
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe-catalyzed Fukuyama-type indole synthesis triggered by hydrogen atom transfer†
Tianze
Zhang
,
Min
Yu
and
Hanmin
Huang
 *
*
Hefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, Center for Excellence in Molecular Synthesis of CAS, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: hanmin@ustc.edu.cn
First published on 6th July 2021
Abstract
Fe, Co, and Mn hydride-initiated radical olefin additions have enjoyed great success in modern synthesis, yet the extension of other hydrogen radicalophiles instead of olefins remains largely elusive. Herein, we report an efficient Fe-catalyzed intramolecular isonitrile–olefin coupling reaction delivering 3-substituted indoles, in which isonitrile was firstly applied as the hydrogen atom acceptor in the radical generation step by MHAT. The protocol features low catalyst loading, mild reaction conditions, and excellent functional group tolerance.
Introduction
Metal hydride species have been a longstanding topic of study in organometallic chemistry. Since the 1960s, noble metal-hydrides have been substantially explored in catalytic homogeneous hydrogenation.1 With the increasing demand for sustainable chemical processes, researchers were prompted to develop reactions catalyzed by earth-abundant metal hydrides.2 A landmark in this continuously growing field was the radical generation initiated by Fe, Co, and Mn hydrides through a metal catalyzed hydrogen atom transfer (MHAT) process, which was first discovered in Drago–Mukaiyama hydrofunctionalization of olefins.3 In this process, the in situ generated metal-hydride is prone to homolysis due to its low bond dissociation enthalpy (BDE), producing a hydrogen radical which further undergoes radical addition to olefins.4 Based on this scenario, a toolkit of catalytic methods has been established, such as Carreira's Co/salen catalysts,5 Boger's Fe/NaBH4 system,6 and Shenvi's dual catalytic platform.7 Remarkably, Baran's Fe(acac)3/phenylsilane system has proved to be a useful methodology for both inter- and intramolecular reductive olefin coupling,8 as well as a general protocol for construction of polycyclic frameworks.9 By means of this catalysis system, a variety of SOMOphiles were further introduced as radical traps, achieving diverse olefin hydrofunctionalization such as hydroamination, hydrogenation, and the Minisci reaction.10While the literature pertaining to the MHAT process is replete with creative advances, we recognized a gap existing in the current research: tremendous efforts have been devoted to the development of extensive SOMOphiles for olefin hydrofunctionalization, however, the variant process of transferring hydrogen atoms to other unsaturated groups remains largely elusive (Scheme 1A). For instance, it is logically reasonable to assume that the isonitrile group could abstract a hydrogen atom from Fe hydride species to form a carbon-centered imidoyl radical, which may further undergo undiscovered radical addition or cyclization reactions. In this context, we envisioned that enlarging the tool-box of the MHAT strategy to include these different hydrogen radicalophiles would open up a new development orientation for metal hydride chemistry.
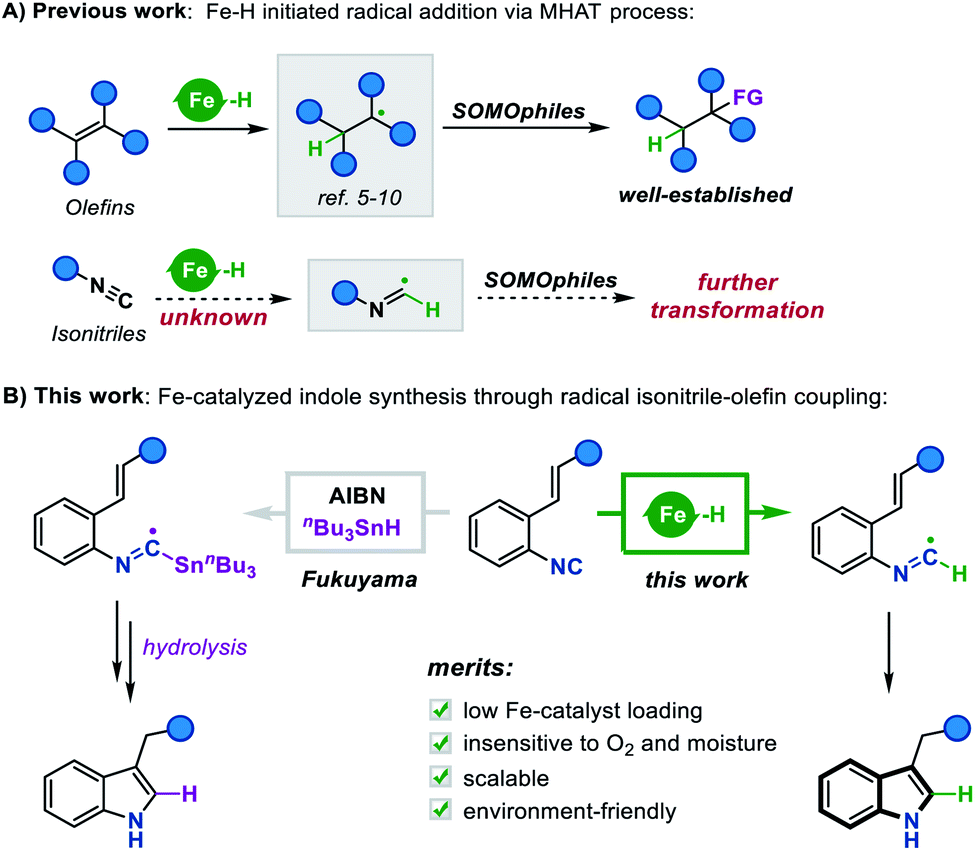 | ||
| Scheme 1 (A) Fe–H initiated radical addition via the MHAT process. (B) This work: Fe-catalyzed indole synthesis through radical isonitrile–olefin coupling. | ||
Isonitriles, as classical radical acceptors, have been widely utilized in a number of imaginative radical cascade reactions for the construction of diverse nitrogen-containing heterocycles.11 Among them, the Fukuyama indole synthesis encompassing radical isonitrile insertion followed by an intramolecular radical cyclization has proved to be efficient for accessing indole skeletons (Scheme 1B).12 Despite its efficiency, the radical generation step often requires an initiator and stoichiometric amounts of toxic Bu3SnH, making the approach neither environment-friendly nor broad-functional group compatible. Although there have been some attempts to overcome these limitations,13 the vast majority lay exclusively on the construction of 2,3-disubstituted indoles, prompting us to explore the idea of merging this radical cyclization with the Fe hydride catalytic system to create a novel platform to 3-monosubstituted indole derivatives. To this end, we herein report an Fe-catalyzed intramolecular isonitrile–olefin coupling reaction with the MHAT process as the radical initiation step, which provides a convenient and rapid approach to 3-substituted indole motifs.
Results and discussion
To test our hypothesis, we initiated the investigation with the reaction by using ethyl o-isonitrile cinnamate (1aa) as the standard substrate. We first examined the reaction conditions previously described by Baran and coworkers for olefin reductive coupling,8b which was carried out in i-PrOH at 60 °C with 30 mol% of Fe(acac)3 and PhSiH3 as the hydrogen source. To our delight, we successfully obtained the desired product in 25% yield (Table 1, entry 1). When other metal acetylacetonates were then evaluated under these experimental conditions, the sterically bulky iron catalyst Fe(dpm)3 was found to be able to significantly improve the yield to 75%, which might result from the high stability and decreased decomposition of the catalyst (Table 1, entries 2 and 3). A higher yield could be obtained even if the loading of Fe(dpm)3 was reduced to 5% equivalent (Table 1, entry 4). Further screening disclosed that other metal acetylacetonates such as Co(dpm)2 and Mn(dpm)3 could also be employed in this system with declined yields (Table 1, entries 5 and 6). No reaction occurred without metal catalysts, excluding the process of direct hydrogen atom transfer from hydrosilane (Table 1, entry 7).14|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Reductant | Yield (%) |
| a Reaction conditions: 1aa (0.2 mmol), catalyst (5 mol%), hydrosilane (0.4 mmol), solvent (1.2 mL), 35 °C, 2 h. Yield determined by GC using n-hexadecane as the internal standard. b Used 30 mol% catalyst. c Isolated yield. d Reaction at 60 °C for 5 h. acac = acetylacetone, dibm = diisobutyrylmethane, dpm = dipivaloylmethane. | ||||
| 1b | Fe(acac)3 | i-PrOH | PhSiH3 | 25 |
| 2b | Fe(dibm)3 | i-PrOH | PhSiH3 | 59 |
| 3b | Fe(dpm)3 | i-PrOH | PhSiH3 | 75 |
| 4 | Fe(dpm)3 | i-PrOH | PhSiH3 | 82 |
| 5b | Co(dpm)3 | i-PrOH | PhSiH3 | 21 |
| 6b | Mn(dpm)3 | i-PrOH | PhSiH3 | 46 |
| 7b | — | i-PrOH | PhSiH3 | NR |
| 8 | Fe(dpm)3 | EtOH | PhSiH3 | 56 |
| 9 | Fe(dpm)3 | t-BuOH | PhSiH3 | 77 |
| 10 | Fe(dpm)3 | DCE | PhSiH3 | NR |
| 11 | Fe(dpm)3 | i-PrOH/DCE | PhSiH3 | 82 (81)c |
| 12 | Fe(dpm)3 | i-PrOH | Et3SiH | 12 |
| 13 | Fe(dpm)3 | i-PrOH | (EtO)3SiH | 28 |
| 14d | Fe(dpm)3 | i-PrOH | DEMS | 81 |
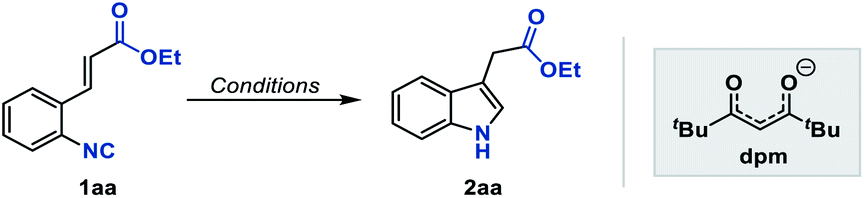
With Fe(dpm)3 set as the optimal catalyst, other reaction parameters were further screened to maximize the efficiency of this reaction. The evaluation with various solvents revealed that the reaction did not proceed at all in aprotic solvents such as THF or DCE, while it worked well in alcohol solvents. In branched alcohols, such as i-PrOH and t-BuOH, the reaction proceeded smoothly, and i-PrOH proved to be the ideal solvent (Table 1, entries 8–10). Notably, the use of DCE as a co-solvent gave a comparable result, which could benefit some insoluble substrates (Table 1, entry 11). Finally, various hydrosilanes were investigated as the reductant, among which the use of Et3SiH and (EtO)3SiH was found to give poor yields (Table 1, entries 12 and 13), whereas inexpensive DEMS ((EtO)2MeSiH) was compatible, providing the desired product in 81% yield.
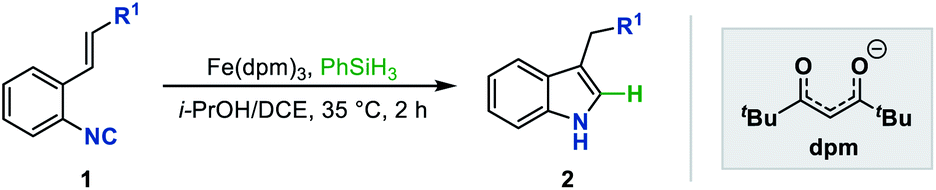
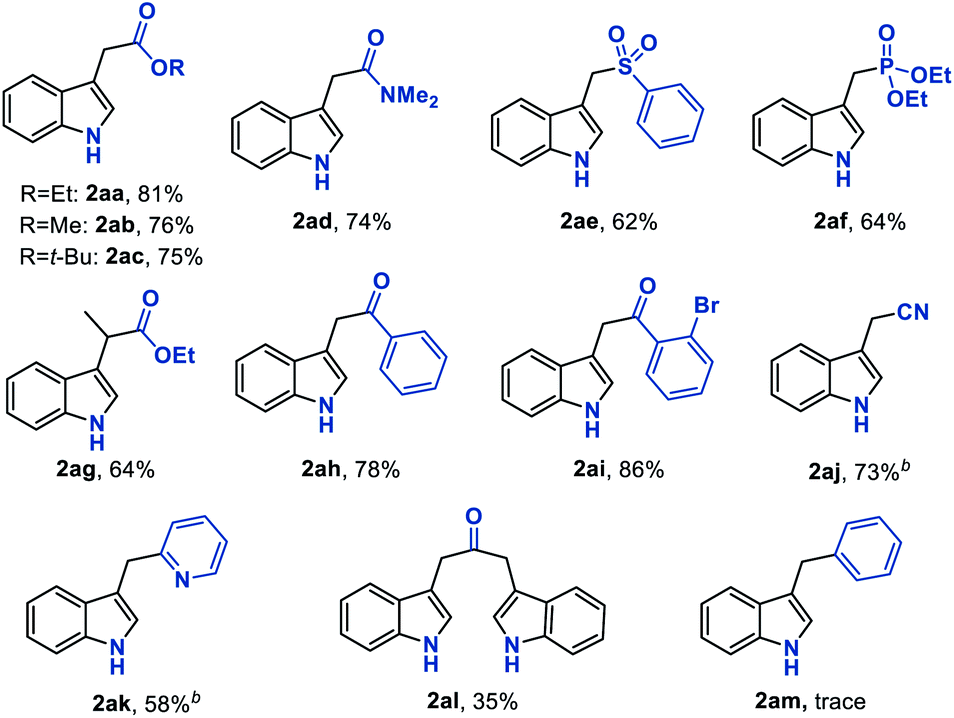
With the established optimum reaction conditions in hand, we then evaluated the substrate generality of this Fe hydride initiated isonitrile–olefin coupling reaction (Table 2). First, the influence of various substituents on the alkene-moiety was explored. To our delight, different esters (2aa–2ac), and amido (2ad), sulfonyl (2ae), and phosphoryl groups (2af) were all compatible with this reaction and delivered the desired products in 62–81% yields. Additionally, the substrate featuring a sterically hindered trisubstituted alkene also underwent the current reaction smoothly, leading to the desired product 2ag in 64% yield. Furthermore, substrates bearing the ketone-functionality (2ah–2ai) showed good compatibility to produce the corresponding indole products as well. Encouraged by these results, we continued to examine the substrates tethered with less electron-withdrawing groups such as cyano (1aj) and pyridyl groups (1ak). The results shown in Table 2 demonstrated that the desired products (2aj and 2ak) could be obtained in good yields by prolonging the reaction time to 12 hours. Malathidone (2al) was also achieved albeit with a lower yield under standard reaction conditions. Notably, the introduction of electron-withdrawing groups into the alkene was necessary for getting acceptable yields of products. Only a trace amount of product (2am) was afforded when using alkene tethered with a phenyl group, which might be ascribed to its less propensity to undergo single-electron reduction with Fe(II) species.
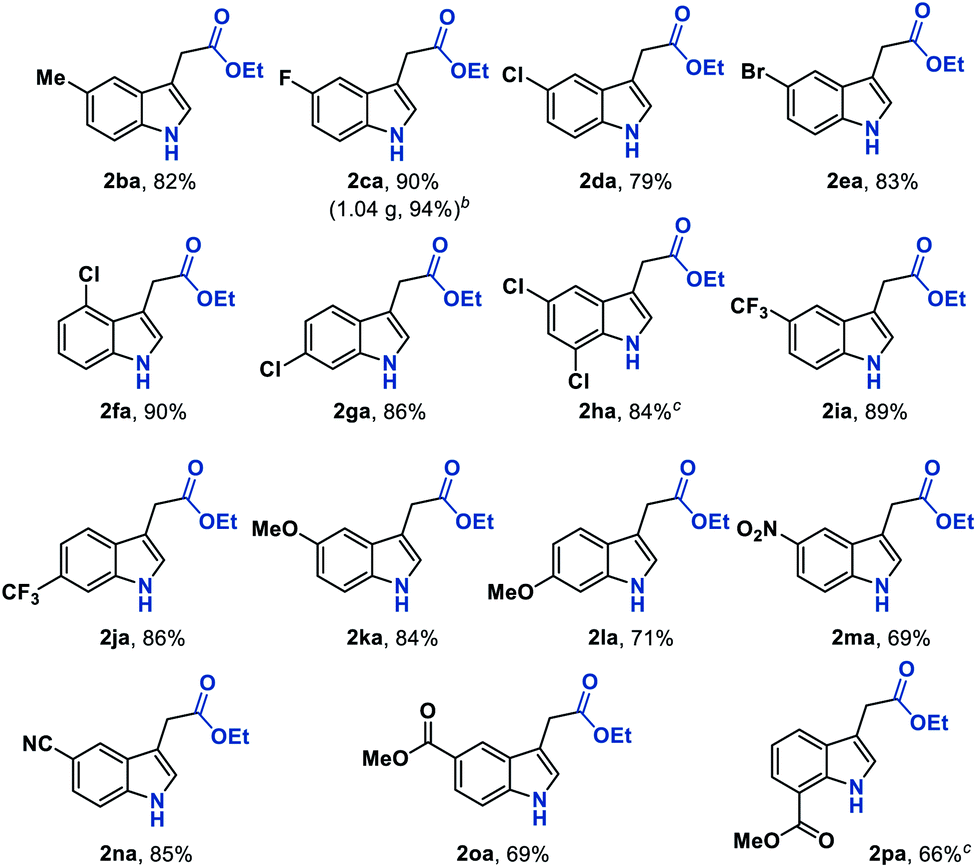
We next sought to explore the generality of this methodology with respect to substituted-aryl isonitriles. To this end, a variety of ethyl o-isonitrile cinnamates were synthesized, and as shown in Table 3, the electronic effects had no obvious influence on this transformation. For both electron-donating (such as Me– and MeO–) and electron-withdrawing (such as F–, Cl–, Br– and CF3–) substituents on the aromatic rings, the substrates were all smoothly converted to the corresponding products (2ba–2la) in good to excellent yields (71–94% yields). Besides, multiple valuable functional groups such as reducible nitro (2ma),10a cyano (2na) and alkoxycarbonyl (2oa, 2pa) groups were all tolerated under the mild reaction conditions, showcasing the strong chemoselectivity of this MHAT approach. It is worth mentioning that both the 6-substituted substrates with significant steric effects close to the isonitrile group (1ha, 1pa) can undergo the catalytic reaction, delivering the corresponding 7-substituted indoles (2ha, 2pa) in good yields (84% and 66% yields, respectively). In particular, with the catalyst loading reduced to 1%, the desired product 2ca could be obtained on a gram-scale in 94% isolated yield, showing the scalability and robustness of this protocol.

Aiming to gain insights into the mechanism of this Fe hydride-initiated isonitrile–olefin coupling reaction, we performed some mechanistic experiments. When a radical scavenger such as TEMPO was added in the standard reaction conditions, the starting material was fully recovered and the byproduct 3 was detected, suggesting that the H-radical might be involved in the present reaction (Scheme 2A). In addition, almost the same yield could be obtained under oxygen-free conditions, disclosing that oxygen was not responsible for reoxidizing the iron catalyst (see the ESI†). The deuterium labeling experiment by using PhSiD3 as the hydrogen source provided deuterated indole 2aa-1 with over 95% D at the 2-position, indicating that the hydrogen atom that added across the isonitrile group originated from PhSiH3 (Scheme 2B). Another deuterium labeling experiment by using EtOD as solvent led to 2aa-2 with over 95% deuteration rate, in which the deuterium atom was incorporated into the position adjacent to the carbonyl group. This is consistent with a polar protonation rather than a radical hydrogen atom abstraction from hydrosilane ending this reaction. It is also worth pointing out that these reactions provide facile access to deuterium-labeled indole compounds, which would be attractive for kinetic studies and drug design.
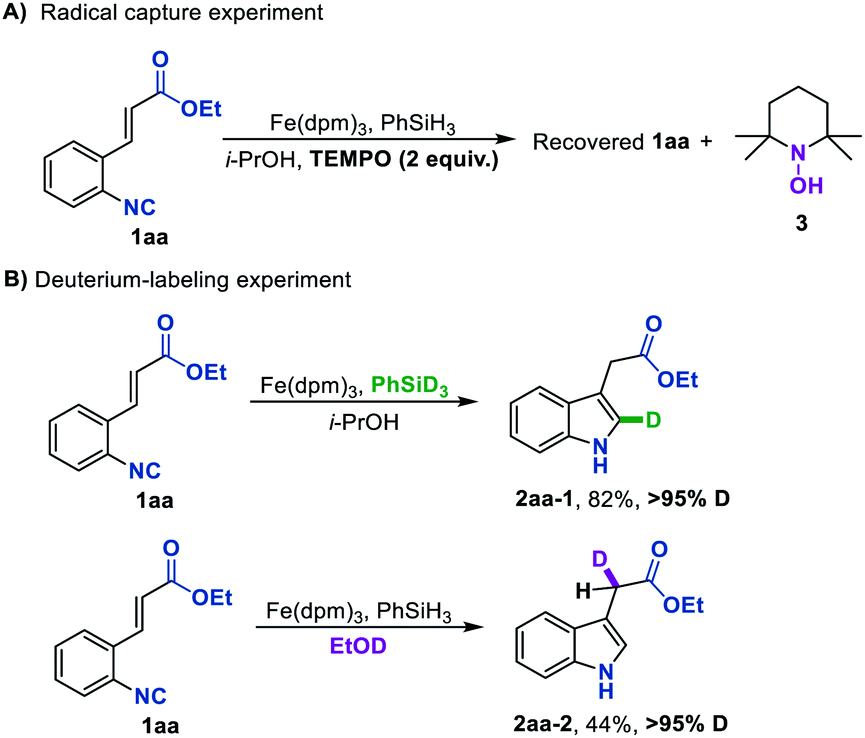 | ||
| Scheme 2 Mechanistic experiments. | ||
On the basis of the above results and precedent reports,4 a plausible reaction mechanism for this process is proposed in Fig. 1. The reaction begins with the generation of Fe hydride from the Fe(III) pre-catalyst and hydrosilane. Then the isonitrile substrate would abstract a hydrogen radical from Fe hydride which is proposed to undergo the metal-catalyzed hydrogen atom transfer (MHAT) process to generate a carbon-centered imidoyl radical A. Next, intramolecular Giese-type addition of the imidoyl radical, followed by aromatization leads to radical intermediate B′. The single-electron transfer process between radical B′ and the Fe(II) intermediate affords the anion C and concurrently regenerates the Fe(III) catalyst. Finally, anion C could be protonated by the solvent to obtain the desired indole product.
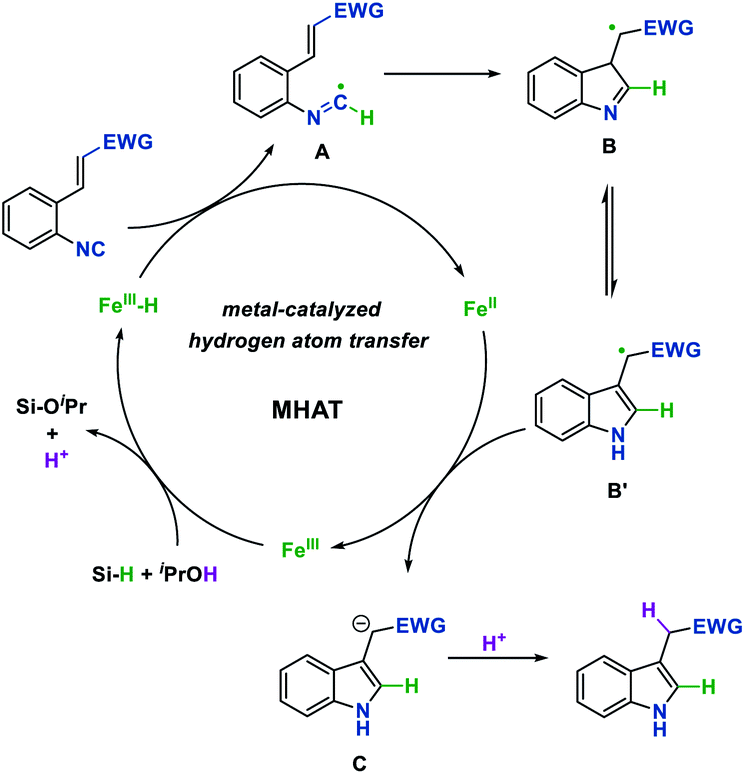 | ||
| Fig. 1 Proposed mechanism of the reaction. | ||
Conclusions
In summary, we have developed a novel and efficient Fe-catalyzed intramolecular reductive isonitrile–olefin coupling reaction to synthesize indole derivatives, which proceeds through a Fe–H initiated hydrogen atom transfer process. The key radical generation step transferring a hydrogen atom to isonitrile via the MHAT process has not previously been reported. The lower catalyst loading and the insensitiveness to air and moisture make this reaction practical and attractive in organic synthesis. Not only is this method a complement to Fukuyama-indole synthesis, but the catalytic patterns open up a new development orientation for metal-hydride chemistry. Further studies on the applications of this MHAT-driven isonitrile hydrogen-radical addition are currently in progress.Data availability
Further details of experimental procedure, characterization and copies of NMR spectra are provided in the ESI.Author contributions
T. Zhang conducted most of the experimental work and wrote the initial manuscript draft. M. Yu conducted the mechanistic study experiments. H. Huang conceptualized and directed the project and finalized the manuscript draft. All authors contributed to discussions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21925111 and 21790333).Notes and references
- For examples and reviews, see: (a) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC; (b) J. Halpern, Science, 1982, 217, 401 CrossRef CAS; (c) S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548 CrossRef CAS.
- For examples and reviews, see: (a) J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211 RSC; (b) X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed; (c) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746 CrossRef CAS PubMed; (d) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912 CrossRef CAS PubMed.
- (a) C. L. Bailey and R. S. Drago, Coord. Chem. Rev., 1987, 79, 321 CrossRef CAS; (b) S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 569, 537 Search PubMed.
- For a detailed study on the catalytic mechanism of metal hydrides, see: (a) H. Jiang, W. Lai and H. Chen, ACS Catal., 2019, 9, 6080 CrossRef CAS; (b) D. Kim, S. M. W. Rahaman, B. Q. Mercado, R. Poli and P. L. Holland, J. Am. Chem. Soc., 2019, 141, 7473 CrossRef CAS PubMed; (c) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401 RSC; (d) J. Choi, L. Tang and J. R. Norton, J. Am. Chem. Soc., 2007, 129, 234 CrossRef CAS.
- (a) J. Waser and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 5676 CrossRef CAS; (b) J. Waser, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 8294 CrossRef CAS; (c) J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2006, 128, 11693 CrossRef CAS PubMed; (d) B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4519 CrossRef CAS; (e) B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 5758 CrossRef CAS PubMed; (f) B. Gaspar and E. M. Carreira, J. Am. Chem. Soc., 2009, 131, 13214 CrossRef CAS PubMed.
- (a) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904 CrossRef CAS; (b) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588 CrossRef CAS PubMed; (c) E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428 CrossRef CAS PubMed.
- (a) S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779 CrossRef CAS PubMed; (b) S. A. Green, S. Vásquez-Céspedes and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 11317 CrossRef CAS PubMed; (c) S. A. Green, T. R. Huffman, R. O. McCourt, V. Puyl and R. A. Shenvi, J. Am. Chem. Soc., 2019, 141, 7709 CrossRef CAS PubMed.
- (a) J. C. Lo, J. Gui, Y. Yabe, C. Pan and P. S. Baran, Nature, 2014, 516, 343 CrossRef CAS; (b) J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304 CrossRef CAS PubMed; (c) J. C. Lo, D. Kim, C. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484 CrossRef CAS PubMed.
- (a) N. A. Godfrey, D. J. Schatz and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 12770 CrossRef CAS; (b) W. P. Thomas, D. J. Schatz, D. T. George and S. V. Pronin, J. Am. Chem. Soc., 2019, 141, 12246 CrossRef CAS PubMed; (c) J. Huang, T. Y. Lauderdale, C. Lin and K. Shia, J. Org. Chem., 2018, 83, 6508 CrossRef CAS PubMed; (d) S. Nagasawa, K. E. Jones and R. Sarpong, J. Org. Chem., 2019, 84, 12209 CrossRef CAS PubMed; (e) P. Chen, C. Wang, R. Yang, H. Xu, J. Wu, H. Jiang, K. Chen and Z. Ma, Angew. Chem., Int. Ed., 2021, 60, 5512 CrossRef CAS PubMed.
- For representative examples, see: (a) J. Gui, C. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886 CrossRef CAS PubMed; (b) H. T. Dao, C. Li, Q. Michaudel, B. D. Maxwell and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 8046 CrossRef CAS PubMed; (c) C. Obradors, R. M. Martinez and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 4962 CrossRef CAS; (d) Y. Shen, J. Qi, Z. Mao and S. Cui, Org. Lett., 2016, 18, 2722 CrossRef CAS PubMed; (e) S. Bordi and J. T. Starr, Org. Lett., 2017, 19, 2290 CrossRef CAS PubMed; (f) Y. Wang and J. W. Bode, J. Am. Chem. Soc., 2019, 141, 9739 CrossRef CAS PubMed; (g) P. V. Kattamuri and J. G. West, J. Am. Chem. Soc., 2020, 142, 19316 CrossRef CAS PubMed; (h) M. Saladrigas, J. Bonjoch and B. Bradshaw, Org. Lett., 2020, 22, 684 CrossRef CAS PubMed; (i) F. Puls, P. Linke, O. Kataeva and H. Knölker, Angew. Chem., Int. Ed., 2021, 60, 2 CrossRef.
- (a) G. P. dos Gomes, Y. Loginova, S. Z. Vatsadze and I. V. Alabugin, J. Am. Chem. Soc., 2018, 140, 14272 CrossRef; (b) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC; (c) J. Lei, J. Huang and Q. Zhu, Org. Biomol. Chem., 2016, 14, 2593 RSC; (d) M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295 RSC; (e) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257 RSC.
- T. Fukuyama, X. Chen and G. Peng, J. Am. Chem. Soc., 1994, 116, 3127 CrossRef CAS.
- For recent reports about indole synthesis from isonitrile substrates, see: (a) B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216 CrossRef CAS PubMed; (b) X. Zhang, P. Zhu, R. Zhang, X. Li and T. Yao, J. Org. Chem., 2020, 85, 9503 CrossRef CAS; (c) C. Wang, Y. Li and S. Yang, Org. Lett., 2018, 20, 2382 CrossRef CAS; (d) L. M. Heckman, Z. He and T. F. Jamison, Org. Lett., 2018, 20, 3263 CrossRef CAS PubMed; (e) M. S. Santos, H. L. I. Betim, C. M. Kisukuri, J. A. C. Delgado, A. G. Corrêa and M. W. Paixão, Org. Lett., 2020, 22, 4266 CrossRef CAS PubMed.
- I. D. Jenkins and E. H. Krenske, ACS Omega, 2020, 5, 7053 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03058b |
| This journal is © The Royal Society of Chemistry 2021 |