
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn in situ masking strategy enables radical monodecarboxylative C–C bond coupling of malonic acid derivatives†
He-Li
Cheng
,
Xian-Hui
Xie
,
Jia-Zheng
Chen
,
Zhen
Wang
and
Jian-Ping
Chen
 *
*
Institute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: ias_jpchen@njtech.edu.cn
First published on 3rd August 2021
Abstract
The utilization of malonic acids in radical decarboxylative functionalization is still underexploited, and the few existing examples are primarily limited to bisdecarboxylative functionalization. While radical monodecarboxylative functionalization is highly desirable, it is challenging because of the difficulty in suppressing the second radical decarboxylation step. Herein, we report the successful radical monodecarboxylative C–C bond coupling of malonic acids with ethynylbenziodoxolone (EBX) reagents enabled by an in situ masking strategy, affording synthetically useful 2(3H)-furanones in satisfactory yields. The keys to the success of this transformation include (1) the dual role of a silver catalyst as a single-electron transfer catalyst to drive the radical decarboxylative alkynylation and as a Lewis acid catalyst to promote the 5-endo-dig cyclization and (2) the dual function of the alkynyl reagent as a radical trapper and as an in situ masking group. Notably, the latent carboxylate group in the furanones could be readily released, which could serve as a versatile synthetic handle for further elaborations. Thus, both carboxylic acid groups in malonic acid derivatives have been well utilized for the rapid construction of molecular complexity.
Introduction
Recently, the radical decarboxylative functionalization (RDF) has appeared as a robust tool to rapidly increase molecular complexity, in which a variety of monocarboxylic acids, such as fatty acids, amino acids and α-oxocarboxylic acids, have been extensively exploited as promising alkyl radical precursors for a diverse array of radical C–C and C–heteroatom coupling reactions (Fig. 1a).1–4 Malonic acid derivatives, which bear two carboxylic acid groups on the same carbon atom, are fundamental building blocks in organic chemistry. However, the development of corresponding RDFs of readily accessible malonic acid derivatives is still in its infancy. The underdevelopment of malonic acids in RDFs probably stems from their increased oxidation potential and the chemoselectivity issue with respect to mono- and bisdecarboxylation.5,6 Prior work demonstrated the latent capacity of utilizing malonic acid derivatives in RDFs, with sporadic examples focusing exclusively on bisdecarboxylative functionalizations, such as the double Hunsdiecker reaction,7 oxidative decarboxylation8 and hydrodecarboxylation reaction,5 by which two identical functional groups could be introduced on the same carbon atom (Fig. 1b). Although radical monodecarboxylative functionalization of malonic acids is highly desirable, it is still largely elusive. Our group recently discovered a chemoselective mono- and bisdecarboxylative fluorination of malonic acids, providing unprecedented entry to either gem-difluoroalkanes or α-fluorocarboxylic acids (Fig. 1b).6 The effectiveness of α-fluorocarboxylic acid products to generate more diverse compounds via another RDF highlighted the benefits of implementing monodecarboxylative functionalization. As such, the development of a novel and chemoselectivity-controlled radical monodecarboxylative functionalization of malonic acid derivatives would find broad appeal, as the remaining carboxylic acid group in the product would serve as a versatile platform for the rapid buildup of molecular complexity.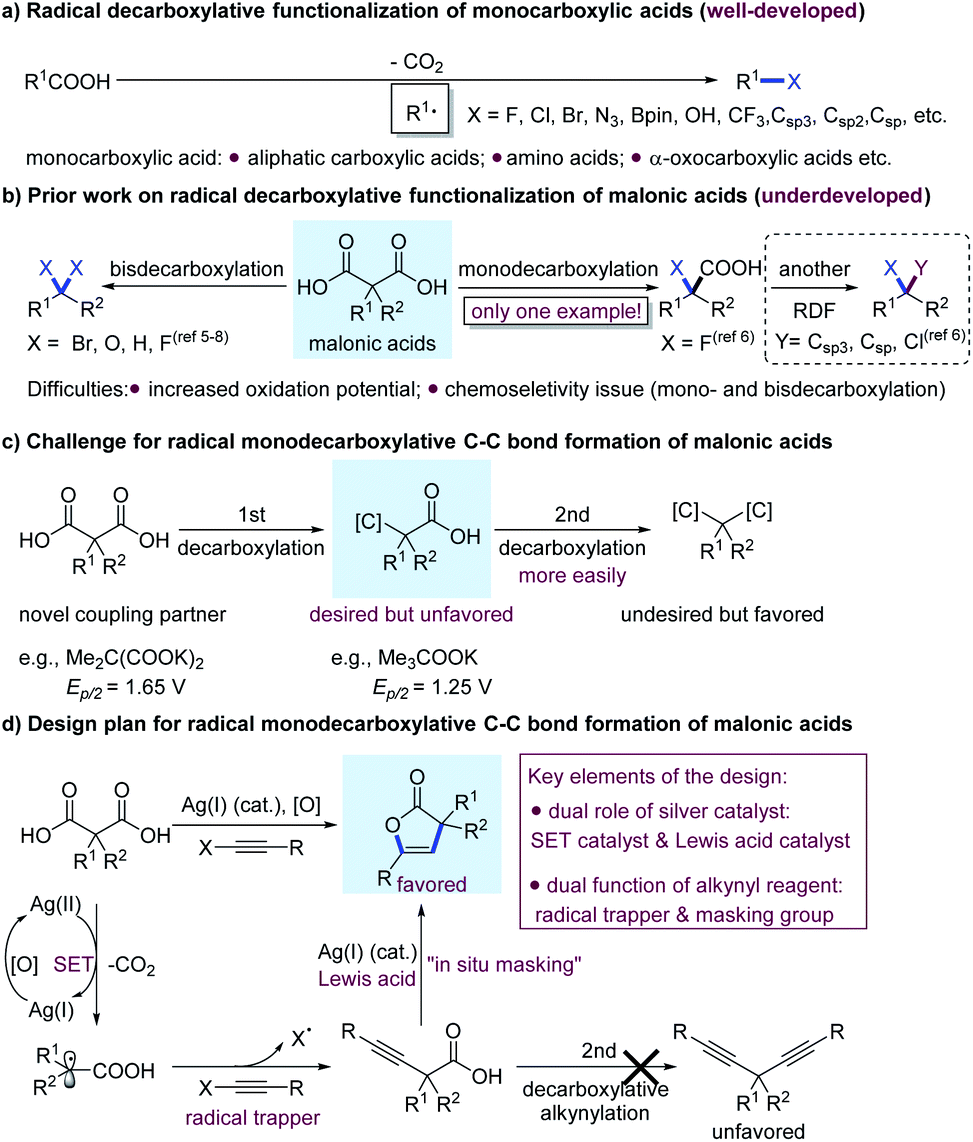 | ||
| Fig. 1 Radical decarboxylative functionalization of carboxylic acids. | ||
Encouraged by the significant advancements of radical decarboxylative C–C coupling of monocarboxylic acids,1,2,4 we are interested in exploiting malonic acid derivatives as novel Csp3 coupling partners for C–C bond formation by taking advantage of a radical strategy. However, in the oxidative decarboxylation scenario, synthetically desired monodecarboxylative functionalization products with relatively low oxidation potential compared with that of malonic acid derivatives9 are highly prone to undergo second radical decarboxylation, affording synthetically less useful bisdecarboxylation products (Fig. 1c). As a result, the realization of controllable radical monodecarboxylative C–C bond coupling of malonic acids represents a challenging goal. To achieve this aim, we hypothesized that the second decarboxylation reaction might be suppressed by masking the remaining carboxylate with an appropriate functionality that was introduced by the first RDF of malonic acids. Inspired by the unique properties of silver catalysts, such as their strong oxidant1f and Lewis acidity, as well as, in particular, their alkynophilicity,10 an in situ masking plan was proposed, as shown in Fig. 1d. Malonic acids could be oxidized by Ag(II) (E° = 1.98 V, Ag2+/Ag+)11via single-electron transfer6 after CO2 extrusion to deliver α-carboxylic acid radicals. Despite the lack of precedent, we proposed that the newly generated nucleophilic radical could be trapped by an alkynyl reagent to afford an α-alkynyl carboxylate.12 The remaining carboxylate would be rapidly intercepted by the alkynyl group in the presence of a silver catalyst via 5-endo-dig cyclization.13 Thus, the undesired second decarboxylation could be well obviated, with 2(3H)-furanones obtained as the final products. Of note, 2(3H)-furanone is not only a structural motif widely found in natural products14 but also an important building block in organic synthesis.15 Precedent reports for their syntheses rely heavily on the transformations of specific precursors, which often require multistep preparation.13a,16 Therefore, the development of practical methods for the assembly of 2(3H)-furanones from readily accessible precursors is also desirable.
Herein, we present the successful controllable radical monodecarboxylative C–C bond coupling of malonic acid derivatives for the first time, providing an unprecedented route to a variety of 2(3H)-furanones bearing all-carbon quaternary centers. Notably, the latent carboxylic acid group in the product could be readily released, which may act as a versatile synthetic handle for further manipulations.
Results and discussion
To validate our hypothesis, we initiated our investigations with the reactions of 2-(4-chlorophenethyl)-2-ethylmalonic acid (1a) and phenylethynylbenziodoxolone (2a) in the presence of AgNO3 (10 mol%) and K2S2O8 (1.0 equiv.) in aqueous acetonitrile, acetone or dimethylformamide (DMF) at 50 °C for 12 h.4a Unfortunately, no desired 2(3H)-furanone derivative 3a was formed in any of the cases (Table 1, entries 1–3). A mass balance study of the reaction in aqueous acetonitrile was carried out, and only 11% yield of 1a was recovered, along with isolation of less than 5% yield of bisdecarboxylative alkynylation product 4a, which suggested that decomposition of starting material 1a occurred. Given the elusive chemistry of malonic acids in RDFs, the generation of 4a is exciting, implying the feasibility of the radical decarboxylative alkynylation of malonic acids. However, to achieve monodecarboxylative C–C bond coupling, a compatible set of reaction conditions between Ag-catalyzed decarboxylative alkynylation and 5-endo-dig cyclization has to be exploited.|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Ph-EBX (equiv.) | Solvent (v/v) | T (°C) | Yield of 3ab (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2–0.3 mmol), AgNO3 (0.02 mmol), K2S2O8 (0.2 mmol), base (0.4 mmol), solvent under N2 at 50 °C or 65 °C for 12 h. b Isolated yield. c Without AgNO3. n-Hex = n-hexane. | |||||
| 1 | — | 1.0 | MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:1 (4 mL) :H2O = 1:1 (4 mL) |
50 | 0 |
| 2 | — | 1.0 | Acetone:H2O = 1:1 (4 mL) |
50 | 0 |
| 3 | — | 1.0 | DMF:H2O = 1:1 (4 mL) |
50 | 0 |
| 4 | PhCOOK | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 5 |
| 5 | KF | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 8 |
| 6 | K2HPO4 | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 30 |
| 7 | K2CO3 | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 10 |
| 8 | KOH | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 42 |
| 9 | K3PO4 | 1.0 | MeCN:H2O = 1:1 (4 mL) |
50 | 42 |
| 10 | K3PO4 | 1.0 | Acetone:H2O = 1:1 (4 mL) |
50 | 27 |
| 11 | K3PO4 | 1.0 | DMF:H2O = 1:1 (4 mL) |
50 | 35 |
| 12 | K3PO4 | 1.0 |
n-Hex:H2O = 1:1 (4 mL) |
50 | 16 |
| 13 | K3PO4 | 1.5 | MeCN:H2O:n-Hex = 1:3:4 (8 mL) |
65 | 72 |
| 14c | K3PO4 | 1.5 | MeCN:H2O:n-Hex = 1:3:4 (8 mL) |
65 | 0 |
Recently, the Nicewicz group and our group demonstrated that bases played a substantial role in the hydrodecarboxylation5 and decarboxylative fluorination6 of malonic acids, respectively. Accordingly, the influence of different bases on the reaction was examined in the present study. To our delight, after the addition of PhCOOK to the reaction (entry 4), product 3a was obtained for the first time, albeit in a low yield. Further screenings revealed that with increasing basicity of the additive, the yield of product 3a also increased (entries 5–9). When KOH or K3PO4 was employed, 3a was produced in 42% yield (entries 8 and 9). With K3PO4 as the optimal base, changing the mixed solvent to acetone/H2O, DMF/H2O or n-hexane/H2O decreased the yield (entries 10–12). Silver catalysts such as Ag2O, AgClO4, Ag2CO3 and AgF were also examined in aqueous acetonitrile with K3PO4 as the base, and the product 3a was obtained in yields ranging from 32–40% (see ESI† for details). Furthermore, replacement of the alkynylating agent 2a with phenylethynyl halides (Cl or Br), or phenyl phenylethynyl sulfone compromised the reaction efficiency dramatically, and only trace amounts of product were observed. Ultimately, careful examinations of the substrate ratio, solvent and temperature revealed that the yield of 3a could be increased to 72% (entry 13); meanwhile, only a trace amount of bisalkynylation product 4a was observed. A control experiment showed that AgNO3 was essential for this reaction (entry 14).
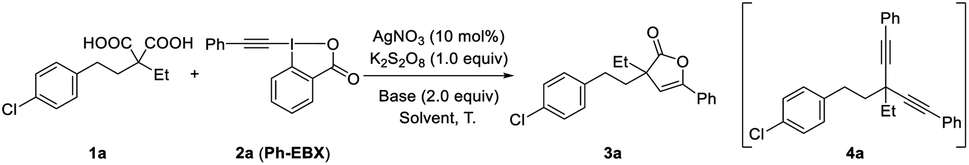
With the optimized conditions in hand, the diversity of malonic acids was first evaluated (Scheme 1). Various dialkyl-substituted malonic acids served as viable C–C bond coupling precursors for this Ag-catalyzed tandem decarboxylative alkynylation and 5-endo-dig cyclization, efficiently affording 2(3H)-furanones containing quaternary carbon centers in moderate to good yields. Even malonic acid with steric hindrance at the α-position was applicable for this reaction, delivering product 3c in 62% yield. Aryl- and alkyl-substituted malonic acids are susceptible to bisdecarboxylative functionalizations due to stabilization of the corresponding benzylic radicals generated under oxidative conditions,6 but with the present in situ masking strategy, 2(3H)-furanones (3h and 3y) instead of bisdecarboxylative alkynylation products were produced exclusively. Additionally, the performance of cyclic malonic acids was examined, and the reaction of cyclohexane-1,1-dicarboxylic acid under the optimized conditions produced rigid spiro-β,γ-unsaturated-γ-lactone 3k in 20% yield, along with a 20% yield of the bisdecarboxylative alkynylation product. This is the first time that we isolated a bisalkynylated product in an appreciable amount. However, simply changing the reaction conditions improved the yield of 3k to 45%, accompanied by a 10% yield of the bisdecarboxylation product. In contrast to the reaction with the six-membered cyclic malonic acid, the reactions of five- and seven-membered cyclic malonic acids proceeded smoothly, delivering the corresponding spiro products 3j and 3l in 58% and 57% yields, respectively; only trace amounts of bisalkynylated products were observed. It is worth noting that the spiro-β,γ-unsaturated-γ-lactone unit is widely present in natural products.14 Furthermore, this mild transformation could accommodate a number of functional groups, including aryl halide (3a–3c, 3i), ether (3n and 3o), phthalimide (3q), distal alkyne (3u), amide (3r and 3s), sulfone (3m), ester (3p) and ketone (3t) groups. Notably, functionalities such as alkenyl and hydroxyl groups susceptible to oxidative conditions remained intact in the system (3v and 3w). The excellent functional group compatibility of this reaction encouraged us to test this protocol in complex molecular settings. Gratifyingly, malonic acids derived from clofibrate and dehydroepiandrosterone were also suitable, producing products 3y and 3z in 65% and 42% yields, respectively. At the present stage, the substrate scope with respect to malonic acid derivatives is restricted mainly to gem-disubstituted derivatives. The reason is presumably that the gem-disubstituents can not only stabilize the α-carboxylic acid radical but also promote 5-endo-dig cyclization via the Thorpe–Ingold effect.17 The reaction of monosubstituted malonic acid with 2a was also examined, but only those with sterically encumbered groups at the α-position could afford product 3x.
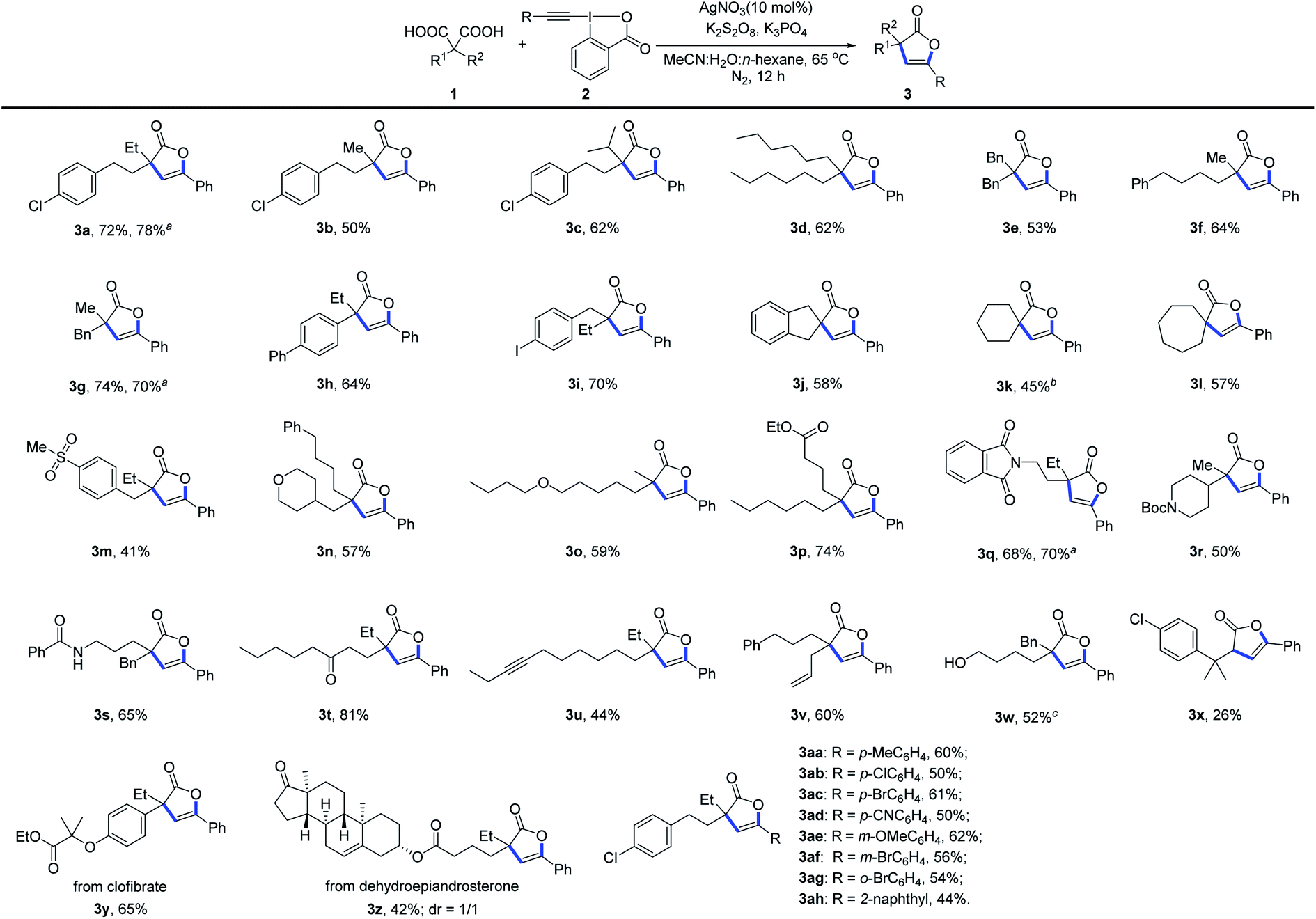 | ||
| Scheme 1 Reaction scope. Unless otherwise noted, all experiments were performed with 1 (0.2 mmol), 2 (0.3 mmol), AgNO3 (0.02 mmol), K2S2O8 (0.2 mmol), K3PO4 (0.4 mmol), CH3CN (1.0 mL), H2O (3.0 mL), and n-hexane (4.0 mL), 65 °C, under N2 for 12 h. Yields of isolated products are listed. aGram-scale reaction. b30 mol% AgF was used instead of AgNO3. c0.1 mmol K2S2O8 was used, and the reaction time was reduced to 6 h. | ||
Subsequently, ethynylbenziodoxolones (EBXs) with various aryl substituents were investigated (Scheme 1, 3aa–3ah). It was found that electron-donating (Me, OMe) and electron-withdrawing (Br, Cl, CN) substituents on the aryl ring were well tolerated, affording products in yields ranging from 50–62%. Additionally, the position of the substitution on the phenyl ring had no obvious effect on the reaction efficiency (3ac, 3af and 3ag). It should be noted that the good compatibility of chloro- and bromo-substituents provides additional opportunities for further derivatizations through cross-coupling techniques. Naphthyl-substituted EBX was also a viable reaction partner, yielding 2(3H)-furanone 3ah in 44% yield. EBXs with alkyl (n-C4H9) and silyl (TIPS) groups were also examined; unfortunately, no desired products were obtained. To demonstrate the scalability of this approach, gram-scale reactions of malonic acids 1a, 1g and 1q were carried out; to our delight, desired products 3a, 3g and 3q, respectively, were produced in yields similar to those obtained on smaller scales (Scheme 1).
Notably, the masked carboxylic acid group in the furanone products could be easily released. Simple treatments of 3g under basic conditions or under Pd(II)/HCOOK produced γ-ketoacid 5 and trialkylacetic acid 6 in 95% and 60% yields, respectively (Scheme 2a). The unmasked carboxylic acid group could function as a useful platform for further elaborations, which was exemplified by facile transformations of γ-ketoacid 5 to β-fluoro-,3b bromo-,3g azido-3d,n and alkynyl-ketones4 (7–10) via radical decarboxylative functionalizations (Scheme 2b). Thus, both carboxylic acid groups in the malonic acid derivatives have been well utilized for the rapid buildup of molecular complexity on the basis of this radical monodecarboxylative C–C bond coupling reaction enabled by an in situ masking strategy.
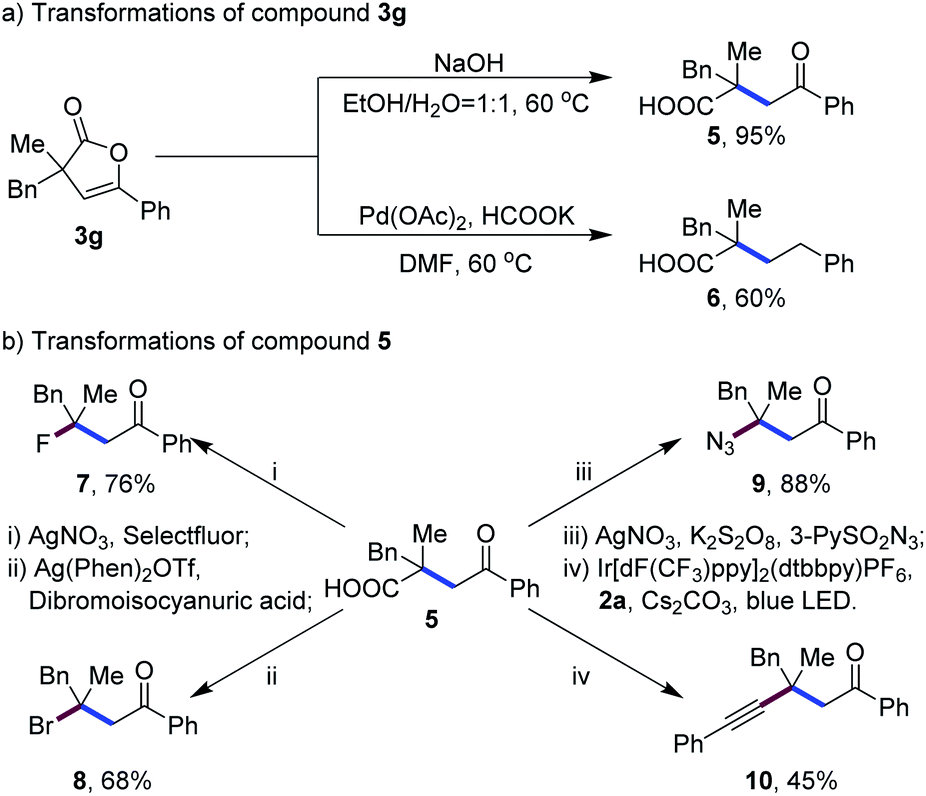 | ||
| Scheme 2 Synthetic applications. | ||
To obtain further insights into the reaction pathway, mechanistic studies were carried out (Scheme 3). Although the generation of alkyl radicals from a monocarboxylic acid in a Ag(I)/S2O82− system has been well-documented,1f,18 the oxidation of malonic acids to produce α-carboxylic acid radicals by Ag(I)/S2O82− is unknown. Thus, the radical clock experiment was carried out. Exposing radical clock substrate 11 to 2a under standard conditions afforded ring-opening product 12, albeit in a low yield, supporting the intermediacy of the α-carboxylic acid radical (Scheme 3a). Moreover, the successful cyclization of 13 to 2(3H)-furanone 3a suggested that α-alkynylcarboxylic acid was the intermediate in the formation of 2(3H)-furanones. Additionally, no cyclized product 3a was found in the absence of AgNO3, but 3a could be obtained in 80% yield under standard conditions without K2S2O8, implying the involvement of the Ag(I) catalyst in the 5-endo-dig cyclization (Scheme 3b). The control experiment of model substrate 1a under standard conditions with D2O instead of H2O as the cosolvent delivered the vinyl position fully deuterated product 3a–d in 60% yield, indicating that the vinyl proton of furanones originated from water (Scheme 3c).
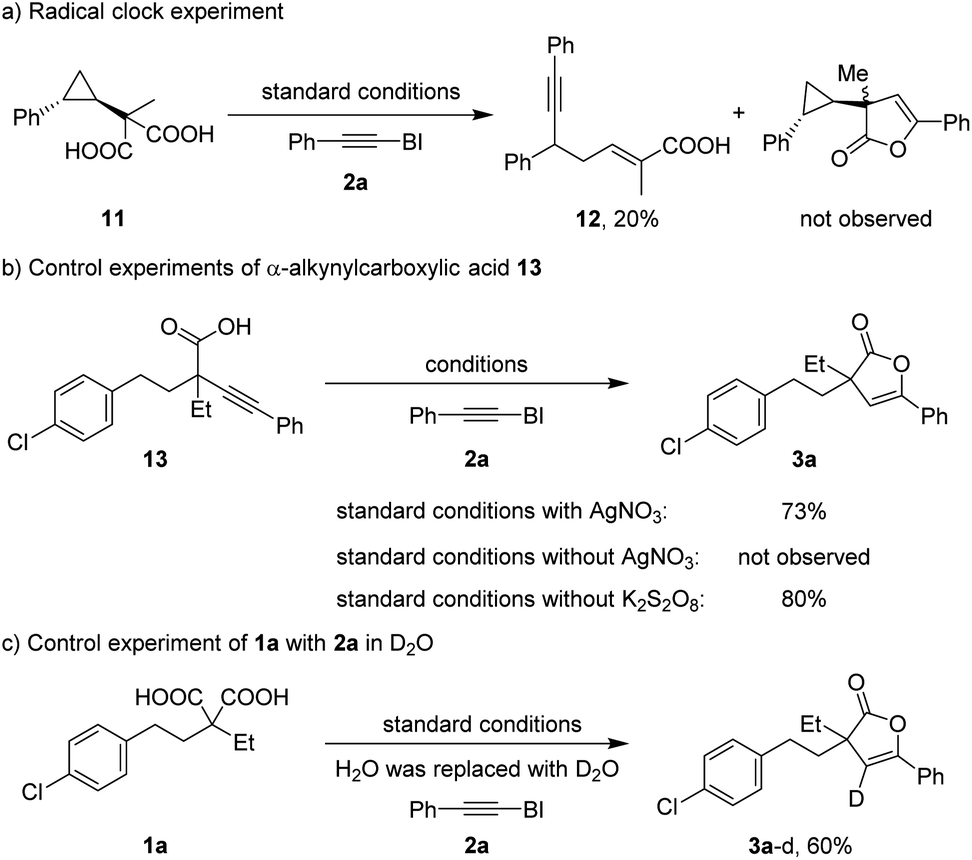 | ||
| Scheme 3 Mechanistic experiments. | ||
On the basis of the above experimental results and literature precedents,4,10,13 a reaction mechanism for this novel transformation of malonic acids has been tentatively proposed (Scheme 4). Initially, Ag(I) is oxidized by persulfate to give Ag(II).18 Then, a single-electron transfer between the resulting Ag(II) and the malonic acid carboxylates occurs, followed by rapid CO2 extrusion, affording the α-carboxylic acid radical 14. Subsequent trapping of the newly generated radical 14 with EBX yields adduct 15, which undergoes radical elimination to generate α-alkynylcarboxylic acid 16 along with the formation of a benziodoxolonyl radical.4,19 The remaining carboxylate can be quickly masked by the Ag-activated alkynyl group via 5-endo-dig cyclization to produce vinyl silver species 18.13 Finally, hydrolysis of the vinyl silver species gives rise to the 2(3H)-furanone product and regenerates the silver catalyst.
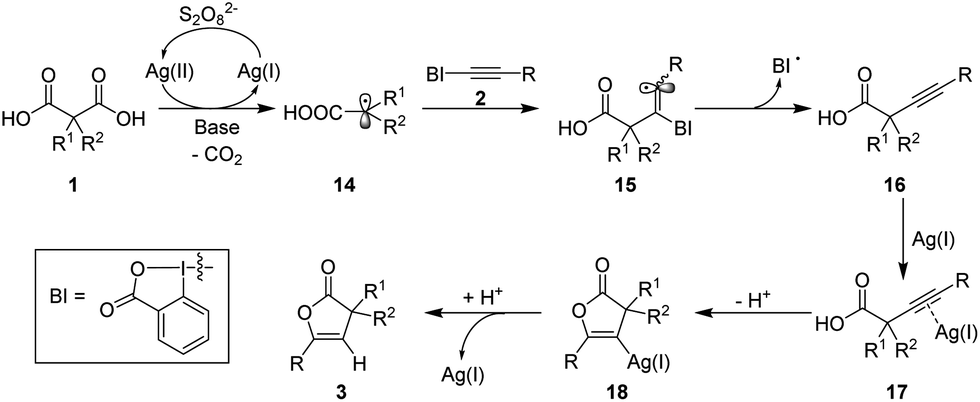 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have accomplished radical monodecarboxylative C–C bond coupling of malonic acid derivatives with alkynylating reagents enabled by a masking strategy, which provides practical entry to synthetically useful 2(3H)-furanone derivatives. The key elements for this in situ masking strategy include (1) the dual role of the silver catalyst as a single-electron transfer catalyst to drive radical decarboxylative alkynylation and as a Lewis acid catalyst to promote 5-endo-dig cyclization and (2) the dual function of the alkynyl reagent as a radical trapper and as an in situ masking group. This reaction features the use of readily available starting materials, excellent functional group compatibility and gram-scale synthetic capability as well as versatile transformations of the latent carboxylate group in the furanone products. Mechanistic studies suggest that α-carboxylic acid radical and α-alkynylcarboxylic acid species are two key intermediates in this transformation. Ultimately, we believe that the successful utilization of malonic acid derivatives as novel Csp3 coupling partners via a radical decarboxylation process will open up a new dimension of application of malonic acids in organic synthesis.Data availability
All experimental procedures, characterization, copies of NMR spectra for all new compounds related to this article can be found in the ESI.†Author contributions
H.-L. C. performed the experiments and prepared the ESI. X.-H. X., J.-Z. C., and Z. W. prepared some substrates and repeated some experiments. J.-P. C. supervised the research and wrote the manuscript with contributions from all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the National Natural Science Foundation of China (21702104), the Natural Science Foundation of Jiangsu Province, China (BK20170983), and a Start-up Grant from Nanjing Tech University (39837120) are gratefully acknowledged.Notes and references
- For reviews on radical decarboxylative functionalizations, see: (a) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS; (b) H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983–4988 CrossRef CAS; (c) G. Fang, X. Cong, G. Zanoni, Q. Liu and X. Bi, Adv. Synth. Catal., 2017, 359, 1422–1502 CrossRef CAS; (d) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (e) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC; (f) L. Zhu and C. Li, in Silver Catalysis in Organic Synthesis, ed. C.-J. Li and X. Bi, Wiley-VCH, Weinheim, 2019, pp 183–269 Search PubMed.
- For selected recent examples of radical decarboxylative carbon–carbon bonds coupling, see: (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS; (b) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS; (c) G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed; (d) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS; (e) L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188–2191 CrossRef CAS; (f) Y. Zhu, X. Wen, S. Song and N. Jiao, ACS Catal., 2016, 6, 6465–6472 CrossRef CAS; (g) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS; (h) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS; (i) A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708–3711 CrossRef CAS; (j) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS; (k) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (l) T.-G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454–2458 CrossRef CAS; (m) T. Yang, Y. Jiang, Y. Luo, J. J. H. Lim, Y. Lan and M. J. Koh, J. Am. Chem. Soc., 2020, 142, 21410–21419 CrossRef CAS; (n) X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS.
- For selected recent examples of radical decarboxylative carbon–heteroatom bonds coupling, see: (a) Z. Wang, L. Zhu, F. Yin, Z. Su, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 4258–4263 CrossRef CAS; (b) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS; (c) F. Hu, X. Shao, D. Zhu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 6105–6109 CrossRef CAS; (d) C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820–9823 CrossRef CAS PubMed; (e) H.-T. Song, W. Ding, Q.-Q. Zhou, J. Liu, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2016, 81, 7250–7255 CrossRef CAS PubMed; (f) L. Candish, E. A. Standley, A. Gómez-Suárez, S. Mukherjee and F. Glorius, Chem.–Eur. J., 2016, 22, 9971–9974 CrossRef CAS; (g) X. Tan, T. Song, Z. Wang, H. Chen, L. Cui and C. Li, Org. Lett., 2017, 19, 1634–1637 CrossRef CAS PubMed; (h) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed; (i) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (j) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS; (k) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS; (l) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS; (m) C. G. Na, D. Ravelli and E. J. Alexanian, J. Am. Chem. Soc., 2020, 142, 44–49 CrossRef CAS PubMed; (n) Y. Zhu, X. Li, X. Wang, X. Huang, T. Shen, Y. Zhang, X. Sun, M. Zou, S. Song and N. Jiao, Org. Lett., 2015, 17, 4702–4705 CrossRef CAS.
- For recent examples of oxidative decarboxylative alkynylation of monocarboxylic acids, see: (a) X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 CrossRef CAS; (b) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 CrossRef CAS; (c) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 CrossRef CAS; (d) C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 CrossRef CAS; (e) M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 CrossRef CAS.
- J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS.
- Z. Wang, C.-Y. Guo, C. Yang and J.-P. Chen, J. Am. Chem. Soc., 2019, 141, 5617–5622 CrossRef CAS.
- C. Blankenship and L. A. Paquette, Synth. Commun., 1984, 14, 983–987 CrossRef CAS.
- (a) J. J. Tufariello and W. J. Kissel, Tetrahedron Lett., 1966, 7, 6145–6150 CrossRef; (b) J. Nokami, T. Yamamoto, M. Kawada, M. Izumi, N. Ochi and R. Okawara, Tetrahedron Lett., 1979, 20, 1047–1048 CrossRef; (c) X. Ma, X. Luo, S. Dochain, C. Mathot and I. E. Markò, Org. Lett., 2015, 17, 4690–4693 CrossRef CAS PubMed.
- For the measurement of oxidation potentials of potassium 2,2-dimethylmalonate and potassium pivalate, see ESI† for details.
- (a) J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS; (b) M. Álvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 108, 3174–3198 CrossRef; (c) G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, FL, 97th edn, 2016–2017, pp. 5–78 Search PubMed.
- Although the radical decarboxylative alkynylation of monocarboxylic acids was well-established (ref. 4), the corresponding chemistry of malonic acids is unknown.
- (a) J. A. Marshall, M. A. Wolf and E. M. Wallace, J. Org. Chem., 1997, 62, 367–371 CrossRef CAS; (b) N. Kikui, T. Hinoue, Y. Usuki and T. Satoh, Chem. Lett., 2018, 47, 141–143 CrossRef CAS.
- (a) M. Kubo, T. Fujii, H. Hioki, M. Tanaka, K. Kawazu and Y. Fukuyama, Tetrahedron Lett., 2001, 42, 1081–1083 CrossRef CAS; (b) J. Marrero, A. D. Rodríguez, P. Baran and R. G. Raptis, Eur. J. Org. Chem., 2004, 3909–3912 CrossRef CAS; (c) K. Ueda, T. Kadekaru, E. R. O. Siwu, M. Kita and D. Uemura, J. Nat. Prod., 2006, 69, 1077–1079 CrossRef CAS PubMed; (d) Q.-L. Liang, Z.-D. Min and Y.-P. Tang, J. Asian Nat. Prod. Res., 2008, 10, 403–407 CrossRef CAS PubMed; (e) M. Tori, Y. Tanio, Y. Okamoto, Y. Saito, X. Gong, C. Kuroda and R. Hanai, Heterocycles, 2008, 75, 2029–2034 CrossRef CAS; (f) D. Liu, M.-J. Xu, L.-J. Wu, Z.-W. Deng and W.-H. Lin, J. Asian Nat. Prod. Res., 2009, 11, 811–816 CrossRef CAS; (g) L. Liu, Y. Han, J. Xiao, L. Li, L. Guo, X. Jiang, L. Kong and Y. Che, J. Nat. Prod., 2016, 79, 2616–2623 CrossRef CAS; (h) J.-J. Wang, H. Y. Chung, Y.-B. Zhang, G.-Q. Li, Y.-L. Li, W.-H. Huang and G.-C. Wang, Phytochemistry, 2016, 122, 270–275 CrossRef CAS PubMed.
- For a comprehensive review see: (a) A. R. Choudhury and S. Mukherjee, Chem. Soc. Rev., 2020, 49, 6755–6788 RSC; for selected recent examples, see: (b) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796–10797 CrossRef CAS; (c) M. S. Manna and S. Mukherjee, Chem.–Eur. J., 2012, 18, 15277–15282 CrossRef CAS PubMed; (d) Y. Wu, R. P. Singh and L. Deng, J. Am. Chem. Soc., 2011, 133, 12458–12461 CrossRef CAS; (e) W. Zhang, D. Tan, R. Lee, G. Tong, W. Chen, B. Qi, K.-W. Huang, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2012, 51, 10069–10073 CrossRef CAS; (f) Q. Tang, L. Lin, J. Ji, H. Hu, X. Liu and X. Feng, Chem.–Eur. J., 2017, 23, 16447–16451 CrossRef CAS; (g) B. M. Trost, C.-I. J. Hung and M. J. Scharf, Angew. Chem., Int. Ed., 2018, 57, 11408–11412 CrossRef CAS; (h) J. A. Griswold and J. S. Johnson, ACS Catal., 2019, 9, 11614–11618 CrossRef CAS; (i) B. M. Trost, E. Gnanamani, C. A. Kalnmals, C.-I. J. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489–1493 CrossRef CAS PubMed.
- (a) Z. W. Just and R. C. Larock, J. Org. Chem., 2008, 73, 2662–2667 CrossRef CAS PubMed; (b) Y.-S. Feng, Z.-Q. Xu, L. Mao, F.-F. Zhang and H.-J. Xu, Org. Lett., 2013, 15, 1472–1475 CrossRef CAS PubMed; (c) K. Endo, S. Yakeishi, R. Takayama and T. Shibata, Chem.–Eur. J., 2014, 20, 8893–8897 CrossRef CAS; (d) X. Wu, S. Shirakawa and K. Maruoka, Org. Biomol. Chem., 2014, 12, 5388–5392 RSC; (e) F. Huang, Z. Liu, Q. Wang, J. Lou and Z. Yu, Org. Lett., 2017, 19, 3660–3663 CrossRef CAS PubMed.
- (a) M. E. Jung, Synlett, 1990, 186–190 CrossRef CAS; (b) M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS.
- (a) J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651–1659 CrossRef CAS; (b) J. M. Anderson and J. K. Kochi, J. Org. Chem., 1970, 35, 986–989 CrossRef CAS.
- The generated benziodoxolonyl radical is reduced through either H-abstraction or reduction/protonation to form the 2-iodobenzoic acid in this process (ref. 4). This was further proved by the isolation of 2-iodobenzoic acid in a yield of 80% (based on the quantity of 2a) from our model reaction.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02642a |
| This journal is © The Royal Society of Chemistry 2021 |