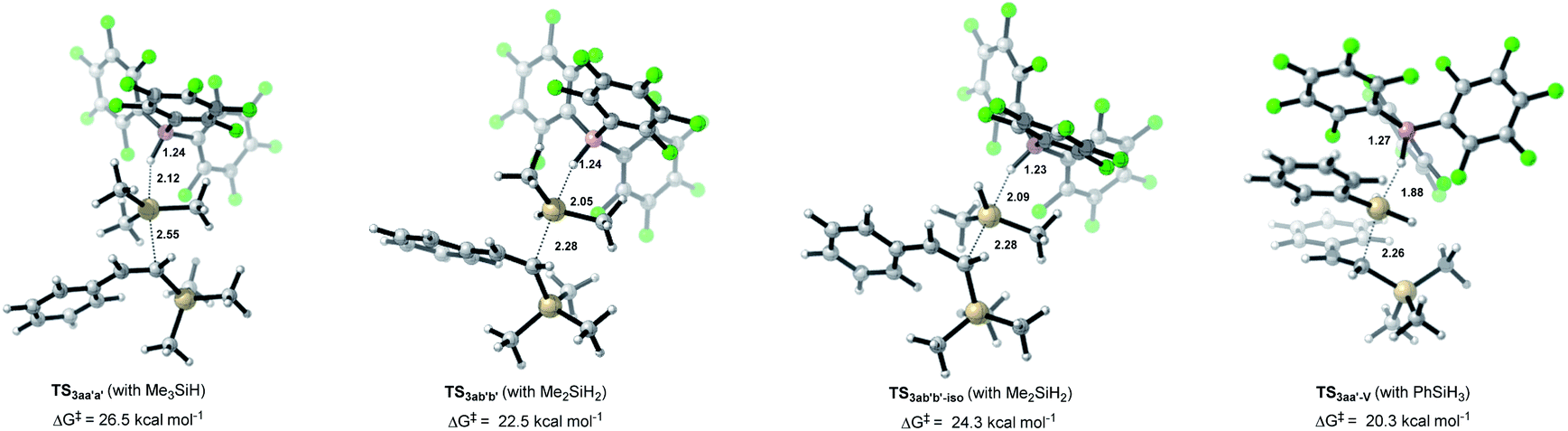
The choice of silane combinations has a significant influence on the dihydrosilylation of alkynes: (1) the use of EtMe2SiH as the sole silane mainly gave the monohydrosilylation product; (2) the use of EtMe2SiH/PhSiH3, or Et2SiH2/PhSiH3, or EtMe2SiH/Et2SiH2 combination could produce the corresponding unsymmetrical geminal bis(silanes), as shown in Table 1 and Scheme 5. Our calculations reveal that the experimentally observed reactivity is in good accordance with the computed activation barriers of the 2nd SN2@Si transition states (vinylsilane hydrosilylation, Fig. 3). As the size of hydrosilane increases, the Gibbs free energy barrier increases from 20.3 kcal mol−1 with the primary silane PhSiH3 to 22.5 kcal mol−1 with the secondary silane Me2SiH2, and to 26.5 kcal mol−1 with the tertiary silane Me3SiH. In TS3aa′a′ (vinylsilane hydrosilylation with Me3SiH), the trimethylsilyl (TMS) group in Me3SiH introduces large steric congestion around B(C6F5)3 to destabilize the transition state.26 This leads to increased distances between the reacting atoms in TS3aa′a′ (dSi–H = 2.12 Å and dSi–C = 2.55 Å) compared to those in TS3ab′b′ (dSi–H = 2.05 Å and dSi–C = 2.28 Å) and those in TS3aa′-V (dSi–H = 1.88 Å and dSi–C = 2.26 Å). In addition, with Me2SiH2 as the substrate, we located another transition state (TS3ab′b′-iso) (an isomer of TS3ab′b′) with the silyl group rotated by 120°. This transition state has a higher activation barrier than TS3ab′b′ (24.3 versus 22.5 kcal mol−1), indicating that the steric effect is a key factor in the 2nd hydrosilylation step.
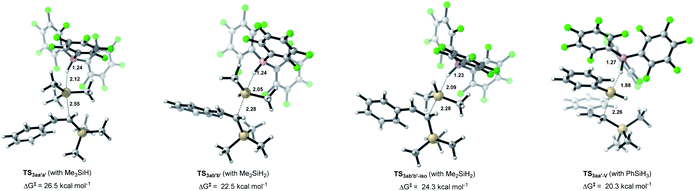 |
| | Fig. 3 Optimized geometries of Si–H cleavage transition states with vinylsilane as the nucleophile. Reaction barriers are given in kcal mol−1. Color code: H, white; C, gray; B, pink; F, green; Si, brown. | |
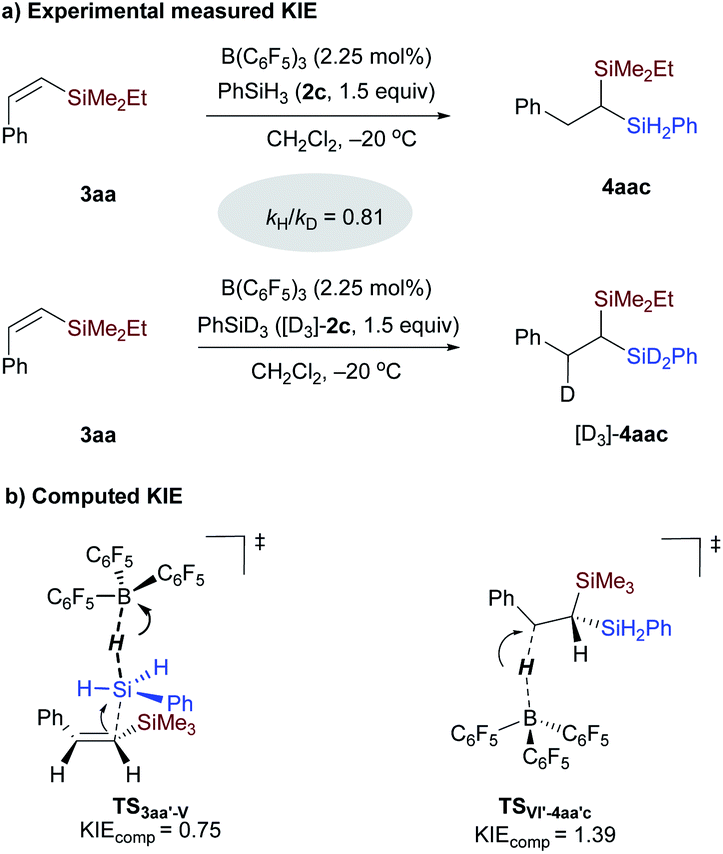
Kinetic isotope effect (KIE) determination for the hydrosilylation of the vinylsilane step.
To further gain insight into the rate-determining step, intermolecular KIE was determined from the initial reaction rates of two parallel reactions of vinylsilane 3aa with PhSiH3 and PhSiD3 (Scheme 8 and Fig. S5†). An inverse secondary deuterium KIE (kH/kD = 0.81) was observed in the hydrosilylation reaction of vinylsilane in the presence of 2.25 mmol% of B(C6F5)3 (Scheme 8a). KIE values of 1.4–1.9 have been reported for the hydrosilylation of carbonyl compounds and heterodehydrocoupling of phosphines and hydrosilanes.13b,27 These values were interpreted in terms of hydride transfer.28 To identify the origin of the observed kinetic isotope effect, KIE calculations using the theory of the Bigeleisen–Mayer equation29 and the rigid-rotor harmonic oscillator approach30 were conducted on the related transition states involved in the hydrosilylation of vinylsilane (see ESI† for Computational details).31 As shown in Scheme 8b, our calculations indicate that the measured inverse secondary KIE is related to the SN2@Si transition state involving the displacement of hydride at the silicon atom with vinylsilane as the nucleophile (TS3aa′-V, KIEcomp = 0.75, Table S4†), rather than the hydride transfer step (KIEcomp = 1.39). The inverse secondary KIE may come from the hybridization change of the silicon center from tetravalent in PhSiH3 to pentavalent in the transition state TS3aa′-V. With these computational and experimental results, we can conclude that the Si–H bond cleavage in adduct IV (viaTS3aa′-V) featuring a pentavalent silicon species is involved in the rate-determining step.
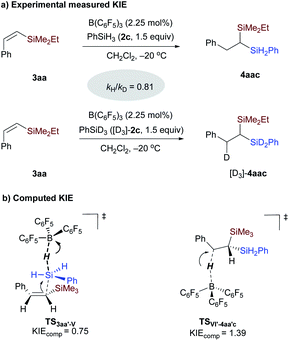 |
| | Scheme 8 Kinetic isotope effect determination for the borane-catalyzed hydrosilylation reaction of vinylsilane 3aa. | |
Based on the experimental and computational studies, we summarized the mechanistic details of the borane-catalyzed dihydrosilylation of alkynes in Scheme 9. The selective formation of unsymmetrical geminal bis(silanes) is a kinetically controlled process, in which the alkyne hydrosilylation can readily occur with EtMe2SiH. In turn, the hydrosilylation of vinylsilane with primary silane is much more efficient than that with EtMe2SiH. The high selectivity toward the unsymmetrical geminal bis(silane) rather than symmetrical products could be rationalized by a combination of steric and electronic effects.
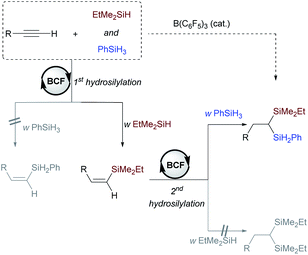 |
| | Scheme 9 Mechanistic proposal for borane-catalyzed dihydrosilylation of terminal alkynes giving rise to unsymmetrical geminal bis(silanes). BCF = B(C6F5)3. | |
Conclusions
We have developed a borane-catalyzed selective dihydrosilylation reaction of terminal alkynes to obtain synthetic valuable unsymmetrical geminal bis(silanes), using a combination of a tertiary silane (or secondary silane) and primary silane. The method exhibits broad substrate scope and can be run in the presence of air and moisture. The resulting silane reagents can be readily functionalized through Si–H bond transformation, increasing the structural diversification of geminal bis(silanes). Combined theoretical calculations and experimental studies shed light on the mechanism, which can account for the high chemoselectivity toward unsymmetrical geminal bis(silanes). We expect our present work to stimulate future studies of boron/silane-related transformations as well as the synthetic utilities of unsymmetrical geminal bis(silane) products obtained through this protocol.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
G. W., X. S., and L. G. performed the experiments. G. W. carried out the theoretical calculations. G. L. computed the KIE values. X. S. and X. L. characterized the compounds. G. W. and S. L. designed the project, analyzed the results and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
G. W. and S. L. thank the National Natural Science Foundation of China (Grant Nos. 22073043, 21903043, 21833002, and 21673110), and the Fundamental Research Funds for the Central Universities (020514380243) for financial support. We thank Prof. Jing Ma and Prof. Xu Cheng for their helpful suggestion on the work. All theoretical calculations were performed on the High-Performance Computing Center (HPCC) of Nanjing University.
Notes and references
-
The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, New York, 1998, vol. 2, p. 1687 Search PubMed.
- For a recent review on geminal bis(silanes), see: L. Gao, Y. Zhang and Z. Song, Synlett, 2013, 24, 139–144 CAS.
- For selected examples, see:
(a) D. R. Williams, A. I. Morales-Ramos and C. M. Williams, Org. Lett., 2006, 8, 4393–4396 CrossRef CAS PubMed;
(b) Z. Yin, Z. Liu, Z. Huang, Y. Chu, Z. Chu, J. Hu, L. Gao and Z. Song, Org. Lett., 2015, 17, 1553–1556 CrossRef CAS PubMed;
(c) Z. Liu, X. Lin, N. Yang, Z. Su, C. Hu, P. Xiao, Y. He and Z. Song, J. Am. Chem. Soc., 2016, 138, 1877–1883 CrossRef CAS;
(d) Z. Chu, K. Wang, L. Gao and Z. Song, Chem. Commun., 2017, 53, 3078–3081 RSC;
(e) Y. Zhang, Q. Guo, X. Sun, J. Lu, Y. Cao, Q. Pu, Z. Chu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2018, 57, 942–946 CrossRef CAS PubMed.
- Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800–12803 CrossRef CAS PubMed.
- H. Hazrati and M. Oestreich, Org. Lett., 2018, 20, 5367–5369 CrossRef CAS PubMed.
-
(a) Z. Cheng, S. Xing, J. Guo, B. Cheng, L.-F. Hu, X.-H. Zhang and Z. Lu, Chin. J. Chem., 2019, 37, 457–461 CrossRef CAS;
(b) J. Guo, H. Wang, S. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881–895 CrossRef CAS.
-
(a) M.-Y. Hu, J. Lian, W. Sun, T.-Z. Qiao and S.-F. Zhu, J. Am. Chem. Soc., 2019, 141, 4579–4583 CrossRef CAS PubMed;
(b) M.-Y. Hu, P. He, T.-Z. Qiao, W. Sun, W.-T. Li, J. Lian, J.-H. Li and S.-F. Zhu, J. Am. Chem. Soc., 2020, 142, 16894–16902 CrossRef CAS PubMed.
- W. Chen, H. Song, J. Li and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 2365–2369 CrossRef CAS PubMed.
-
(a) R. J. P. Corriu, M. Granier and G. F. Lanneau, J. Organomet. Chem., 1998, 562, 79–88 CrossRef CAS;
(b) P.-F. Fu, J. Mol. Catal. A: Chem., 2006, 243, 253–257 CrossRef CAS.
- For reviews on the chemistry of electron-deficient boron Lewis acids:
(a) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC;
(b) M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef.
- Application of B(C6F5)3 in Frustrated Lewis Pair (FLP) chemistry, see:
(a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed;
(b) P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed;
(c) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed;
(d) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS.
- For recent reviews, see:
(a)
D. Weber and M. R. Gagné, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 33–85 Search PubMed;
(b) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC;
(c) W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed;
(d) T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432 CrossRef PubMed;
(e) H. Fang and M. Oestreich, Chem. Sci., 2020, 11, 12604–12615 RSC.
-
(a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9941 CrossRef CAS;
(b) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed;
(c) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed;
(d) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed;
(e) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- Selected examples of defunctionalization reactions with borane/silane combinations:
(a) V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed;
(b) L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576–581 CrossRef CAS PubMed;
(c) T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS;
(d) J. Lowe, B. Bowers, Y. Seo and M. R. Gagné, Angew. Chem., Int. Ed., 2020, 59, 17297–17300 CrossRef CAS.
- For some recent transformations based on the borane/silane catalytic system, see:
(a) N. Gandhamsetty, S. Joung, S.-W. Park, S. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 16780–16783 CrossRef CAS PubMed;
(b) C. K. Hazra, N. Gandhamsetty, S. Park and S. Chang, Nat. Commun., 2016, 7, 13431 CrossRef CAS PubMed;
(c) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS PubMed;
(d) Y. Ma, L. Zhang, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 12434–12437 CrossRef CAS PubMed;
(e) Z.-Y. Zhang, Z.-Y. Liu, R.-T. Guo, Y.-Q. Zhao, X. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2017, 56, 4028–4032 CrossRef CAS PubMed;
(f) C. K. Hazra, J. Jeong, H. Kim, M. H. Baik, S. Park and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 2692–2696 CrossRef CAS PubMedZ.-Y. Liu, M. Zhang and X.-C. Wang, J. Am. Chem. Soc., 2020, 142, 581–588 CrossRef CAS PubMed.
-
(a) C. Chen, F. Eweiner, B. Wibbeling, R. Fröhlich, S. Senda, Y. Ohki, K. Tatsumi, S. Grimme, G. Kehr and G. Erker, Chem.–Asian J., 2010, 5, 2199–2208 CrossRef CAS PubMed;
(b) C. Chen, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2010, 132, 13594–13595 CrossRef CAS PubMed;
(c) C. Chen, T. Voss, G. Kehr, R. Fröhlich and G. Erker, Org. Lett., 2011, 13, 62–65 CrossRef CAS PubMed;
(d) G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839–1850 RSC;
(e) G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 RSC.
- A. Bismuto, G. S. Nichol, F. Duarte, M. J. Cowley and S. P. Thomas, Angew. Chem., Int. Ed., 2020, 59, 12731–12735 CrossRef CAS PubMed.
- Although borane-catalyzed hydrosilylation of internal alkynes is known, its application in the hydrosilylation of terminal alkynes has not been disclosed. For the hydrosilylation of internal alkynes with the borane/silane system, see:
(a) L. D. Curless and M. J. Ingleson, Organometallics, 2014, 33, 7241–7246 CrossRef CAS;
(b) Y. Kim, B. Ramesh, R. B. Dateer and S. Chang, Org. Lett., 2017, 19, 190–193 CrossRef CAS PubMed;
(c) X. Zhao, D. Yang, Y. Zhang, B. Wang and J. Qu, Org. Lett., 2020, 22, 970–975 CrossRef PubMed.
- For computational analysis, see: K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed.
- Lewis acids, such as AlCl3 and EtAlCl2, which were used in the hydrosilylation of alkynes to produce vinylsilanes, see: T. Sudo, N. Asao, V. Gevorgyan and Y. Yamamoto, J. Org. Chem., 1999, 64, 2494–2499 CrossRef CAS . However, AlCl3 did not produce 1,1-bis(silane) 4aac using a combination of EtMe2SiH and PhSiH3..
- For air- or moisture-tolerant borane Lewis acid-catalyzed reactions, see:
(a) K. Ishihara, N. Hanaki, M. Funahashi, M. Miyata and H. Yamamoto, Bull. Chem. Soc. Jpn., 1995, 68, 1721–1730 CrossRef CAS;
(b) V. Fasano, J. E. Radcliffe, A. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef;
(c) D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed;
(d) V. Fasano, J. E. Radcliffe and M. J. Ingleson, ACS Catal., 2016, 6, 1793–1798 CrossRef CAS.
-
(a) H. Mayr and G. Hagen, J. Chem. Soc., Chem. Commun., 1989, 91–92 RSC;
(b) M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem. -Eur. J., 2013, 19, 249–263 CrossRef CAS PubMed.
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed;
(b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
-
C. Y. Legault, CYLview, 1.0b, Universitéde Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- Morandi et al elegantly proved that the type of silane is also critical to the regioselective deoxygenation of 1,2-Diols, see:
(a) N. Drosos and B. Morandi, Angew. Chem., Int. Ed., 2015, 54, 8814–8818 CrossRef CAS PubMed;
(b) G.-J. Cheng, N. Drosos, B. Morandi and W. Thiel, ACS Catal., 2018, 8, 1697–1702 CrossRef CAS.
- L. Wu, S. S. Chitnis, H. Jiao, V. T. Annibale and I. Manners, J. Am. Chem. Soc., 2017, 139, 16780–16790 CrossRef CAS PubMed.
- H. Mayr, N. Basso and G. Hagen, J. Am. Chem. Soc., 1992, 114, 3060–3066 CrossRef CAS.
- J. Bigeleisen and M. G. Mayer, J. Chem. Phys., 1947, 15, 261–267 CrossRef CAS.
-
(a) D. J. O'Leary, P. R. Rablen and M. P. Meyer, Angew. Chem., Int. Ed., 2011, 50, 2564–2567 CrossRef PubMed;
(b) A. Fong, M. P. Meyer and D. J. O'Leary, Molecules, 2013, 18, 2281–2296 CrossRef CAS PubMed.
- Quantum mechanical tunneling effects were also considered for both methods using the one-dimensional parabolic approximation.
(a) R. P. Bell, Chem. Soc. Rev., 1974, 3, 513–544 RSC;
(b)
R. P. Bell, The Tunnel Effect in Chemistry; Chapman and Hall: New York, 1980 CrossRef;
(c) D. B. Northrop, J. Am. Chem. Soc., 1999, 121, 3521–3524 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization, and computational data. See DOI: 10.1039/d1sc02769g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Xiaoshi
Su‡
,
Liuzhou
Gao‡
,
Xueting
Liu
,
Guoao
Li
and
Shuhua
Li
,
Xiaoshi
Su‡
,
Liuzhou
Gao‡
,
Xueting
Liu
,
Guoao
Li
and
Shuhua
Li