
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the limit of proton-coupled electronic doping in a Ti(IV)-containing MOF†
Jenna L.
Mancuso
,
Kevin
Fabrizio
 ,
Carl K.
Brozek
* and
Christopher H.
Hendon
*
,
Carl K.
Brozek
* and
Christopher H.
Hendon
*
Department of Chemistry and Biochemistry, University of Oregon, Eugene, OR 97403, USA. E-mail: cbrozek@uoregon.edu; chendon@uoregon.edu
First published on 30th July 2021
Abstract
TiIV-containing metal–organic frameworks are known to accumulate electrons in their conduction bands, accompanied by protons, when irradiated in the presence of alcohols. The archetypal system, MIL-125, was recently shown to reach a limit of 2e− per Ti8 octomeric node. However, the origin of this limit and the broader applicability of this unique chemistry relies not only on the presence of TiIV, but also access to inorganic inner-sphere Lewis basic anions in the MOF nodes. Here, we study the loading of protons and electrons in MIL-125, and assess the thermodynamic limit of doping these materials. We find that the limit is determined by the reduction potential of protons: in high charging regimes the MOF exceeds the H+/H2 potential. Generally, we offer the design principle that inorganic anions in MOF nodes can host adatomic protons, which may stabilize meta-stable low valent transition metals. This approach highlights the unique chemistry afforded by MOFs built from inorganic clusters, and provides one avenue to developing novel catalytic scaffolds for hydrogen evolution and transfer hydrogenation.
Introduction
By coupling to the motion of protons, electrons can enter reactivity patterns, charge storage, and electron transport pathways that would be difficult to access otherwise through a mechanism known as proton-coupled electron transfer (PCET).1 In net, the reaction can be thought of as a hydrogen atom transfer, and is consequently invoked in various chemical processes found in enzymes,2,3 chloroplasts,4,5 hydrogen fuel cells,1,6 and reductive catalysts.7–9 However, achieving selective PCET depends on knowledge of the relative redox potentials of the reagents. With the appropriate frontier energy level alignments, PCET can be either thermally or photochemically promoted in both molecular and solid-state manifolds.10–14 Mismatched energetics create pitfalls ranging from sluggish kinetics to undesirable H2 formation, or back-hydrogenation to reform reactants.15 Hence, there is a desire to leverage the PCET manifold to, for example, suppress the hydrogen evolution reaction (HER) by stabilizing the e−–H+ pair (H*) below that of the NHE (−4.4 eV relative to vacuum16).In conventional solid-state scaffolds, the ill-defined nature of surface-active sites hinders catalyst design for selective PCET reactivity. For instance, the surface of TiO2 — a semiconductor known to perform HER17 — features a broader array of active sites, beyond the idealized (101)-Anatase surface presented in Fig. 1,1,18,19 limiting efforts to identify and tune specific localities for selective reactivity other than H2 formation. Conversely, molecular catalysts offer well-defined single-atom active sites with wide synthetic tunability, but suffer from poor recyclability.20 Combining the best of both approaches, metal–organic frameworks (MOFs) merge the benefits of heterogeneous catalysis and synthetic modularity.21–24 Previous studies in MOF catalysis have primarily focused on thermally driven reactions at either the linker or node using intrinsic25–27 and extrinsic28–30 catalytically active metals. Conversely, MOF photoredox chemistry31–33 typically focuses on electron transfer between the inorganic and organic MOF components,34 shuttling electrons to interstitial molecular catalysts35–37 or guests.38–41
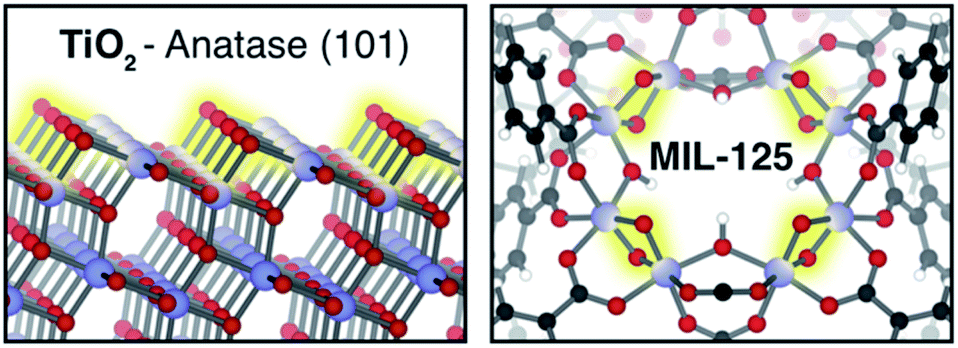 | ||
| Fig. 1 TiO2-based materials can be reduced via PCET, with a corresponding TiIV/III reduction, and O–H formation. Depicted are Lewis basic μ2-oxo sites on TiO2 (shown is the (101)-Anatase surface). In a MOF analogue, octameric Ti8(μ2-O)8(μ2-OH)4 clusters offer four isolated pairs of bridging O per node (highlighted in yellow). These sites are thought to host extrinsic cations (e.g. H+, Na+, etc.), affecting the reduction potential of vicinal TiIV centers. | ||
However, one burgeoning area of MOF research has been the exploration of the chemistry afforded by the compositional similarity between inorganic clusters in MOFs, and their conventional dense-phase semiconducting counterparts, Fig. 1.42,43 In particular, a central theme of our research is the study of chemical opportunities enabled by the Lewis basic inorganic ligands in the inorganic clusters. Such anions are prevalent in both Ti-MOFs and others, and it is these scaffolds that have been shown to stabilize H* at molecularly precise active sites.44–48
In the seminal report, UV irradiation in the presence of alcohols caused MIL-125 to turn from colorless to black.49 The color was shown to persist indefinitely, only returning to colorless upon exposure to air. The authors attributed the apparent bandgap reduction to the formation of TiIII centers stabilized by protons liberated from the oxidation of alcohol by the photogenerated holes, i.e., PCET from the solvent to the framework, Scheme 1. EPR later validated the mechanism of TiIII formation.50 Paired with electronic structure calculations, TiIII-formation was rationalized by identifying the conduction band minimum (CBM) origin as vacant Ti d-states in the pristine MOF — a general property extending to all reported TiIV-containing MOFs,25 and likely a general consideration when designing photoactive MOFs with inorganic-centered conduction bands.
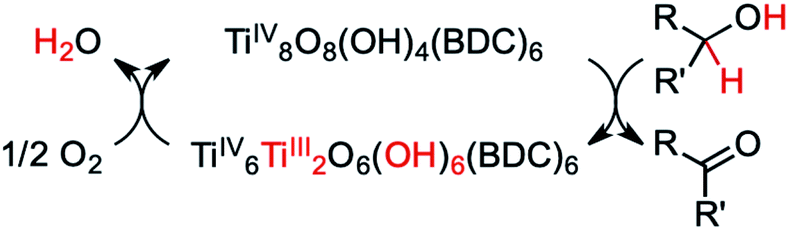 | ||
| Scheme 1 As posited by Dan-Hardi et al.,49 MIL-125 forms TiIII upon photo-promoted reaction with primary and secondary alcohols (R′ = H or alkyl, respectively). The active catalyst can then proceed to transfer two hydrogen atoms to oxygen, forming water. | ||
The reversible colorless-to-black transformation, spectroscopic evidence of TiIII, and aldehyde formation observed in irradiated MIL-125 (ref. 49) demonstrates that the MOF is undergoing photochemical electronic doping, i.e., photodoping. Kinetically, this process requires that rate of exciton recombination be slower than hole quenching, wherein a sacrificial reductant (alcohol) diffuses to the photogenerated hole. Although subsequent reports have detected small quantities of H2 evolved from this process,51,52 these data suggest that a significant portion of the TiIII–OH+ pairs generated through photodoping remain indefinitely stable,49,53,54 suppressing H2 formation. This is further supported by the requirement for MIL-125 and other Ti-containing MOFs to use a co-catalyst to promote H2 formation.39,55
The maximum charge capacity, i.e., relative stability of TiIII centers, of MIL-125 shows a strong cation dependence. Recent studies by the Mayer, Saouma and colleagues demonstrated that Na+-based chemical reductants enable stoichiometric reduction of all eight Ti centers per node,53 while the PCET-based photodoping strategy only reaches two H* per Ti octamer.54 Since Na+ is undoubtedly less stabilizing than H+ (which presumably forms a covalent bond with bridging oxos, a process itself known to affect the electronic structure of Ti-containing MOFs73), we hypothesize that H2 formation sets the thermodynamic limit to PCET-based charge storage in MIL-125: in the low-charging regime, the energies of electron–proton pairs are more stable relative to HER, but exceed the redox potential of HER in the high-charging regime. Further, stoichiometric reduction of the Ti centers can only be achieved with Na+ because the Na+/Na redox couple lays far more negative (closer to the vacuum level) than the new conduction band of the all-TiIII-MOF. Consequently, we surmise that the charge storage capacity of MOF nodes depends on both the redox potentials of the metal ions and the node nuclearity or, more specifically, the presence of inorganic anions to accept protons.
Here, we sought to understand the limits to PCET-based charge storage in MIL-125. To do so, we have combined electronic structure calculations and experiments to identify the microscopic origin of (1) the stability of TiIII–OH+ pairs towards H2 evolution in the low-charging regime, and (2) the thermodynamic limit to the number of TiIII–OH+ that can accumulate. We find that the inorganic oxo units in the MIL-125 clusters accept the protons liberated from alcohols during PCET photodoping, thereby stabilizing the e−/H+ pairs against H2 formation by spatially separating them on Ti and O atoms. From this study, we offer a more general design principle for forming MOFs with the potential to act as transfer hydrogenation catalysts, but not dehydrogenation catalysts: the incorporation of Lewis-basic inorganic inner sphere ligands paired with redox-active metals may achieve controllable PCET reactions in MOFs. In other words, we offer a general approach to accessing non-equilibrium reactivity in MOFs featuring metal oxide clusters.
Results and discussion
The CBM of a semiconductor provides an estimate for the reduction potential of that material (i.e., the electron affinity).56,57 Yet, the CBM alone is insufficient to assess the thermodynamic tendency to host additional electrons because the ground state structure does not account for local reorganization (for example, distortions due to ion pairing, or reduction in bond order) upon conduction band population. Computational chemistry is well-suited to study both instantaneous reduction of the MOF, as well as the emergent structural deviations. Using plane-wave DFT we are able to compute the ground state electronic structure for MIL-125 and subsequent PCET products. Using a vacuum level alignment,58 the conduction band minimum of MIL-125 sits at −3.7 eV, closer to the vacuum level than H2 formation from H+ (−4.4 eV, Fig. 2a). Here, we elect to use the standard hydrogen electrode gas phase value of −4.4 eV,59,60 but note that solvent and proximity of reagents likely affect this value significantly (up to 200 meV in some cases61,62). We also note while ligand functionalization may in principle affect d-orbital splitting of the Ti-centers, our calculations thus far suggest that aminating does not dramatically affect the CBM position, as it is composed of inorganic metal-oxo orbitals. Together, one might anticipate that electrons occupying the conduction band would be sufficiently high in potential to reduce protons.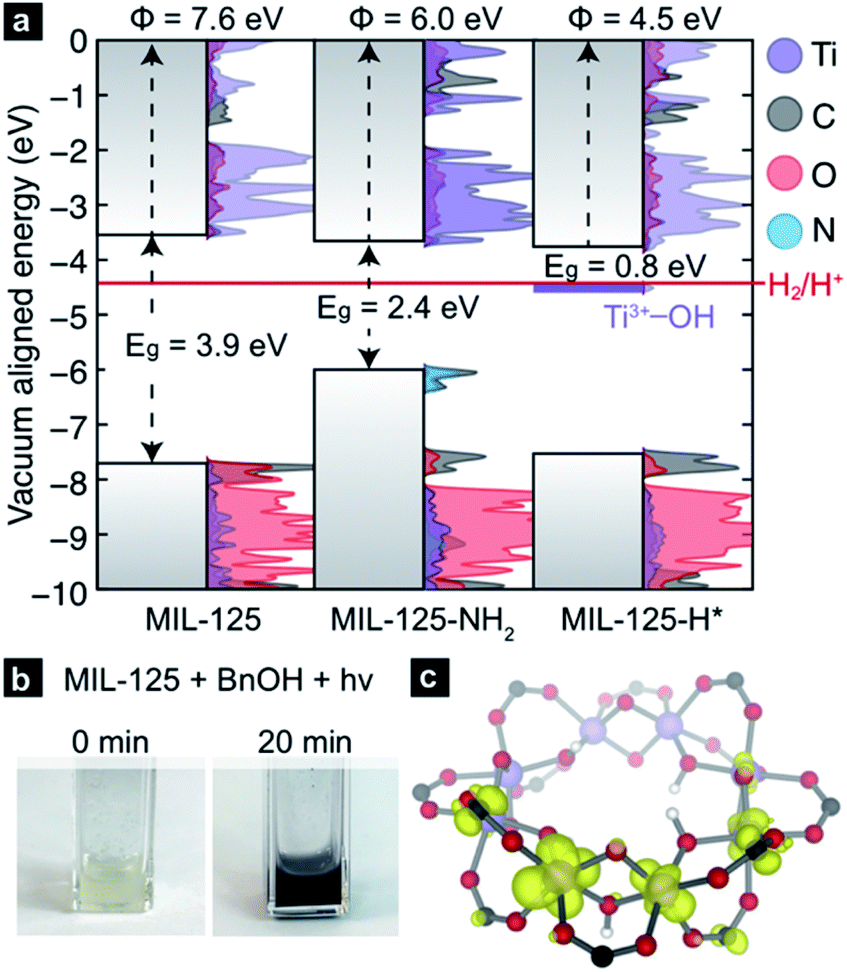 | ||
| Fig. 2 (a) Vacuum-aligned DOS plots show that both the bulk and organo-functionalized derivatives of MIL-125 have CBMs above H+/H2 (−4.44 eV, shown in red). A new mid-gap state emerges upon H* addition, with a potential 100 mV below that of H+/H2. (b) The addition of a proton and electron via photoillumination can be evidenced by a change in colour, which persists as long as the MOF is kept free from oxidants. (c) Electronic structure calculations of the H* adduct reveals that the additional electron localizes on a Ti-center near the adatomic proton, bound to one of the μ2-oxo ligands. | ||
However, upon irradiation in the presence of a primary or secondary alcohol, the MOF becomes black, Fig. 2b. Due to the persistent color change, these experiments indicate that there is at least one doped configuration that suppressed H2 formation, otherwise MIL-125 would fade back to colorless over time without exposure to air. We hence surmise that MIL-125-H* is a thermodynamic minimum on the potential energy surface of hydrogen transfer. In the simplest case, where a single H* is added to a bridging oxo, our calculation reveals that the newly formed “mid-gap” state, a half-occupied Ti-centered orbital (Fig. 2c), has an electronic potential of −4.5 eV, ∼100 meV more positive of H+/H2 reduction potential (shown in purple, Fig. 2a). Hence, we offer that the PCET product should be thought of as a stable TiIII–OH+ moiety rather than an ill-defined H*.
However, the recent experimentation by Saouma et al. revealed an apparent thermodynamic limit of two protons and two electrons per Ti8 node.54 It remains unclear whether the two protons would preferentially adhere to vicinal μ2-oxos or in some other configuration. It is further unclear why only two reductive events can occur in excess alcohol. Lastly, we wanted to understand the thermodynamic origin of that two-electron limit. To assess this energetic landscape, we developed an assortment of models containing two hydrogen atoms at various positions around the nodes, Fig. 3. From these calculations, we find that the addition of two hydrogen atoms to MIL-125 yields consistently HER-stable mid-gap TiIII states. The most stable configuration arises from the addition of two H* on geminal μ2-oxos (Fig. 3c, second column). The addition of two H* to μ2-oxos on different nodes (Fig. 3c, fifth column), connected by benzene dicarboxylate (BDC), is disfavored by up to 11.3 kcal mol−1, Fig. 3d. In that case, the TiIII radicals delocalize through the π*-system of the linker (see Fig. S1†). We find, however, that if the two hydrogen atoms are added to different nodes and not connected by a common linker, the reduction potential is stabilized to the same level as a single addition (approximately 60 meV positive of HER).
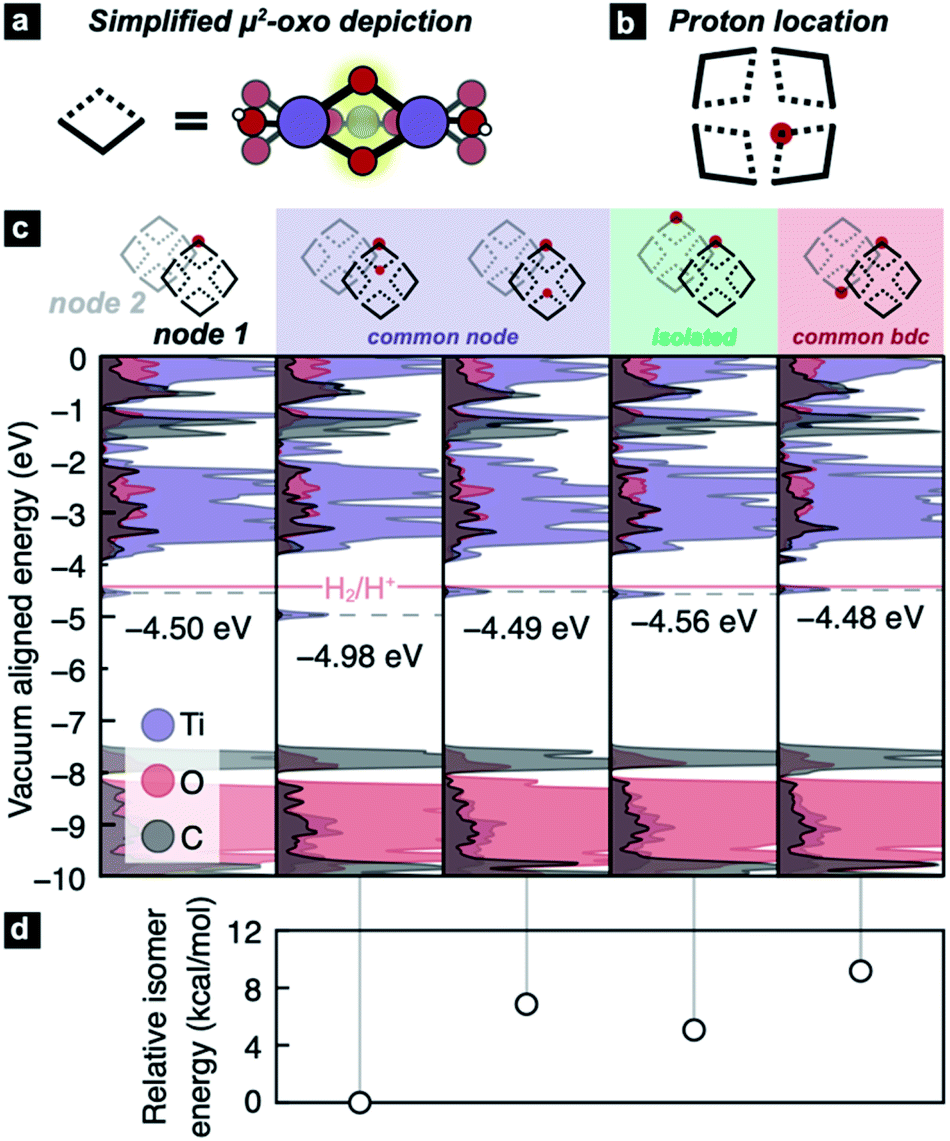 | ||
| Fig. 3 (a) A schematic of a portion of the Ti8 node, highlighting the μ2-oxos, and simplifying them to diamonds, whose central apexes represent the bridging oxos. (b) A node represented by our simplified notation. The unit cell of MIL-125 contains two nodes, thus (c) the vacuum-aligned atom-projected DOS plots contain two nodes per DOS plot, with the depicted PCET product shown above. The Fermi level (i.e. the gas phase reduction potential) is denoted by the dotted gray lines. | ||
We also examined the 2H*-per-node limit presented by Saouma, Fig. 4. Practically, this amounts to four hydrogen atoms per computational cell. Several observations may be made from these calculations. First, we note that isolated vicinal pairs of protons/electrons result in additive density of the mid-gap states. Second, same-node-separated protons result in splitting of the mid-gap DOS, and this process is energetically disfavored both in terms of formation energy (Fig. 4, lower panel) and potential to reduce protons. Third, we note that the four separated protons on a single node is more than twice the energy of two protons on a single node (from Fig. 4, column four, ∼21 kcal mol−1, and Fig. 3, column three, ∼8 kcal mol−1). We can hence make the general conclusion that the adatomic hydrogenic atoms thermodynamically prefer to add across pairs of bridging oxos. Kinetically, we also expect that alcohol radicals would react with pairs of bridging oxos before diffusing unreacted to distant oxo units.
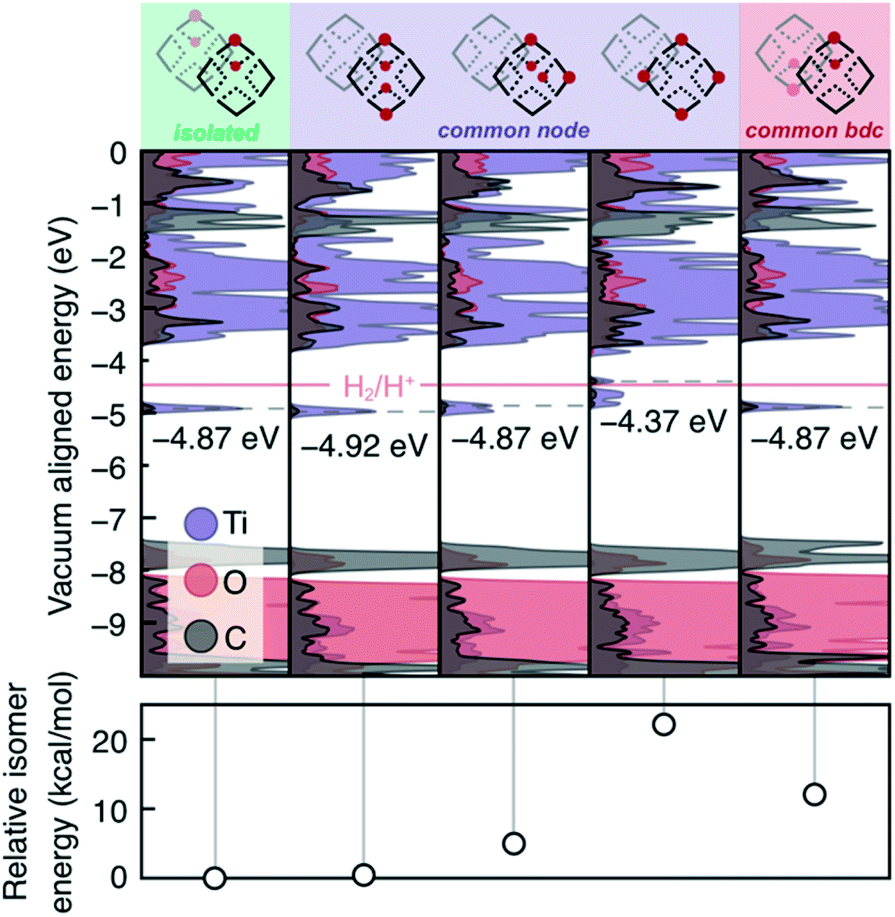 | ||
| Fig. 4 At the experimental limit presented by Saouma et al., two hydrogen atoms per node, the MOF appears electronically indistinguishable from one another, unless a single hydrogen atom is added to each one μ2-oxo per pair in a single node. While this structure may still be accessible, it is only kinetically stable compared to H2 formation. | ||
As yet, these calculations have not identified the chemical origin of the MIL-125 doping limit of two H* per node. Thus, we further explored higher H* loadings, up to the stoichiometric hydrogenation of the μ2-oxo units, and we determined a thermodynamic doping limit of four e−/H+ pairs per node within certain configurational parameters. In order for HER to remain suppressed with more than four PCET events at the same node, neighboring nodes must be in the fully oxidized TiIV-state (Fig. S3 and S4†). HER remains suppressed when six H* are added to a single node, but the presence of a hydrogenated μ2-oxo pair on the adjacent node facilitates H2 formation. Meanwhile, four H* per node is stable with respect to HER as long as two pairs coexist on each node, or all four localize on one, Fig. S3 and S4.† Higher loadings become progressively less stable (Table S1†), and beyond 4 electrons per the node (i.e., 50% doping), the occupied states are split such that many configurations exceed the reduction potential of H+, an extreme example is presented in Fig. S3.†
Finally, we also examined stabilizing the TiIII with adatomic Na+, Fig. S5.† A similar effect is observed where a metastable mid-gap TiIII–ONa+ state is half populated. In this case, the electron sits closer to vacuum than HER, but should not form H2 since there is no H+ available to reduce. Our calculations therefore support the previous observation that stoichiometric reduction of TiIII is achievable in the presence of Na+.
In summary, computations have uncovered two possible thermodynamic explanations for the observed limit to PCET-based charge storage in MIL-125: (1) e−/H+ pairs accumulate until protons are reduced, generating H2 or (2) e−/H+ pairs accumulate until saturating the available cluster orbitals, at which point alcohol oxidation would cease. To test these possibilities, we sought to experimentally quantify MOF cluster photo-charging and associated H2 evolution by irradiating MIL-125 for varying durations in the presence of (1) excess of alcohol and (2) stoichiometric quantities (four alcohol molecules per cluster). Scenario 1 serves to test whether MIL-125 can continuously consume alcohol, while scenario 2 serves to test whether the clusters cease charging after complete reduction of the clusters.
In the former, the reaction of benzyl alcohol with MIL-125 was monitored by comparing benzylaldehyde 1H NMR signals with those of an internal standard (see ESI† for further experimental detail). Quantification by 1H NMR of unreacted alcohol at each time point relative to the amount of aldehyde generated provides indirect measure of H2 evolution and other side-reactions, i.e., any deviation from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 would imply an e−/H+ “leak”. After 20 minutes of irradiation in the presence of stoichiometric quantities of benzylic alcohol, an average of 2% of the clusters in MIL-125 accumulated one-electron/one-proton. Importantly, stoichiometric conversion of each alcohol to the corresponding aldehyde was observed. In a separate experiment, excess alcohol and a 2 h irradiation led to just 6% of the clusters storing an average of one electron, whereas irradiation for 18 h led to reduction of 20% of the clusters, or ∼2 TiIII per cluster, aligning with the observations made by Saouma and colleagues.54
:1 would imply an e−/H+ “leak”. After 20 minutes of irradiation in the presence of stoichiometric quantities of benzylic alcohol, an average of 2% of the clusters in MIL-125 accumulated one-electron/one-proton. Importantly, stoichiometric conversion of each alcohol to the corresponding aldehyde was observed. In a separate experiment, excess alcohol and a 2 h irradiation led to just 6% of the clusters storing an average of one electron, whereas irradiation for 18 h led to reduction of 20% of the clusters, or ∼2 TiIII per cluster, aligning with the observations made by Saouma and colleagues.54
All materials retained the white-to-black color change after irradiation, supporting the computational insights that e−/H+ pairs remain stable towards HER in the low-charge regime. Because photocharging ceases prior to complete cluster reduction, we surmise that HER must serve as the thermodynamic limit, and yet scarce H2 can be tracked by us and other groups, and most of the alcohol remains unreacted. Kinetic factors therefore likely prevent further reduction and subsequent H2 generation. Specifically, depletion of alcohol near Ti-oxo clusters during photodoping would prevent continued accumulation of charges because exciton recombination would outcompete hole quenching due to the slow diffusion of new alcohol molecules. This kinetic-based explanation remains the focus of current studies in our labs.
To further test the hypothesis that e−/H+ pairs are stabilized against H2 evolution in MIL-125 during photodoping, we sought to measure their redox potentials during the photodoping process. Considering that decamethylferrocenium 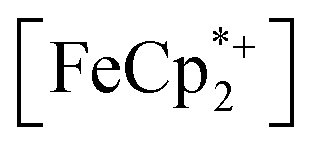 is known to have a similar redox potential to the computed value for photodoped MIL-125,54 we sought to use the former as an optical redox indicator of the photodoped MOF suspension. By measuring the concentration of
is known to have a similar redox potential to the computed value for photodoped MIL-125,54 we sought to use the former as an optical redox indicator of the photodoped MOF suspension. By measuring the concentration of 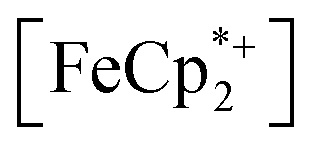 in situ through monitoring the absorption feature at 778 nm, attributable to a LMCT transition,63 the ratio of
in situ through monitoring the absorption feature at 778 nm, attributable to a LMCT transition,63 the ratio of 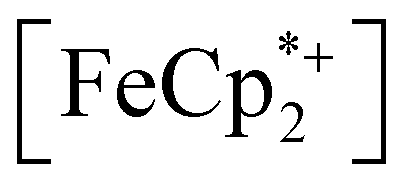 to
to 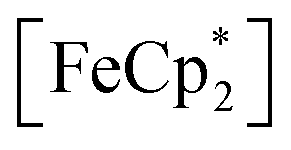 can be determined and related through the Nernst equation to calculate the solution Fermi level, Fig. 5.64,65 Assuming redox equilibration between all species in the suspension, this value also reports the redox potential of the photodoped MOF at that time point. Hence, we can compare this value to the known redox potential of HER in acetonitrile (−624 mV vs. Fc+/Fc)66 as further proof that the electron–proton pair is stabilized against recombination. Toward this aim, the mixture was dispersed in an air-free quartz cuvette, irradiated with a broadband photolysis lamp, and stopped intermittently to measure UV-vis spectra.
can be determined and related through the Nernst equation to calculate the solution Fermi level, Fig. 5.64,65 Assuming redox equilibration between all species in the suspension, this value also reports the redox potential of the photodoped MOF at that time point. Hence, we can compare this value to the known redox potential of HER in acetonitrile (−624 mV vs. Fc+/Fc)66 as further proof that the electron–proton pair is stabilized against recombination. Toward this aim, the mixture was dispersed in an air-free quartz cuvette, irradiated with a broadband photolysis lamp, and stopped intermittently to measure UV-vis spectra.
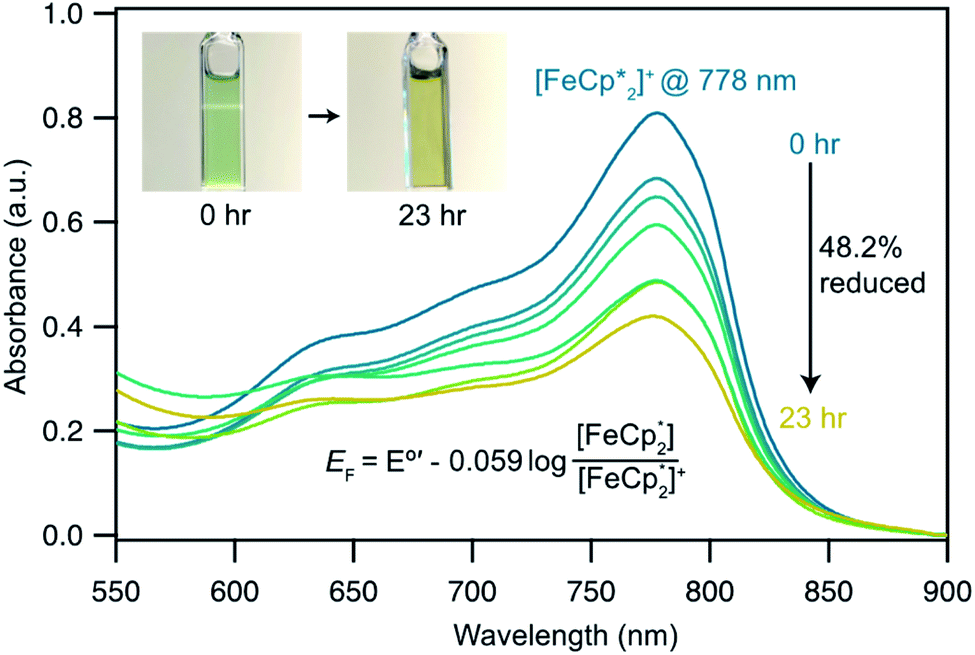 | ||
Fig. 5 Electronic absorption spectra collected at various illumination times between 0 and 23 h during photochemical reduction of MIL-125 powder nanocrystals in the presence of 25 mM 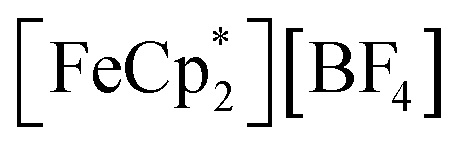 in a MeCN/EtOH mixture. in a MeCN/EtOH mixture. | ||
In this case, ethanol was selected as the hydrogen atom source due to its smaller size relative to benzyl alcohol, facilitating the inclusion of 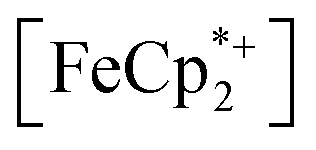 into the pores of MIL-125. Fig. 5 shows that irradiation of the MOF-ethanol mixture induces a decrease in the
into the pores of MIL-125. Fig. 5 shows that irradiation of the MOF-ethanol mixture induces a decrease in the 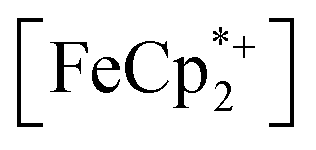 absorption bands, signaling reduction by photodoped MIL-125. As a control,
absorption bands, signaling reduction by photodoped MIL-125. As a control,  does not undergo photoreduction in the absence of MIL-125 (Fig. S9†). After 23 h of irradiation, the absorption bands of
does not undergo photoreduction in the absence of MIL-125 (Fig. S9†). After 23 h of irradiation, the absorption bands of 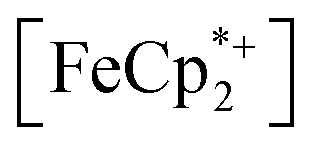 stopped decreasing, indicating a steady-state concentration of all redox-active species in the mixture. Analysis of these spectra indicate MIL-125 photoreduced 48.2% of the original
stopped decreasing, indicating a steady-state concentration of all redox-active species in the mixture. Analysis of these spectra indicate MIL-125 photoreduced 48.2% of the original 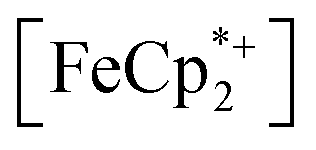 , yielding a steady-state redox potential of −588 mV vs. Fc+/Fc, just +36 mV milder than that of the electrochemical potential of HER. The comparatively positive potential of photodoped MIL-125 helps explain the lack of detectable H2 generation and supports our computational results that estimate the TiIII–OH+ mid-gap state to sit ∼100 meV more stable than HER. The slight difference between experimental and computational results can be easily attributed to accounting for the true H+ activity of the suspension, which is considerably lower than the pH = 0 conditions defined for NHE. In other words, the true redox potentials of photodoped MIL-125 sit energetically even farther from vacuum than HER and closer to the calculated results. Remarkably, given that 1.25 equivalents of Fc*+ per Ti atom were introduced for this experiment, 48.2% reduction would imply that each MIL-125 cluster supports approximately five TiIII–OH+ pairs, on average, in excellent agreement with our computational predictions of the thermodynamic limit to MIL-125 charge storage.
, yielding a steady-state redox potential of −588 mV vs. Fc+/Fc, just +36 mV milder than that of the electrochemical potential of HER. The comparatively positive potential of photodoped MIL-125 helps explain the lack of detectable H2 generation and supports our computational results that estimate the TiIII–OH+ mid-gap state to sit ∼100 meV more stable than HER. The slight difference between experimental and computational results can be easily attributed to accounting for the true H+ activity of the suspension, which is considerably lower than the pH = 0 conditions defined for NHE. In other words, the true redox potentials of photodoped MIL-125 sit energetically even farther from vacuum than HER and closer to the calculated results. Remarkably, given that 1.25 equivalents of Fc*+ per Ti atom were introduced for this experiment, 48.2% reduction would imply that each MIL-125 cluster supports approximately five TiIII–OH+ pairs, on average, in excellent agreement with our computational predictions of the thermodynamic limit to MIL-125 charge storage.
Cation-induced electrostatic stabilization of the MIL-125 clusters can account for the considerable increase in charge storage when performed with Fc*+ compared to PCET photodoping conditions. During the optical redox indicator experiment, each electron that transfers from MIL-125 to Fc*+ must be associated with a H+ liberated from alcohol oxidation. Therefore, when the MOF-Fc*+ reaches steady-state conditions, the H+ activity is higher than during PCET photodoping conditions. We expect that these additional cations impart strong electrostatic stabilization of the TiIV/III redox couples. Additionally, we expect that the increased concentration of H+ increases the likelihood of achieving a stable arrangement, as outlined above by our calculations, that permit up to four TiIII–OH+ pairs per cluster. The impact of cations on the redox potentials of Ti-MOFs has been recently investigated by us and will be a fruitful strategy for photoredox reactivity.
Conclusions
In sum, a combined computational and experimental study has been used to explain the microscopic origin of PCET-based charge storage in MIL-125 arising from the inorganic oxo ligands in the nodes. These motifs accept H+ liberated by alcohols during the photodoping reaction, thereby providing coulombic stabilization and separation from the otherwise highly reducing photogenerated TiIII center. Computational and experimental results place the redox potential of these TiIII–OH+ pairs at reduction potentials more stable than HER in the low-charging regime.The lack of gaseous byproduct from the oxidation of alcohol molecules within MIL-125 pores is seen to result from electron delocalization across TiIII/IV redox couples and the covalent association of a functional counter-ion. Single PCET events, or those that occur at remote active sites, yield conduction band electrons too low in energy to afford molecular hydrogen formation from the associated protons. Paired events at geminal μ2-oxo ligands further suppress HER. We identified routes for hydrogen loading up to 4H* per node such that certain spatial configurations allowed for up to 50% reduction of Ti-sites. With this in mind, the spatial distribution of PCET sites formed under kinetic conditions is likely to inhibit access to this configuration, as evidenced by experiment. Irradiation of MIL-125 in the presence of benzylic alcohol showed negligible hydrogen formation, consistent with HER suppression. Quantitative 1H NMR suggests, nevertheless, that photodopant concentrations of <2H* per node were obtained with excess reductant over 18 h, but as many as five TiIII could be stabilized in each cluster when performed with an optical redox indicator.
The extent of e−/H+ accumulation is therefore likely due to a thermodynamic limit set by HER and electrostatic interactions of the electrolyte medium. While these results suggest H2 must evolve during steady-state charge accumulation, this work and previous studies imply minimal H2 generation. We therefore expect kinetics to be slow for accessing the states that are unstable towards HER. It is also notable that a few photodoped states in the low-charge regime were computed to be unstable towards HER, while the vast majority are stable. Therefore, the accumulation of e−/H+ pairs and minimal H2 generation may stem from the greater probability of e−/H+ finding stable arrangements. While this particular study focused on Ti-containing conduction bands, we suspect that the general concept of accessing low valent transition metals may apply to other MOFs with equally accessible redox couples (e.g. FeIII/II/2+, CoIII/II, etc.). In sum, this study indicates MOFs featuring inorganic clusters with redox-active early transition metals and inorganic basic sites could offer a platform to stabilize excited electrons in the conduction band, broadening the gamut of photochargeable materials.
Computational methods
Beginning with the experimentally obtained crystal structure, all geometric equilibrations were performed with PBEsol67 as implemented in the Vienna ab initio Software Package.68–70 A projector-augmented plane-wave basis set71 with a 500 eV plane-wave cutoff was used, and ionic and electronic convergence criteria were set to 0.005 eV and 10−6 eV, respectively. A Γ-centered 2 × 2 × 2 k-grid was employed for geometry equilibration of both MIL-125 and the amino-derivative. Subsequent single proton, single-electron derivatives were optimized with a Γ-only sampling of the first Brillouin zone. Electronic properties were then computed using HSEsol06 (ref. 72) (PBEsol + 25% HF), a hybrid GGA functional. The same convergence criteria were employed. Atom projected density of states were aligned to vacuum potential by determining the background potential of the VASP unit cell from the pore center using a previously reported method.58Experimental methods
MIL-125 was synthesized according to an adapted literature procedure48 wherein, terephthalic acid, N,N-dimethylformamide and methanol were added to a Pyrex jar and sonicated for ca. 5 min to ensure complete dissolution. Under an N2 environment, titanium(IV) ethoxide was added to the mixture. The reaction mixture was sealed under N2 and heated at 150 °C for 18 h. Next, the solid product was collected via sequential centrifugation, and subsequent washing with DMF, methanol, and acetone to remove pore-trapped solvent. All q-1H NMR experiments were referenced to 1,3,5-trimethoxybenzene in DMSO-d6 and irradiated using a mercury arc lamp. The photoreduction of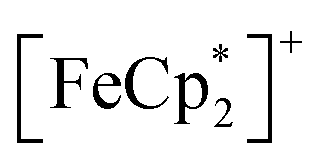 took place in a MeCN/EtOH mixture such that the ratio of TiIV was
took place in a MeCN/EtOH mixture such that the ratio of TiIV was 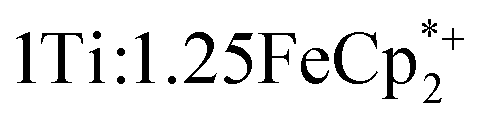 Full details of all experimental procedures can be found in the ESI.†
Full details of all experimental procedures can be found in the ESI.†
Data availability
All equilibrated computational structures are provided in the ESI.†Author contributions
JLM and CHH performed the DFT calculations, KF and CKB performed the synthesis. All authors contributed to all drafts of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the insightful comments made by Prof. A. Cook and C. R. Marshall. This material is based upon work supported by the National Science Foundation through the Division of Materials Research under grant nos. DMR-1956403 and DMR-2114430. C. H. H. acknowledges the Research Corporation for Science Advances (Cottrell Award). We also acknowledge the continued support from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation [ACI-1548562] and the PICS Coeus High Performance Computer, which is supported by the National Science Foundation [1624776], and the generous startup funding from the University of Oregon.Notes and references
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS.
- P. E. M. Siegbahn and M. R. A. Blomberg, Chem. Rev., 2010, 110, 7040–7061 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- G. Feher, J. P. Allen, M. Y. Okamura and D. C. Rees, Nature, 1989, 339, 111–116 CrossRef CAS.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Coord. Chem. Rev., 2010, 254, 2459–2471 CrossRef CAS.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS.
- N. Hoffmann, Eur. J. Org. Chem., 2017, 2017, 1982–1992 CrossRef CAS.
- H. Yayla and R. Knowles, Synlett, 2014, 25, 2819–2826 CrossRef CAS.
- C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2016, 138, 1377–1385 CrossRef CAS PubMed.
- Y. Song, U. Sanyal, D. Pangotra, J. D. Holladay, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, J. Catal., 2018, 359, 68–75 CrossRef CAS.
- J. Feng, Y. He, Y. Liu, Y. Du and D. Li, Chem. Soc. Rev., 2015, 44, 5291–5319 RSC.
- N. Dubouis and A. Grimaud, Chem. Sci., 2019, 10, 9165–9181 RSC.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS.
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS.
- IUPAC Compendium of Chemical Terminology: Gold Book, ed. M. Nič, J. Jirát, B. Košata, A. Jenkins and A. McNaught, IUPAC, Research Triagle Park, NC, 2.1.0., 2009 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- M. Batzill, Energy Environ. Sci., 2011, 4, 3275 RSC.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- K. K. Tanabe and S. M. Cohen, Angew. Chem., Int. Ed., 2009, 48, 7424–7427 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef CAS.
- J. L. Mancuso and C. H. Hendon, Adv. Theory Simul., 2019, 2, 1900126 CrossRef CAS.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef CAS.
- K. Otake, J. Ye, M. Mandal, T. Islamoglu, C. T. Buru, J. T. Hupp, M. Delferro, D. G. Truhlar, C. J. Cramer and O. K. Farha, ACS Catal., 2019, 9, 5383–5390 CrossRef CAS.
- P. Ji, Y. Song, T. Drake, S. S. Veroneau, Z. Lin, X. Pan and W. Lin, J. Am. Chem. Soc., 2018, 140, 433–440 CrossRef CAS PubMed.
- X. Feng, Y. Song, J. S. Chen, Z. Xu, S. J. Dunn and W. Lin, J. Am. Chem. Soc., 2021, 143, 1107–1118 CrossRef CAS PubMed.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng, Z. Wu, L. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- Y. Fang, Y. Ma, M. Zheng, P. Yang, A. M. Asiri and X. Wang, Coord. Chem. Rev., 2018, 373, 83–115 CrossRef CAS.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 CrossRef CAS PubMed.
- Y. Fan, Y. Ren, J. Li, C. Yue and H. Jiang, Inorg. Chem., 2018, 57, 11986–11994 CrossRef CAS.
- R. Huang, Y. Peng, C. Wang, Z. Shi and W. Lin, Eur. J. Inorg. Chem., 2016, 2016, 4358–4362 CrossRef CAS.
- P. J. Celis-Salazar, C. C. Epley, S. R. Ahrenholtz, W. A. Maza, P. M. Usov and A. J. Morris, Inorg. Chem., 2017, 56, 13741–13747 CrossRef CAS.
- C. A. Gaggioli, J. Sauer and L. Gagliardi, J. Am. Chem. Soc., 2019, 141, 14603–14611 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CrossRef CAS.
- Z. Shao, Q. Wu, X. Han, Y. Zhao, Q. Xie, H. Wang and H. Hou, Chem. Commun., 2019, 55, 10948–10951 RSC.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS.
- F. X. Llabrés i Xamena, A. Corma and H. Garcia, J. Phys. Chem. C, 2007, 111, 80–85 CrossRef.
- J. L. Mancuso, A. M. Mroz, K. N. Le and C. H. Hendon, Chem. Rev., 2020, 120, 8641–8715 CrossRef CAS PubMed.
- C.-W. Ding, W. Luo, J.-Y. Zhou, X.-J. Ma, G.-H. Chen, X.-P. Zhou and D. Li, ACS Appl. Mater. Interfaces, 2019, 11, 45621–45628 CrossRef CAS.
- S. Ghosh, J. Castillo-Lora, A. V. Soudackov, J. M. Mayer and S. Hammes-Schiffer, Nano Lett., 2017, 17, 5762–5767 CrossRef CAS.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS.
- H. Hirao, W. K. H. Ng, A. M. P. Moeljadi and S. Bureekaew, ACS Catal., 2015, 5, 3287–3291 CrossRef CAS.
- D. M. Kurtz, Chem. Rev., 1990, 90, 585–606 CrossRef CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- Y. An, B. Xu, Y. Liu, Z. Wang, P. Wang, Y. Dai, X. Qin, X. Zhang and B. Huang, ChemistryOpen, 2017, 6, 701–705 CrossRef CAS PubMed.
- L. Shen, M. Luo, L. Huang, P. Feng and L. Wu, Inorg. Chem., 2015, 54, 1191–1193 CrossRef CAS PubMed.
- C. T. Saouma, C.-C. Tsou, S. Richard, R. Ameloot, F. Vermoortele, S. Smolders, B. Bueken, A. G. DiPasquale, W. Kaminsky, C. N. Valdez, D. E. De Vos and J. M. Mayer, Chem. Sci., 2019, 10, 1322–1331 RSC.
- C. T. Saouma, S. Richard, S. Smolders, M. F. Delley, R. Ameloot, F. Vermoortele, D. E. De Vos and J. M. Mayer, J. Am. Chem. Soc., 2018, 140, 16184–16189 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, H.-Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H.-L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 105–111 CrossRef CAS PubMed.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- J. F. Janak, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 7165–7168 CrossRef CAS.
- K. T. Butler, C. H. Hendon and A. Walsh, J. Am. Chem. Soc., 2014, 136, 2703–2706 CrossRef CAS PubMed.
- H. Reiss and A. Heller, J. Phys. Chem., 1985, 89, 4207–4213 CrossRef CAS.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CAS.
- J. A. S. Roberts and R. M. Bullock, Inorg. Chem., 2013, 52, 3823–3835 CrossRef CAS PubMed.
- M. L. Pegis, J. A. S. Roberts, D. J. Wasylenko, E. A. Mader, A. M. Appel and J. M. Mayer, Inorg. Chem., 2015, 54, 11883–11888 CrossRef CAS PubMed.
- A. Paul, R. Borrelli, H. Bouyanfif, S. Gottis and F. Sauvage, ACS Omega, 2019, 4, 14780–14789 CrossRef CAS.
- G. M. Carroll, C. K. Brozek, K. H. Hartstein, E. Y. Tsui and D. R. Gamelin, J. Am. Chem. Soc., 2016, 138, 4310–4313 CrossRef CAS PubMed.
- G. M. Carroll, A. M. Schimpf, E. Y. Tsui and D. R. Gamelin, J. Am. Chem. Soc., 2015, 137, 11163–11169 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- K. Fabrizio, K. A. Lazarou, L. I. Payne, L. P. Twight, S. Golledge, C. H. Hendon and C. K. Brozek, J. Am. Chem. Soc., 2021 DOI:10.1021/jacs.1c04808.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03019a |
| This journal is © The Royal Society of Chemistry 2021 |