
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural products dereplication by diffusion ordered NMR spectroscopy (DOSY)†
Guy
Kleks
 ab,
Darren C.
Holland
ab,
Joshua
Porter
ab and
Anthony R.
Carroll
*ab
ab,
Darren C.
Holland
ab,
Joshua
Porter
ab and
Anthony R.
Carroll
*ab
aSchool of Environment and Science, Griffith University, Gold Coast, QLD 4222, Australia. E-mail: a.carroll@griffith.edu.au
bGriffith Institute for Drug Discovery, Griffith University, Brisbane, QLD 4111, Australia
First published on 15th July 2021
Abstract
Diffusion-ordered NMR spectroscopy (DOSY) can be used to analyze mixtures of compounds since resonances deriving from different compounds are distinguished by their diffusion coefficients (D). Previously, DOSY has mostly been used for organometallic and polymer analysis, we have now applied DOSY to investigate diffusion coefficients of structurally diverse organic compounds such as natural products (NP). The experimental Ds derived from 55 diverse NPs has allowed us to establish a power law relationship between D and molecular weight (MW) and therefore predict MW from experimental D. We have shown that D is also affected by factors such as hydrogen bonding, molar density and molecular shape of the compound and we have generated new models that incorporate experimentally derived variables for these factors so that more accurate predictions of MW can be calculated from experimental D. The recognition that multiple physicochemical properties affect D has allowed us to generate a polynomial equation based on multiple linear regression analysis of eight calculated physicochemical properties from 63 compounds to accurately correlate predicted D with experimental D for any known organic compound. This equation has been used to calculate predicted D for 217![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 043 compounds present in a publicly available natural product database (DEREP-NP) and to dereplicate known NPs in a mixture based on matching of experimental D and structural features derived from NMR analysis with predicted D and calculated structural features in the database. These models have been validated by the dereplication of a mixture of two known sesquiterpenes obtained from Tasmannia xerophila and the identification of new alkaloids from the bryozoan Amathia lamourouxi. These new methodologies allow the MW of compounds in mixtures to be predicted without the need for MS analysis, the dereplication of known compounds and identification of new compounds based solely on parameters derived by DOSY NMR.
043 compounds present in a publicly available natural product database (DEREP-NP) and to dereplicate known NPs in a mixture based on matching of experimental D and structural features derived from NMR analysis with predicted D and calculated structural features in the database. These models have been validated by the dereplication of a mixture of two known sesquiterpenes obtained from Tasmannia xerophila and the identification of new alkaloids from the bryozoan Amathia lamourouxi. These new methodologies allow the MW of compounds in mixtures to be predicted without the need for MS analysis, the dereplication of known compounds and identification of new compounds based solely on parameters derived by DOSY NMR.
Introduction
One of the challenges in natural product (NP) discovery is the re-isolation and identification of known compounds. The process of identifying these known compounds early in the discovery process is known as dereplication. Using dereplication strategies, the time consuming process involved in re-isolation and structure elucidation of known compounds is avoided allowing the isolation and structure elucidation efforts to focus solely on new compounds.1 In NP discovery, LC-MS is a common tool for dereplication. This requires comparison of data acquired by MS or tandem MS (MS/MS) with that of known NPs found in databases. This has been facilitated by the use of molecular networking via, for instance, the free online community-based platform Global Natural Products Social Molecular Networking (GNPS), in which MS/MS data is used to identify a network of chemically related NPs.2,3 Unfortunately, because fragmentation patterns, ion intensities and the experimental parameters used to acquire data can vary across instruments, mis-identification and/or failure to identify compounds can still occur.4–6While the most common NP dereplication techniques are MS-based, the main tool for structure elucidation of NPs is NMR spectroscopy. The only approach currently used to correlate NMR and MS data is by hyphenated NMR techniques such as LC-NMR-MS. This technique allows real-time acquisition of combined MS and NMR data to be obtained for compounds eluting from a LC system.7 While hyphenated NMR is a powerful tool for dereplication, it is time consuming and requires specialized and expensive hardware not available in most laboratories. An alternative approach is to perform LCMS separation first, followed by fraction analysis (after evaporation and reconstitution in an appropriate NMR solvent) either in NMR tubes or using a flow NMR probe.8 This technique is also labour intensive and time consuming.
The difference between MS and NMR gives rise to several problems:
(1) Since MS is several orders of magnitude more sensitive than NMR, compounds that are marked as potential new compounds by MS-based dereplication techniques may be present in concentrations too small for isolation and subsequent NMR identification.
(2) Compounds that are not easily ionizable may not be visible by MS. Furthermore, those with low signal intensities may be overlooked.
(3) Structural/configurational isomers can be misidentified as known compounds by MS, while NMR analysis can delineate the difference between isomers more clearly.
(4) There are approximately 30000 unique accurate masses that account for >215000 published NPs. However, many of these masses are represented by hundreds of different NPs. For example 264.13615 da represents the MW of 640 different NPs.9
These points highlight the need for more accessible NMR-based dereplication methods that allow for the identification of compounds in mixtures. Recent developments in this area include a large open access NP database that is functional group annotated for NMR feature matching (DEREP-NP),9 a HSQC-TOCSY analysis method that has been used to identify molecular fragments in complex mixtures,10 SMART 2.0 (Small Molecule Accurate Recognition Technology) a machine learning tool to identify compounds in mixtures based on HSQC data11 and MADByTE (Metabolomics and Dereplication by Two-Dimensional Experiments) a tool that associates HSQC and TOCSY data from complex mixtures to allow identification of molecular networks.12 A significant limitation of these methods is that none have the ability to predict the MWs of resonances computationally annotated by NMR.
Diffusion-ordered spectroscopy (DOSY) is an NMR technique that allows resonances associated with individual components in a mixture to be separated in the NMR tube based on their size. This non-destructive technique does not require any special equipment and can be performed on any modern NMR spectrometer.
Based on pulsed field gradient (PFG) NMR, the diffusion rate of a compound in a solvent is measured by acquiring a series of spectra at incrementing gradient strengths, resulting in signal attenuation that is used to calculate a diffusion coefficient (D). This provides a DOSY spectrum, a pseudo-2D spectrum in which the resonances are separated in a derived second dimension according to the diffusion rate of the molecule they emanate from. The diffusion of a molecule in solvent is described by the Stokes–Einstein equation as the diffusion coefficient (D):
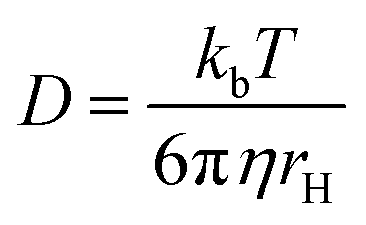
This equation assumes a molecule possesses a sphere-like shape and is dissolved in a continuous fluid. In the numerator kb is the Boltzmann constant and T is the temperature. The denominator represents the friction experienced by the molecule, in which η is the solvent viscosity and rH is the hydrodynamic radius of the molecule.13
Differences in T between samples will result in variation of Ds and the temperature should be kept constant in order to compare Ds of different samples. Sample temperature should also be regulated, since a temperature gradient in the sample could lead to convection, resulting in erroneous Ds.14 While low viscosity solvents, such as chloroform (0.54 cP at 298 K), are more prone to the formation of convection, DMSO is more viscous (1.99 cP at 298 K), making it a good candidate for DOSY.14 The solvent viscosity (η) changes with sample concentration,15 affecting the diffusion rate of all the components in the sample. To be able to compare the Ds of compounds measured in different samples, an internal reference must be used. While an obvious choice for an internal reference would be the residual NMR solvent peak, previous research has shown ambiguous results, with some advising caution with referencing Ds to the solvent signal,15 while others have reported excellent results.16
The Stokes–Einstein equation states that the D of the molecule possesses an inverse relationship to its hydrodynamic radius. In other words, a small molecule diffuses faster than a large one, thereby displaying a larger D value. Therefore, DOSY provides a spectroscopic separation of the signals associated with compounds in a mixture without any physical separation, making DOSY a powerful technique for mixture analysis.
A significant limitation of DOSY is signal overlap, complicating the extraction of the D since the observed signal attenuation is derived from two or more components with different diffusion rates. While there are several techniques that attempt to resolve this problem, it remains a very complicated task.16,17 Clearly, this poses a problem for complex mixtures such as those commonly encountered in NP research. However, DOSY can be implemented with 2D NMR experiments such as DOSY-COSY and DOSY-HSQC,18,19 expanding the diffusion into another dimension thus minimizing the possibility of signal overlap. Alternatively, the likelihood of overlap could be reduced by collapsing all signal multiplets to singlets using pure shift DOSY.20
As DOSY separates the resonances in a mixture according to their D, corresponding to molecular size, the D can be correlated to molecular weight (MW). There are many examples in which DOSY was used to determine MW from D of polymers,21,22 simple organic compounds15 and organometallics.14,23 However, in NP research DOSY has only been used for separation of resonances by their D, and this information was not correlated to any other physical or chemical properties. To our knowledge, there are only two examples in which DOSY has been used in NP discovery for the dereplication and identification of unknown compounds. The first was the dereplication of a chromatographically inseparable mixture of NPs from a marine cyanobacterium,24 and while the second was used to aid in the identification of a mixture of bromopyrroles from the marine sponge Agelas sp.25 Since the literature contains very little information about D of NPs, dereplication of NPs by DOSY cannot currently rely solely on experimental D. Therefore, experimentally derived D need to be investigated in more detail to determine if this property correlates to a common physical property used for dereplication, such as MW or if it can be used as surrogate for MW.
In this paper, we investigate correlations between experimental D of NPs to various structural and chemical properties. This has allowed us to develop models that can be used to dereplicate known NPs and identify new NPs through application of DOSY NMR techniques.
Results and discussions
Referencing DOSY data
As described above, the viscosity of the NMR solvent can affect D, and changes in the concentration of dissolved analytes can affect the viscosity of the solvent. To avoid these issues we have used relative diffusivity of an internal reference compound to account for variations of viscosity between samples.26 The “standard” D of the reference (Dstand) was determined as the D of the reference in a blank sample (containing only the reference compound and the NMR solvent at 350 μM), and the ratio between the observed D of the reference (Dref) in each sample to that of the standard was used to standardize the D of the compound (Dcomp) using the following equation: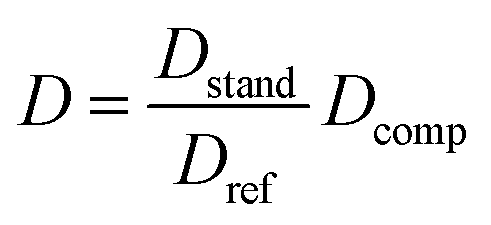
This referencing method enables the DOSY data to be reproducible (Fig. S1†), allowing D values acquired for compounds to be compared even on different spectrometers using different pulse sequences. An ideal reference should resonate at a chemical shift that rarely overlaps with analyte resonance and should have a diffusion co-efficient like that of the analytes.
Since diffusion parameters in the DOSY NMR pulse sequence should be set to provide up to ∼90% signal attenuation for analytes, and since the average MW of reported NPs is 414 amu,27 we chose tetrakis(trimethylsilyloxy)silane (TTMS), 384.84 amu, Dstand 3.157 10−10 m2 s−1 at 298 K as a more suitable internal reference. TTMS shows good solubility in DMSO, is a liquid at room temperature making it easy to handle, and with a boiling point of 103–106 °C, can be evaporated upon removal of DMSO (189 °C) from the sample. TTMS displays a single proton resonance at δH 0.09 ppm derived from its 12 constituent methyl groups (36 protons) meaning only a small concentration (0.5% v/w) is enough to produce an intense signal.
The relationship between D to MW
The relationship between D to MW is non-linear, and has been shown to possess a power-law relationship:21| D = A × MWα |
To achieve a linear relationship, the logarithms of both the D and MW should be used:
| log(D) = αlog(MW) + log(A) | (1) |
However, it has been shown that a specific power-law relationship needs to be established for each compound class and each solvent.28 After a correlation between D to MW is determined, it can be used to predict the MW of an unknown compound in a specific compound class from its experimental D. While this model has been shown to produce accurate MW predictions for specific compound classes,14,23 variation between structure classes has been shown to produce large errors in MW estimation.15,29
We have acquired 1H DOSY spectra in DMSO-d6 for 55 individual compounds with a MW range of 123–1486 amu, including 38 NPs, four NP derivatives, and 13 synthetic compounds of which eight are drugs (Table S1,† for structures and numerical structure codes see ESI†). The NPs in the dataset consist of diverse structure classes such as oxygenated linear and cyclic terpenes, alkaloids and their TFA salts, anthracycline, a β-triketone, a coumarin derivative, a saponin, and macrocyclic compounds such as macrolide antibiotics.
The least-squares fit of the experimental log D vs. the log MW of each compound generated model 1 (n = 55, R2 = 0.852) with coefficient values of −0.6057 and −8.0952 for α and log(A), respectively.
model 1:
| MWpre = 10((log(D)+8.0952)/(−0.6057)) |
To quantify the accuracy of the MW prediction (MWpre), the MWpre error as the percentage of the residual MWpre from the true MW (MWtrue) for each compound was determined (eqn (2)):
| MWpre error = (MWpre − MWtrue)/MWtrue | (2) |
This provides an intuitive error scale in which compounds that diffuse slower than their predicted D (Dpre – derived from the calibration curve) will translate to a MWpre value that is greater than their true MW and will therefore display a positive error, and underestimation of MWpre will show a negative error.
In previous research, calibration curves generated using the power law relationship in CDCl3 and D2O displayed MWpre errors mostly within a range of ±30%.29 While analysis of data previously reported for compounds in THF-d8 and toluene-d8 (Tol-d8) showed MWpre errors within a range of ±20%.14 Our results in DMSO-d6 showed an average +20.6% MWpre error for overestimated compounds, and −14.7% for underestimated compounds. However, the total MWpre error range for our dataset was larger than for CDCl3, D2O, THF-d8 or Tol-d8 with a maximum error of +55% for overestimated compounds and −32% for underestimated compounds (Fig. 1).
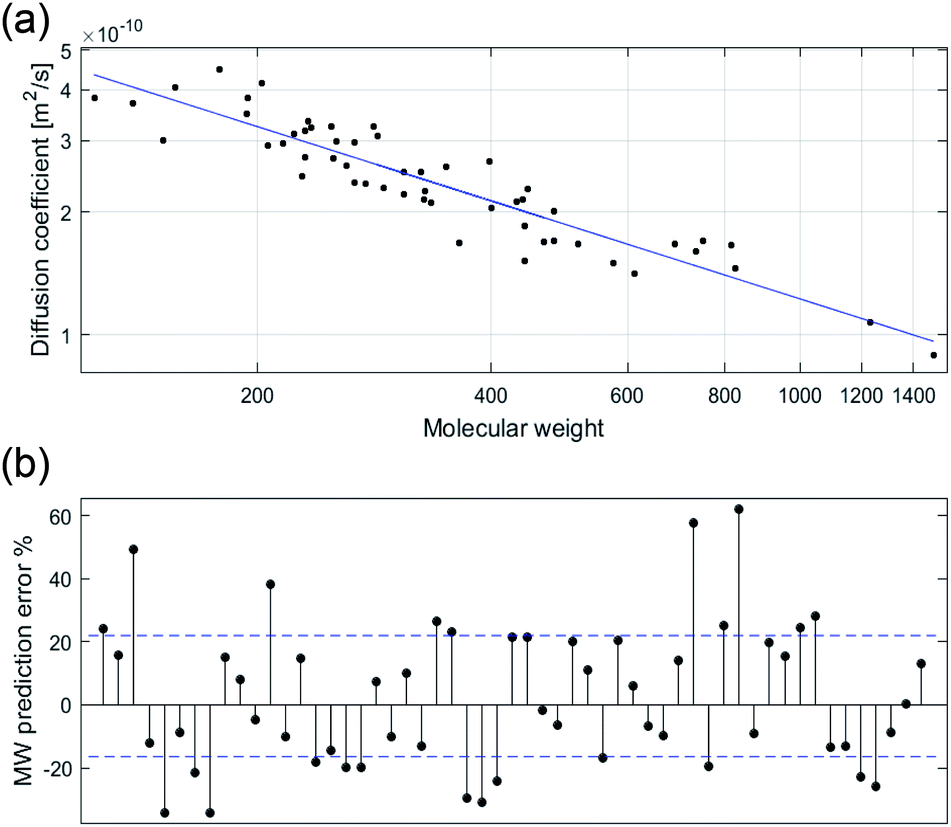 | ||
| Fig. 1 log–log plot of the experimental D in DMSO-d6 against MW for each individual compound (n = 55) (a). The residual error for each compound as percentage of MW prediction from the true MW with mean error range in blue (b). | ||
The difference in the MWpre error range observed for compounds previously studied in CDCl3 and D2O (although none of these compounds structures were disclosed in the paper)29 compared to DMSO can be attributed to the wider dissolution range of DMSO, capable of dissolving more polar compounds than CDCl3 and less polar compounds than D2O. All of the compounds previously studied in THF-d8 and Tol-d8 are lipophilic with low structural diversity.15 In contrast, our dataset consists of structurally diverse compounds displaying a wide polarity range, with clogP ranging from −7.0 to 9.6 (see Fig. S2† for comparison of physicochemical properties). Several NPs in our dataset such as carboxylic acids, aminoglycoside and flavonoids are too polar to be dissolved in CDCl3. Since the high structural diversity of the compounds used in this study to directly correlate D with MW resulted in an inaccurate power law (model 1) relationship, we next investigated the factors that contributed to these variations with the aim to establish a more accurate DOSY MW prediction model.
Molecular shape
Molecular shape affects diffusion rates with expanded disc-like (ED) or compact spherical molecules (CS) displaying faster diffusion trends relative to other molecules classified as dissipated spheres and ellipsoids (DSE).15 Pyrene (8), anthracene and acridine have previously been classified as EDs.15 Using model 1, the predicted MWs of the ED compounds 8 and phenanthrene (5) (Fig. 2) show significantly faster diffusion rates relative to that predicted by their MW, corresponding to a −34% MWpre error for both (Fig. S3†). In addition, artemisinin (25), arborinine (27), clarithromycin (51) and oleandomycin-triacetate (52) all showed significantly faster diffusion rates than predicted by their MWs and this might also be attributed to their shape. TTMS, the reference compound for our dataset, displayed the most underestimated MWpre (error of −46%) using model 1. This agreed with the literature since similar compounds, tetrakis(trimethylsilyl)silane and tris(trimethylsilyl)amine, classified as CS display faster diffusion compared to DSE compounds.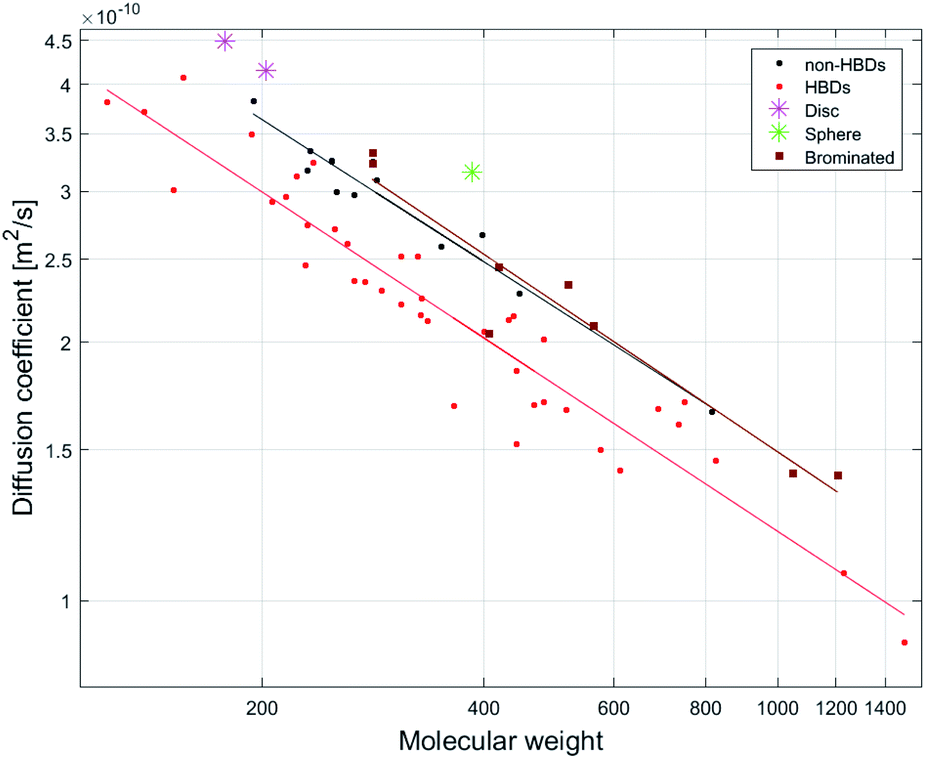 | ||
| Fig. 2 Log–log plot of relationships between D to MW for three compound groups: HBD-containing compounds (n = 40, R2 = 0.91), non-HBD containing compounds (not including disc or sphere-like compounds) (n = 13, R2 = 0.94) and brominated compounds (n = 7, R2 = 0.99). | ||
Stalke and co-workers have suggested shape-specific MW prediction, generating power-law relationships between D and MW for three shape types: (1) CS, (2) ED and (3) DSE.15,30 While this improved their MW prediction, for compound mixtures it requires a priori knowledge of the different shape-classes that are present in the mixture, reported D for these shape classes, and a method to match the resonances in the mixture with the appropriate shape class. The distinction between CS, DSE and ED shape classes is also problematic because compounds lie on a continuum between the three shape-classes. Furthermore, analogues or stereoisomers can display different hydrodynamic radii, resulting in different MWpre errors within a specific shape-class and these shape-specific power law models do not work for compounds with similar shapes but containing hydrogen bond donor (HBD) or no HBD groups.
Hydrogen bonding
Intermolecular hydrogen bonding (H-bonding) can result in a slower diffusion rate than predicted by MW (i.e. smaller diffusion coefficient) due to an increase in the hydrodynamic radius (rH) of the compound.31,32 Since DMSO is a hydrogen bond acceptor (HBA), it is capable of hydrogen bonding with a hydrogen bond donor (HBD). Inspection of Fig S3† shows that most compounds with MWpre errors >0 from model 1 (26 of 27) are compounds containing HBDs and those with MWpre errors <0 are non-HBD containing compounds.The effect of H-bonding on diffusion is clearly visualized by the linear least-squares fit of MW vs. D for HBD-containing compounds (n = 40, R2 = 0.91) and non-HBD containing compounds (n = 13, R2 = 0.94), as the latter display faster diffusion rates since they are not involved in H-bonding (Fig. 2).
Since the NP dataset consists of both HBD-containing compounds and non-HBD containing compounds, MW prediction using model 1 provides intermediate MWpre values for these two groups. Power-law based MW prediction (i.e. model 1) therefore has a wide error range and will result in an overestimated MWpre for many HBD-containing compounds, and an underestimated MWpre for many non-HBD containing compounds. This is demonstrated by predicting MW using model 1 for the HBD compound gemfibrozil (19), and the non-HBD compound parthenolide (18) (250.3 and 248.3 amu, respectively). Model 1 predicts MWpre values that differ by 70 amu, with a MWpre of 269 for 19 and a 199 MWpre for 18 (Fig. S3†). Therefore, using model 1 to predict the MW for 19 from its experimental D provides an overestimated MWpre of +7% (MWpre error). This overestimation derives from an increase in the rH of 19 as its HBD group is intermolecularly H-bonded to DMSO, resulting in a slower diffusion rate (smaller D). Conversely, the MWpre for 18 is underestimated by −20% as it does not contain any HBDs and is therefore not involved in H-bonding with DMSO. The increase in the effective size of a compound through H-bonding with DMSO therefore relates to the equilibrium constant for the H-bonding interaction and the resident time spent as one molecular system.
Cabrita and co-workers have shown that DOSY can be used to qualitatively evaluate H-bond strength by comparing the D of HBD-containing compounds in the presence and absence of a HBA.31,32 A HBD acidity scale (αH2) has been used to correlate H-bond acidity to the increase of rH observed by DOSY for acidic compounds.31 The αH2 scale ranges from 0 (no H-bonding) to 1 (strongest H-bonding).33,34 While this scale has been established in CCl4, it has been shown that H-bond acidity 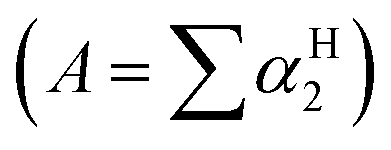 can also be predicted with high accuracy by the 1H NMR chemical shift difference for a protic hydrogen between DMSO to CDCl3.35 The H-bond acidity of many classes of compounds have been determined and predictable trends relating to functional groups have been obtained. In summary, phenols, carboxylic acids, 1° amides, the amide NHs in imidazol-2-one, pyrimidine-2,4(1H,3H)-dione and related structures show comparable and strong H-bond acidity, 1° alcohols, benzylic 1° and 2° alcohols, acyclic 2° alcohols, anilides, 1° anilines, indole, pyrrole (and related aromatic amines) possess intermediate H-bond acidity, 2° anilines and 2° and 3° cyclic alcohols and vicinol diols possess weak H-bond acidity, while alkyl amines possess extremely weak H-bond acidity.33,36 It is known that the acidity of HBD groups are also affected by factors such as electron donating or withdrawing substituents and steric hindrance and these factors also affect H-bond strength, although in many cases deviations within functional group classes are minimal.33 Cabrita and co-workers have shown that the faster diffusion rate of 2,6-di-tert-butylphenol relative to its structural isomer, 2,4-di-tert-butylphenol, for example, is due to hindering by the second bulky group ortho to the phenol and this results in weaker H-bonding with the HBA.31
can also be predicted with high accuracy by the 1H NMR chemical shift difference for a protic hydrogen between DMSO to CDCl3.35 The H-bond acidity of many classes of compounds have been determined and predictable trends relating to functional groups have been obtained. In summary, phenols, carboxylic acids, 1° amides, the amide NHs in imidazol-2-one, pyrimidine-2,4(1H,3H)-dione and related structures show comparable and strong H-bond acidity, 1° alcohols, benzylic 1° and 2° alcohols, acyclic 2° alcohols, anilides, 1° anilines, indole, pyrrole (and related aromatic amines) possess intermediate H-bond acidity, 2° anilines and 2° and 3° cyclic alcohols and vicinol diols possess weak H-bond acidity, while alkyl amines possess extremely weak H-bond acidity.33,36 It is known that the acidity of HBD groups are also affected by factors such as electron donating or withdrawing substituents and steric hindrance and these factors also affect H-bond strength, although in many cases deviations within functional group classes are minimal.33 Cabrita and co-workers have shown that the faster diffusion rate of 2,6-di-tert-butylphenol relative to its structural isomer, 2,4-di-tert-butylphenol, for example, is due to hindering by the second bulky group ortho to the phenol and this results in weaker H-bonding with the HBA.31
Unsurprisingly, compounds containing strongly acidic HBD groups such as carboxylic acids and phenols showed the largest MWpre errors in the NP dataset when model 1 was used.
Intramolecular H-bonding between an acidic proton to an adjacent carbonyl oxygen is manifested in 1H NMR spectra with the observation of the acidic proton resonating as a sharp signal at a significantly deshielded chemical shift (δH >12 ppm).37 Unlike all other carboxylic acid containing compounds, nalidixic acid (15) displayed an underestimated MWpre of −18%. This alongside a deshielded and sharp resonance at δH 14.89 suggested that the carboxylic acid proton is intramolecularly H-bonded and not participating in intermolecular H-bonding and this is in contrast with other compounds containing a carboxylic acid in the NP dataset. Arborinine (27) shows an underestimated MWpre of −24% that is different to the other phenolic compounds in the dataset. A sharp and deshielded resonance at δH 14.86 suggests that the phenolic proton in 27 does not undergo intermolecular H-bonding because it is intramolecular H-bonded to the adjacent carbonyl oxygen. Salicylic acid (2) also contains an intramolecular H-bonded phenol as well as a carboxylic acid and it shows an overestimated MWpre of +16% that is likely to be associated with only an H-bond between the carboxylic acid proton and DMSO.
Contribution of H-bonded DMSO to total MW
Applying the power law least squares fit equation derived from analysis of the non-HBD compounds (n = 13, α = −0.562, logA = −8.151, R2 = 0.95) to the compounds possessing HBDs (n = 40) provided a predictive tool to estimate their intermolecular extended H-bonded MWs (EHBMWpre model 1a).
model 1a:
| EHBMWpre = 10((logD+8.151)/(−0.562)) |
Subtraction of MWtrue from EHBMWpre and dividing this mass by the MW of DMSO-d6 (84 amu) (eqn (3)) then allowed a good estimate of the number of intermolecular H-bonding interactions with DMSO per molecule for compounds containing functional groups with strong H-bond acidity (Fig. 3).
| Number of H-bonds = (EHBMWpre − MWtrue/MWDMSO d6) | (3) |
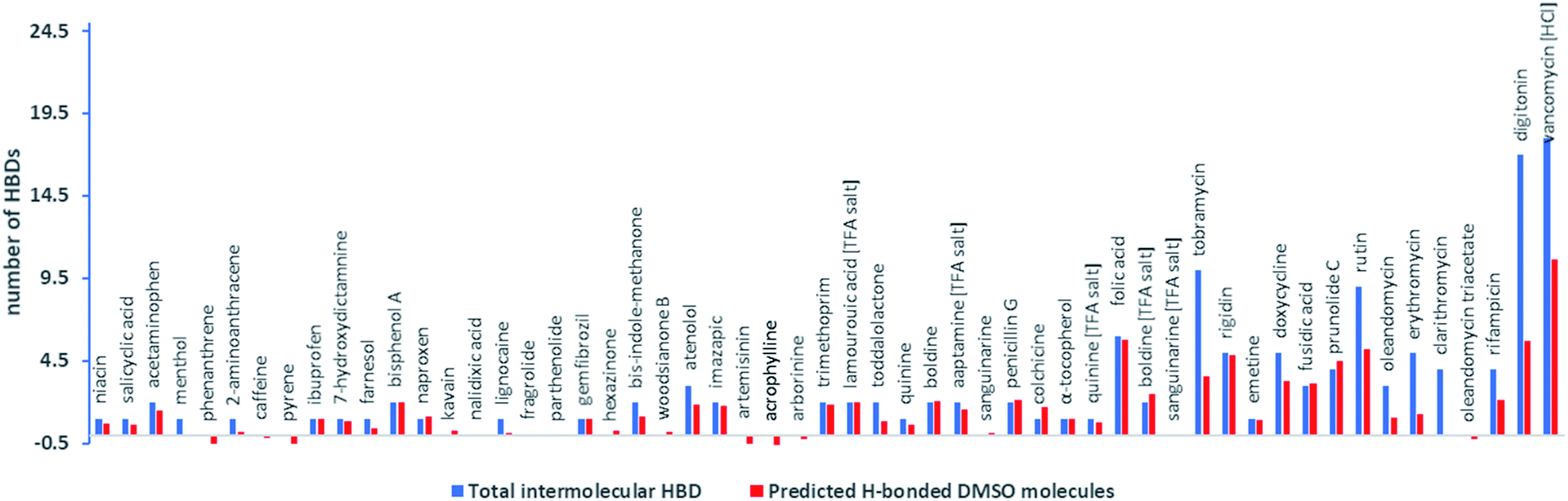 | ||
| Fig. 3 Total intermolecular HBDs per compound and predicted number of DMSO molecules associated with intermolecular H-bonding interactions per molecule derived from model 1a and eqn (3). | ||
Most compounds (27 out of 37) that have between one and five intermolecular HBDs based on HBD count (but excluding intramolecular H-bonds) have EHBMWpre calculated from D within ±12% of their expected EHBMWs. This indicated that the MW errors generated by model 1 are exaggerated in small MW compounds because the mass of DMSO in one H-bonding interaction can contribute significantly to the overall mass.
Carboxylic acids
Using model 1, compounds containing a carboxylic acid as the sole intermolecular HBD group showed overestimated MWpre: niacin (1) (+24%), 2 (+16%), ibuprofen (9) (15%), naproxen (13) (15%) and 19 (+7%). These MWpre errors agree with a single DMSO H-bonding interaction and the variation in error is the result of the different mass contribution of the compound and DMSO to the overall EHBMW.Phenols
Compounds that contain phenolic groups as the only HBD group also display overestimated MWpre using model 1. While boldine (32) and prunolide C (47) show similar overestimated MWpre of +20% and +24%, bisphenol A (12) and 7-hydroxydictamnine (10) display variation in MWpre error with +38% and +8% respectively. Analysis of the result from eqn (3) shows that the overestimated MWpre for 47, 32, 12 and 10 obtained using model 1 correlates with the ratio of compound MW to number of phenols, again indicating that the magnitude of the error correlates with the mass of the combined DMSO interactions relative to the MW of the compound. The phenol, α-tocopherol (37), displays an underestimated MWpre (−7%) using model 1 but applying eqn (3) shows that this error can still be explained by one DMSO molecule H-bonding with 37.Even though 37 displays a slightly lower H-bond acidity (A = 0.43) compared to 12 and 32 (A = 0.48 and A = 0.52 respectively), which can be attributed to steric hindrance by the adjacent methyls in the 2,6-dimethylphenol moiety, this does not significantly reduce its H-bonding interaction with DMSO.
Nitrogen HBD groups
Some compounds that contain only protons attached to nitrogen as the sole HBD group displayed less significant MWpre overestimation than carboxylic acids and phenols using model 1. Penicillin G (35) and colchicine (36) both containing a secondary amide, displayed MWpre errors of +21% and +6%. Analysis of the results from eqn (3) suggests that 35 has two H-bonding interactions with DMSO (via its carboxylic acid and amide protons) and 36 has 1.5 interactions, with the tropanone ring in 36 likely acting as a weak second HBD. Two additional compounds, imazapic (24) and lamouroic acid (29), both containing carboxylic acid and amide groups display similar MWpre errors (+23% and +21%) were also predicted to have two H-bonding interactions with DMSO (based on analysis of eqn (3)). Trimethoprim (28), containing two 1° amines attached to a pyrimidine, displayed the largest MWpre overestimate (+22% error using model 1) for a compound containing an N HBD group in the dataset however the estimated number of H-bonding interactions with DMSO (using eqn (3)) is two, as expected. Emetine (44), containing a secondary amine displayed an underestimated MWpre of −9% and a predicted 0.8 interaction with DMSO. These results are in line with literature expectations since amides show significantly higher H-bond acidity than secondary amines.33,34 The bis-indole, di(1H-indol-3-yl)methanone (21) displayed a MWpre error of +10% but the result from eqn (3) suggests that only 1.2 DMSO molecules are predicted to H-bond to it. Likewise, 2-aminoanthracene (6) containing an aniline moiety had an underestimated MWpre of −9% and an estimated DMSO H-bonding contribution of 0.3. These results are also in line with the predicted H-bond acidity scale since indoles are more acidic than anilines, but less acidic than amides.Lignocaine (16) contains an acetanilide moiety but displays an underestimated MWpre of −14% that is indicative of a non-HBD containing compound. The predicted H-bonding contribution of DMSO is also low (0.2). This does not agree with the literature as acetanilides displays high hydrogen bond acidity (αH2 = 0.50) compared to other nitrogen functional groups.34 Therefore, this suggests that the amide proton is sterically hindered by the methyls of the 2,6-dimethylphenyl moiety and this conclusion is reinforced by a very low measured H-bond acidity (A = 0.04) for the amide proton in 16.
Alcohol HBD groups
Of the remaining eight compounds with EHBMWpre lower than expected all but one contains alcohols or aniline HBDs, functional groups known to possess weak H-bond acidity. Four compounds, oleandomycin (49), erythromycin (50), 51 and rifampicin (53) are macrocycles containing three to five alcohols that most likely take part in cross ring intramolecular H-bonding, one is a vicinal diol toddalolactone and two compounds are small MW mono-alcohols. Compounds containing sugar moieties (n = 4) have significantly lower EHBMWpre and this is also in line with the literature since 1,2-diols have weak H-bond acidities resulting in H-bonding in a non-equimolar ratio with DMSO. Digitonin (54) for example has 17 HBDs associated with a pentasaccharide and hydroxy groups in the triterpene moiety, but it appears that an additional mass equivalent to only three DMSO molecules H-bond to this compound. These observations affirm that DOSY is a powerful tool to quantify the extent of H-bonding between the analyte and NMR solvent and that hydrogen bond acidity contributes to the residence time for hydrogen bonding interactions (and thus the average hydrodynamic radius).Based on these observations, classifying HBD groups into three categories: phenol/carboxylic acid HBD (OH), nitrogen HBD (NH) and alcohol HBD (aOH) provided an opportunity to predict the contribution of additional MW derived from each type of HBD group.
Multiple linear regression analysis of counts of HBDs in each compound using the three HBD categories, and predicted EHBMWpre (model 1a) vs. the actual MW of the compounds established a highly predictable MW estimation (R2 = 0.98, n = 55, model 1b) and indicated that, on average, phenol/carboxylic HBDs (OH) contribute 86.081 amu, nitrogen HBDs (NH) 44.584 amu, and alcohol HBDs (aOH) 19.44 amu per HBD to the total predicted MW (Fig. 3).
model 1b:
| MWpre = 0.932 × 10EHBMWpre − 86.081 × OH − 44.584×NH − 19.44×aOH + 21.088 |
| MWpre = 0.932 × 10((logD+8.151)/−0.562) − 86.081 × OH − 44.584 × NH − 19.44 × aOH + 21.088 |
These results are in line with H-bond acidity trends reported in the literature for acidic, phenolic and alcoholic HBDs. The reported H-bond acidity of nitrogen HBDs are quite variable and combining all counts of nitrogen HBDs into one category produced an average prediction for the MW contribution for amides, amines and aromatic NH and sub-categorizing these groups will likely improve the MW prediction further (Fig. 4).
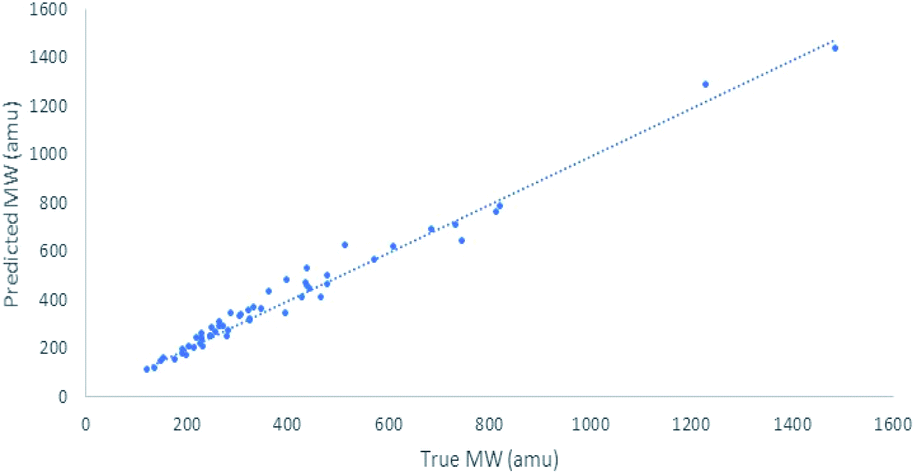 | ||
| Fig. 4 Plot of relationships between MWtrue and MWpre based on model 1b with factors for acidic OH (phenols/carboxylic acids), nitrogen HBDs and alcoholic HBDs that contribute to hydrodynamic radii used to predict MW of 55 natural products and their derivatives. (n = 55, R2 = 0.98) based on their diffusion co-efficients. | ||
Molecular density
Molar density is another factor affecting diffusion rates. Stalke and co-workers have shown that compounds containing heavy atoms such as bromine and iodine display fast diffusion rates relative to their MWs.38 The molar density of the Br atom (calculated by dividing its mass by its van der Waals volume)15 shows a 5.2/3.4/2.8-fold higher density than C/N/O atoms respectively. The linear least-squares fit of D vs. MW for a group of brominated NPs (n = 7, R2 = 0.99) displayed a different diffusion trend to other compounds in the dataset (Fig. 2). Although all of the brominated compounds contain HBDs, they show faster diffusion rates compared to non-HBD containing compounds. Calculation of the Br ratio shows that the compounds that displayed fast diffusion rates possess a Br ratio >28%. The exception was botryllamide C (56), displaying a diffusion rate like that of other HBDs molecules (MWpre error of +6%) due to a lower Br ratio of 20% (and was therefore excluded from the linear least-squares fit displayed in Fig. 2).Although we did not use the brominated compounds to construct the calibration curve for model 1, the effect of bromine density on diffusion is demonstrated by comparing the MWpre errors generated by applying model 1 to brominated analogues of compounds in the NP dataset. While 21 showed a MWpre error of +10% due to H-bonding, its 6,6′-dibromo analogue showed a faster diffusion rate that translated to a MWpre error of −24%. 47 displayed a MWpre error of +24%, while its brominated analogues prunolide B (59) (containing 6 bromines) and prunolide A (60) (containing 8 bromines) displayed MWpre errors of −24% and −34% respectively (Fig. S3†). The faster diffusion rate of 60 correlates to the increase in bromine ratio for 60 compared to 59 (45.8% and 53.0%), suggesting that a higher bromine ratio will result in a faster diffusion rate relative to MW, corresponding to an underestimation in MWpre using model 1.
While the Br atom is significantly denser than the C atom (5.2-fold), more common elements in NPs, such as O and N, are only 1.9 and 1.5-fold denser than C. Neufeld and Stalke suggested measuring the density of a compound by dividing its MW by the sum of all van der Waals volumes of the atoms.15 This however provided a poor correlation for the non-HBD containing compounds in our dataset due to the fact that the variation in diffusion rates relative to MW derive also from molecular shape and not just from molar density. The β-triketone woodsianone B (22) for example,39 has a higher O content (24%) compared to fragrolide (17) and 18 (19%) and slightly lower O content than 25 but displayed a slower diffusion rate relative to its MW as it has a different molecular shape. The structurally related compounds, the sesquiterpenes 17, 18 and 25 displayed an opposite trend. While 17 and 18 (both containing 19% O) displayed a MWpre error of −20%, 25 (28% O) showed a much larger MWpre error of −30%. However, this difference might also be associated with a more compact spherical shape of 25.
A clear example where an increase in molar density is observed is for the TFA salts of alkaloids. Ion pairing is observed by DOSY as a decrease in diffusion rate (smaller D) as a result of an increase in the rH of the compound.40 Our dataset contain three alkaloids (quinine (31), 32 and sanguinarine (34)) that displayed an increased diffusion rate as TFA salts relative to their MWs (including 114 amu for TFA) compared to diffusion rates of these compounds without TFA. This increase in diffusion rate, corresponding to underestimation in MWpre by model 1, indicates an increase in molar density as TFA contributes two oxygens and three fluorines to the overall density of the compound.
While Stalke and co-workers have suggested that molar density has a larger impact on diffusion rate than molecular shape,38 the ED compounds in our dataset, 8 and 5, showed larger MWpre errors using model 1 than brominated compounds. However, unlike in the Stalke dataset in which the majority of compounds did not contain HBDs (57 of 60),38 all of the brominated compounds in our dataset contained at least one HBD suggesting that higher molar density counterbalances the effect of H-bonding in predicting MW. This suggests that the factors affecting MW prediction involve a combination of H-bonding, molar density and molecular shape. The correlations observed between D to MW for each separate group of compounds (HBDs, non-HBDs and brominated) in our dataset are more accurate than the combined relationship of HBDs and non-HBDs (n = 55, R2 = 0.85) and therefore predicting MW from D for each specific HBD, non-HBD and high density compound groups is advisable. However, unless there is a method to associate the resonances observed in a mixture with compounds that do or do not contain HBDs and/or heavy atoms such as bromine then these MW prediction equations are irrelevant. For non-halogenated compounds our analysis indicates that H-bonding generates the greatest error for MW prediction by DOSY in DMSO.
Improved MW prediction models
As discussed earlier, intramolecular H-bonded protons are characteristically deshielded, resonating at frequencies greater than δH 12 ppm and do not usually contribute to intermolecular H-bonding. The compounds 15 and 27 display very deshielded (>δH 14.00) and sharp resonances that are clear indicators of intramolecular H-bonding. Therefore, in developing a HBD model, we have set an upper limit for the chemical shift of the effδH parameter so that only the chemical shifts of intermolecularly H-bonded protons are used. This ideally meant that the effδH parameter only captures HBD's that lead to an increase in a compound's rH, and therefore affect diffusion. Since not all NPs contain HBDs, a value of effδH = 0.00 has been set for compounds containing no HBDs. Therefore, effδH was limited to a chemical shifts range between 0 and 14, where effδH equals the most deshielded exchangeable proton resonance observed between δH 0 and 14.
Multiple linear regression analysis of 55 compounds afforded the following relationship:
model 2:
| logD = −0.6077logMW − 0.0102 effδH − 8.0282 |
This model displayed a very significant improvement (R2 = 0.95) in MW prediction relative to a simple power-law relationship of D vs. MW using model 1 (n = 55, R2 = 0.85) (Fig S4 and S5†). The effδH parameter showed an excellent correlation (P-value = 1.3 10−13) to the variation between logD vs. logMW.
The effδH parameter in model 2 is a surrogate for the MW of DMSO in the extended MW of HBD compounds. The more deshielded the exchangeable signal, the greater the mass that is subtracted from the total MWpre, resulting in the MWpre being closer to that expected for the MW of a compound alone (without a contribution of DMSO in HBD/HBA interactions). When the effδH parameter is set to 0 for any compound, the resulting MWpre are almost identical to the EHBMWpre generated using model 1a. Model 2 improved the MWpre for the polar carboxylic acids 1, 24 and 2 displaying a very accurate MWpre (−4%, −1% and −3% error compared to +24% + 23% and +16% respectively from model 1). However, only two of three lipophilic carboxylic acids 9, gemfibrozil (19) and 13 had improved MWpre with model 2 vs. model 1 (−7% vs. +15%, −8% vs. +15%, −13% vs. +7% respectively). Some compounds containing more than one strong HDB (such as bis phenol A, acetaminophen (3), atenolol (23), 28 and folic acid (39)) had improved MWpre but were still >20% in error while tobramycin (42) and fusidic acid (46) had MWpre errors increased to >20%. This suggested that using the exchangeable proton chemical shift of only the most deshielded resonance in the model ignored the contribution of other strongly acidic HBDs.
The amide proton in 16 is deshielded leading to a significant reduction in its MWpre using model 2. This led to a MWpre error for 16 of −23% (an increased underestimation of its MWpre compared to model 1). The chemical shift of the NH proton does not account for the steric hindrance it experiences from the two ortho methyl groups.
While this model displays a very good correlation between the chemical shift of acidic exchangeable protons and variations in diffusion, it has some significant practical limitations. First, chemical exchange causes signal broadening. Resonances associated with acidic protons such as those present in carboxylic acids or phenols can undergo chemical exchange with residual H2O, resulting in signal broadening up to a point where these resonances cannot be seen in a 1H NMR spectrum. Second, several compounds in our dataset (doxycycline hyclate (45) 53 and rutin (48)) displayed resonances for intramolecular H-bonded phenols below the effδH limit of 14.00 ppm (δH 11.48, 12.44 and 12.60 ppm respectively). These intramolecular H-bonded phenolic resonances are identifiable as they are sharper than other phenols in their spectra that can undergo chemical exchange with H2O. Third, a proton which is a HBD can undergo chemical exchange with H2O and will display a larger D value than the other proton resonances in the same compound.41 These exchangeable proton resonances therefore display an average D value somewhere between that of the compound and that of H2O and this D is dependent on the diffusion delay used in the pulse sequence.41 These caveats could make it difficult to distinguish exchangeable proton resonances from specific molecules in a mixture.
Unfortunately, practical application of model 2 for MW prediction of compounds in mixtures requires the effδH for each compound in the mixture to be quantified. Using model 2 with a mixture containing a compound capable of intermolecular H-bonding and a compound that cannot, will significantly increase the MWpre error for one of these compounds. For example, assuming a mixture contains a flavonoid and a non-polar terpene and setting effδH to match the exchangeable chemical shift of the phenol would decrease the MWpre error for the flavonoid but increase the terpene's MWpre substantially, and vice versa.
Therefore, application of model 2 is only applicable to mixtures that have undergone initial partitioning, such as an acid/base extraction to obtain separate fractions containing either acidic, neutral, or basic components from an extract. Alternatively, if exchangeable proton resonances observed in the spectrum of mixtures can be associated to specific groups of other resonances in the mixture based on integral intensities then different effδH values can be applied to each group of resonance at specific experimental D.
P is a common parameter to measure lipophilicity, we examined the correlation between D and calculated logP (n = 55, R2 = 0.870). This was an improvement from model 1 (n = 55, R2 = 0.852), and the P-value for calculated logP (0.009) implied a correlation to D (Table S4†). We therefore examined the correlation between the RP HPLC retention of compounds in our dataset to their D. While RP HPLC elution vs. % MeCN has been used to predict logP with good accuracy,43,44 since MeOH is a much more common solvent used for RP HPLC separation of NPs we therefore investigated the relationship between D and NP elution using H2O/MeOH gradients on C18 silica gel.
The % MeOH elution of 41 compounds were measured by positive or negative ESI LC-MS with a H2O (10 mM ammonium acetate)/MeOH gradient. The dwell time was subtracted from the retention time to calculate the % MeOH at which each compound eluted. Multiple linear regression analysis using the % MeOH elution for 41 compounds has provided the following relationship:
model 3:
| logD = −0.6497logMW + 0.1906 (% MeOH) − 8.0979 |
This model displayed a significant improvement in MW prediction (R2 = 0.91) relative to the power-law MW prediction for the same compounds (n = 41, R2 = 0.84). The % MeOH at which compounds eluted by RP HPLC also showed a much better correlation to D (P-value = 2.2 × 10−6) than calculated logP (P-value = 0.009) for the same compounds (Fig. S6†).
In general, model 3 reduced the MWpre error compared to results from model 1 for non HBD compounds as well as many of the compounds that contained a higher proportion of HBD groups relative to their MW. Most compounds containing no HBDs still have underestimated MWpre while a smaller proportion of compounds containing HBD still had overestimated MWpre. This suggested that the % MeOH parameter partially accounted for the MW contribution of DMSO for many HBD containing compounds leading to a reduction in the difference in the logA term in the power law relationship for compounds containing no HBDs compared to HBD compounds. The % MeOH term in model 3 is therefore a surrogate for the contribution of H-bonded DMSO to the NPs hydrodynamic radius and thus MWpre. As the proportion of % MeOH required to elute a NP by RP HPLC increases, the reduction in DMSO MW contribution to MWpre for HBD compounds decreases. Unfortunately, this meant that for compounds containing mainly lipophilic moieties but also containing a HBD group, and that interact with C18 solely through hydrophobic interactions, thus eluting in a high proportion of MeOH, the % MeOH term in model 3 does not provide a compensatory factor to reduce the MW contribution of DMSO to the overall MWpre for these compounds.
Model 3 improved the MWpre for the polar acids 1, 2 and 24 displaying a MWpre errors of −9%, −6% and −3% respectively. It also improved the MW prediction for 3, 28, 23, 30, 31, 32, 36, 44, 50, 48 and 53. However, the lipophilic acids (9, 19 and 13), phenols (37, 47, 10 and 12) and 21 (eluting at 68%, 57% 78%, 100%, 64%, 62%, 67% and 63% MeOH respectively) each displayed an increased MWpre error (+25%, +22% + 24%, +21%, 22%, 13%, 46% and 14% respectively). This suggested that model 3 was ineffective at improving the MWpre for lipophilic compounds with a strongly acidic HBD group. Model 3 significantly improved the MWpre error for 16 (−5%) suggesting that the sterically hindered amide proton does not contribute to polar interactions with the HPLC solvent (Fig. S6†).
Model 3 therefore provides a compromise MW prediction between HBD and non HBD containing compounds. It is important to note that this model is not suitable for RP HPLC fractions obtained using a H2O/MeOH gradient with an acid modifier since the retention times of acidic and/or basic compounds will change at low pH.43
Predicting D by compound structural properties
The complexity of physicochemical properties that contribute to D in highly functionalised molecules such as NPs led us to the conclusion that one predictive model to correlate D with MW for complex mixtures of NPs was futile. However, through the generation of models 2 and 3 we have clearly demonstrated that with some prior knowledge of a molecular structure, accurate predictions of D can be made. Some failings were also recognized, with molecular shape or molar density proving to be contributing factors to D as we observed for brominated compounds. Since the aim of this study is to develop an orthogonal NMR dereplication method that can correlate structural information derived from NMR with MW, we decided to see if D could be predicted based on structural features and use experimental D as a surrogate for MW. There are several public and proprietary databases that contain structures of published NPs and cheminformatics platforms provide tools to calculate a multitude of parameters that could be used to aid in the prediction of D. We have previously used the universal natural product database UNPD (containing 217043 publicly available NPs reported prior to 2013) to develop a platform (DEREP-NP) for rapid identification of known NPs based on structural features derived from experimental MS and NMR data.9 DEREP-NP was established in DataWarrior,45 an open-source software that also allows other structural and chemical properties for compounds to be calculated. Based on our observations of factors that contribute to predicting accurate D, we calculated seven additional properties for all 217049 compounds in the database. These properties accounted for:
(a) molar density through a count of oxygens which are not attached to hydrogens (heavy O), and by a Br ratio (% Br) which is calculated as the sum of the Br atoms mass as a % of the total compound MW.
(b) Molecular shape Index (shape), generated by DataWarrior, provides values close to 0 for spherical compounds and values close to 1 for linear compounds, thus taking molecular shape into account.
(c) Lipophilicity by the relative polar surface area (polarity), providing a much better correlation than clogP.
(d) Intermolecular H-bonding generated as the total mass (17 amu × tally of free phenols and carboxylic acids) as a % of total compound MW (% OH). Counts of free phenols and carboxylic acids were generated by subtracting counts of phenols and carboxylic acids that were intramolecularly H-bonded (as determined using substructure count feature). The ratio of nitrogen HBDs (% NHBD) and alcohol HBDs (% AHBD) were calculated in the same manner.
Multiple linear regression analysis of eight structural and chemical properties for the 63 compounds that we have acquired experimental D has provided the following relationship:
model 4:
| logD = −7.6365–0.7403logMW + 0.139 shape + 0.0069 heavy O − 0.8506 (% phenol/acid) − 0.2586 (% Br) − 0.4016 (% NHBD) −0.0947 × (% AHBD) − 0.1282 polarity |
This model provides a very accurate prediction of diffusion coefficients (n = 63, R2 = 0.99), with predicted D (Dpre) displaying a small average error range of −3.7% to +3.0% from experimental D, and a maximum error range of −9.3% to +7.9% (Fig. 5 and S7†).
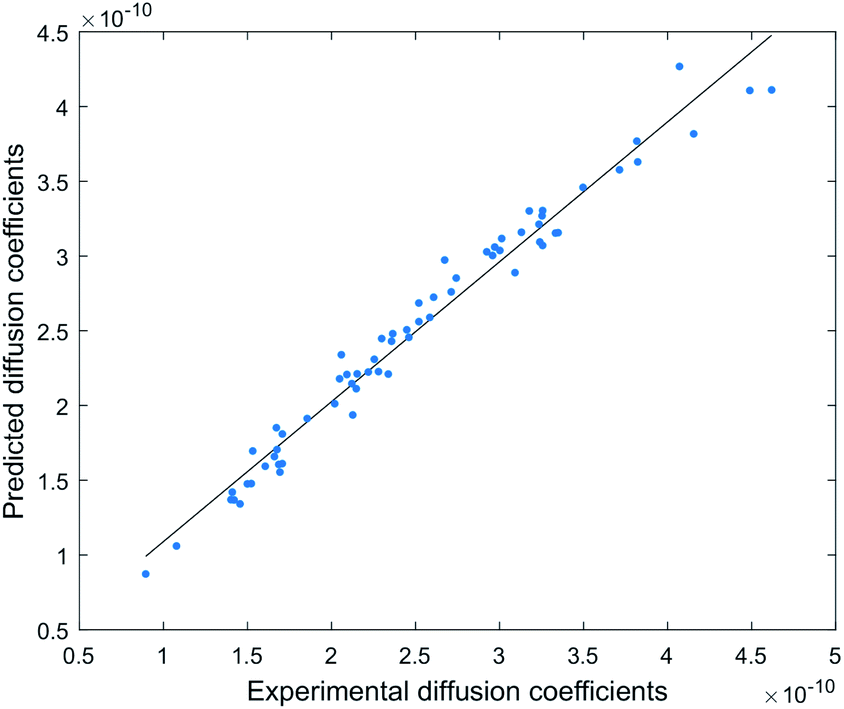 | ||
| Fig. 5 Correlation between experimental and predicted diffusion coefficients (n = 63, R2 = 0.99). | ||
This model can be used to predict diffusion coefficients (Dpre) for any compound in the DEREP-NP database (or for any other database of compounds) in DataWarrior‡ and with the structural fragment tools already embedded in DEREP-NP this orthogonal Dpre data can replace MW data for structure matching. The histograms for counts of log(MW) and log(Dpre) were also very similar (Fig. 6). This again highlights the role of H-bond acceptors interacting with the DMSO solvent.
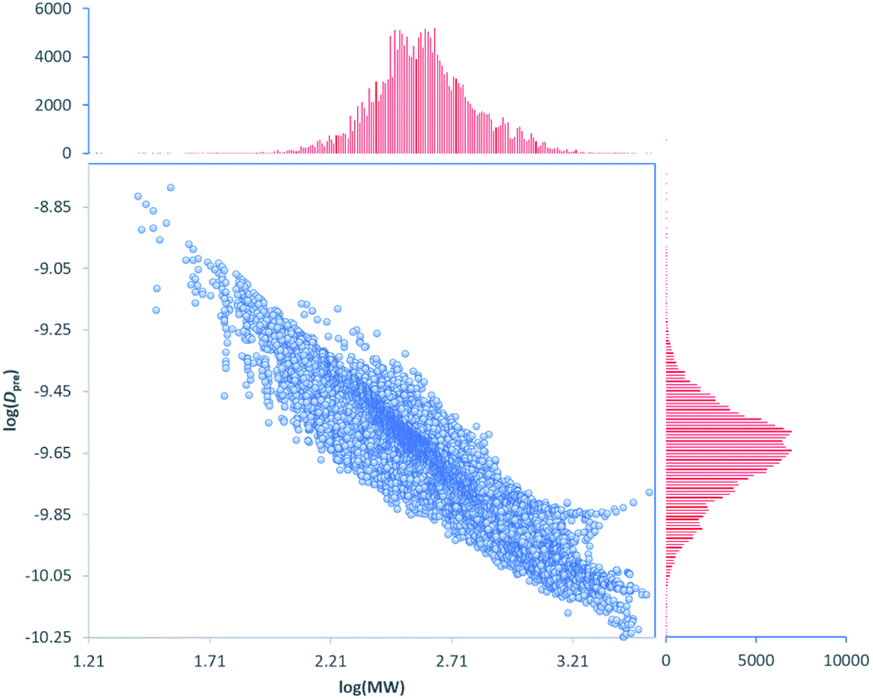 | ||
| Fig. 6 Correlation between log(Dpre) vs. log(MW) and histograms of counts of log(Dpre) and log(MW) for all 217043 compounds in the DEREP-NP database. | ||
In addition, counts of the total number of basic N in a molecule allowed us to predict D (Dpre_TFA) for TFA salts of all alkaloids in the NP-DEREP database.
This is a different approach to use DOSY-based dereplication than that suggested with models 2 and 3 as the D obtained experimentally are compared against computationally predicted Dpre. This provides a useful tool for dereplication, allowing a Dpre filter based on the experimental D to be used in conjunction with other structural filters (such as counts of methyl, sp2 proton etc) derived from the observed proton resonances in the 1H NMR spectrum. We compared the Dpre parameter to experimental D (Dexp) for an additional four NPs not used in the generation of the model. The TFA salts of aerophobin-2 (64), 19-bromoisoeudistomin U (65), and aplysamine 2 (66) all have Dpre within 4% error of their experimental D, while aerothionin (67) had a 9% error.
Validation of models
The accuracy of models 1–4 was tested on a mixture of chromatographically inseparable compounds obtained from the Australian shrub Tasmannia xerophila, also known as Alpine Pepper. The ground leaf material of T. xerophila was extracted with MeOH and CH2Cl2, both fractions were combined and separated by RP HPLC and NP HPLC.The RP HPLC fractions from T. xerophila was found to contained complex mixtures. Analysis of the residuals from the monoexponential fitting of the 1H DOSY data showed a curvature pattern for all residuals in the δH 0.50–7.50 region, indicating signal decay deriving from more than one component.16 This overlap is the major limitation of 1H DOSY experiments, rendering inaccurate D values. Individual D values for overlapping resonances can potentially be extracted using multiexponential processing, however this method cannot distinguish similar D.16 The multiexponential fitting of the 1H DOSY data for the RP HPLC fraction did not result in any improvement, leading to erroneous D for resonances in the δH 0.50–7.50 region.
Baseline interference can be detected prior to acquiring 1H DOSY data through examining the 1H NMR spectrum baseline at high signal intensity. In the case of the RP HPLC fractions (Fig. S8†) there was an uneven broad baseline suggesting that the fractions contained mixtures of larger molecular weight tannins as well as small molecules. To analyze complex mixtures such as this requires a 3D DOSY methodology and this is outside of the scope of this manuscript.
The NP HPLC fractions on the other hand had a cleaner baseline, devoid of tannin signals responsible for the broad baseline. Fraction 10 (F10) predominately contained 1H NMR resonances associated with a mixture of two compounds (i, and ii) in a ratio of 2:1 that coeluted in 70% MeOH by RP HPLC. 1H DOSY data separated these resonances into two bands in the diffusion dimension (Table 1). Applying the power-law MW prediction (model 1) to this data established MWpre = 160 and 185 amu for i, and ii respectively (Table 1 and Fig. 7). The 1H NMR spectrum of the mixture contained a resonance for an exchangeable proton at δH 3.95 that, based on its integral size, could be assigned to compound (ii). Therefore, applying MW prediction using model 2 with effδH values of 0 for i and 3.95 for ii generated MWpre = 202 and 202 amu for i and ii respectively (Table 1 and Fig. 7).
| Compound name | D exp | D pre (% error) | MW amu (% error) | DEREP-NP hitsb | |||||
|---|---|---|---|---|---|---|---|---|---|
| (10−10 m2 s−1) | True | Model 1 | Model 2 | Model 3 | D pre (±5%) | NMR/Dpred | NMRe | ||
| a TFA salt. b DEREP-NP hit compounds obtained using. c Only Dpre set to ±5% of Dexp. d Combination of NMR features and Dpre set to ±5% of Dexp. e Only NMR features. | |||||||||
| Spathulenol (68) | 3.40 | 3.55 (+4.6) | 220 | 185 (−16) | 202 (−8) | 207 (−6) | 8089 | 24 | 914 |
| Cyclocolorenone (69) | 3.72 | 3.70 (−0.6) | 218 | 160 (−27) | 202 (−7) | 180 (−18) | 4612 | 8 | 64 |
| Convolutamine K (70) | 2.65 | 2.76 (+4.1) | 408 | 280 (−31) | 353 (−13) | 22460 |
0 | 1 | |
| Convolutamine Ka (71) | 1.96 | 1.94 (−0.8) | 636 | 460 (−28) | 580 (−9) | 26470 |
0 | 1 | |
| Convolutamine L (72) | 3.26 | 3.44 (+5.4) | 323 | 198 (−39) | 250 (−23) | 9817 | 0 | 4 | |
| Convolutamine La (73) | 2.51 | 2.63 (+4.8) | 437 | 306 (−30) | 386 (−12) | 23272 |
0 | 4 | |
| Volutamine F (74) | 1.77 | 1.70 (−4.1) | 828 | 542 (−35) | 683 (−18) | 19792 |
0 | 0 | |
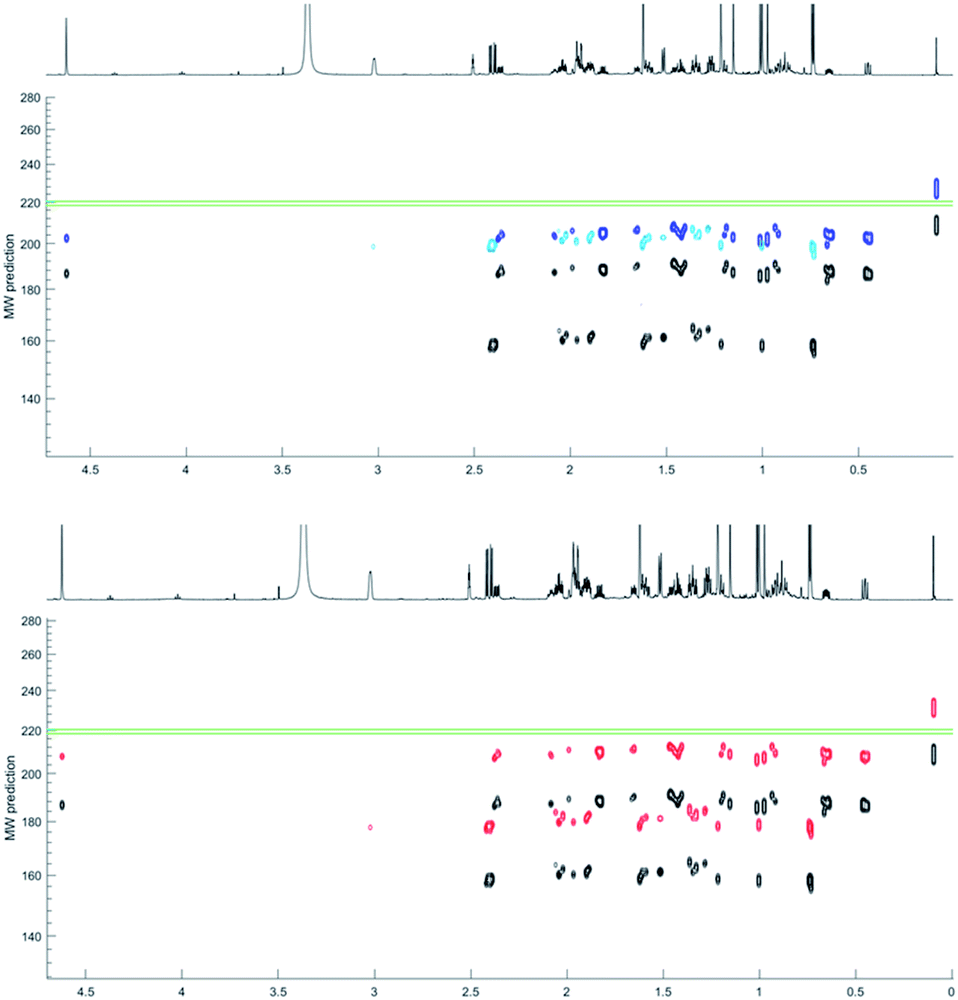 | ||
| Fig. 7 MW prediction improvement with model 2 (upper blue) and model 3 (bottom red) compared to model 1 (both black) for i (faster diffusing) and ii (slower diffusing). Green lines represent true MWs. | ||
Since the two compounds eluted with 70% MeOH by RP HPLC model 3 predicted an average MWpre = 180, and 207 amu for i, and ii respectively (Table 1 and Fig. 7).
Matching Dpre in the DEREP-NP database to Dexp (±5% error) for i generated 4612 hits. Since DEREP-NP was specifically developed as a tool to identify compounds based on NMR features, the additional structural features identified for the faster diffusing compound (i) in the 1H and DOSY NMR spectra included four methyls, a methyl doublet (CH3–CH) two methyl singlets (CH3–Cq) and a deshielded methyl (CH3–C sp2), and incorporating these features as structural filters reduced the number of potential structures to 96. A pair of aliphatic protons with a large mutual 18.5 Hz coupling constant, suggesting a methylene group adjacent to a carbonyl, was present in the molecule, and a CO filter was added reducing the number of hits to 28. Since no protonated sp2 hybridized proton was observed, setting a non-aromatic CH sp2 filter to zero further reduced the number of hit structures to eight.
For ii, the Dpre (±5% Dexp) filter resulted in 8089 hit structures. Other structural filters were then applied based on the observed proton resonances: CH3-Cq for 3 methyl singlets, zero CH sp2 hybridized protons, >3 CH sp3 hybridized protons and a sp2 hybridized CH2 group and a OH, reducing the number of hit structures to 24.
To verify the accuracy of the MW prediction generated by the four models the structures associated with the two diffusion bands of resonance were identified from analysis of 2D NMR data, to be the known sesquiterpenes spathulenol (ii = 68) (MW 220.35) first isolated from Eucalyptus spathulata,46 and cyclocolorenone (i = 69) (MW 218.33) first isolated from Pseudowintera colorata.47
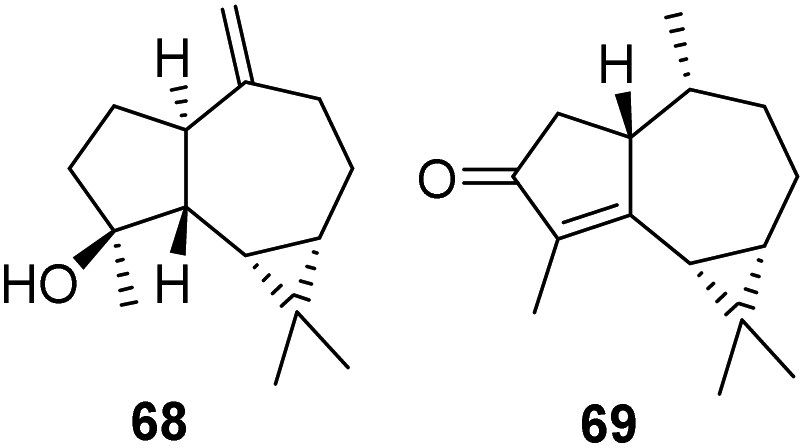
The hydroxyl proton in 68 shows H-bonding with DMSO leading to slower diffusion compared to 69 even though the two compounds have almost identical MW. Each of these compounds were present in the respective hits identified through the DEREP-NP database search. Of the eight hits matched to i three are 69 or its stereoisomer, while five of the 24 possible matched structures for the slower diffusing compound are 68 or its configurational isomers. For both compounds all other matches are sesquiterpenes with molecular weights ±16 amu. Keeping the structural filters determined by 1H NMR but eliminating the Dpre filter (±5% error range) increases the number of possible structures for i from eight to 64, and for ii from 24 to 914 (Table 1), demonstrating the potential of this method for dereplication. Clearly, acquiring a quick 2D experiment, such as an edited HSQC, which provides more structural information, would help to further reduce the number of hits and this approach should be considered as a possible extension for 1H DOSY-based dereplication. To our knowledge, this is the first database to contain predicted D data for compounds.
This real-life example validates that models 2 and 3 more accurately predict MWs than a power-law model (Table 1). H-bonding contributes to the variation in D relative to MW, providing a clear separation in the diffusion dimension for two compounds with only a 2 amu difference in MW. We have recently published the structures of several new brominated alkaloids isolated from the bryozoan Amathia lamourouxi.48 To further test the application of this dereplication methodology, we acquired DOSY data for both the free bases and TFA salts of (70 and 71) and K (72 and 73) and the free base of volutamine F (74). Their Dprevs. Dexp were all within 5.5% error. Applying the Dpre filter (±5%) in DEREP-NP and adding filters for counts of aromatic singlets, aromatic methoxys and N-methyls produced no hits indicating that the compounds were not, as expected, in the database and were thus considered to be new.
Conclusions
The quest to develop a tool to predict MW by NMR has ultimately led us to produce a highly accurate model to predict diffusion coefficients based on structural features. Experimental D can be correlated to predicted D (a surrogate for MW) and this orthogonal physicochemical property along with structural features, both of which are derived from NMR can be used to dereplicate known structures found in databases without the need to acquire mass spectroscopic data. Furthermore, the acquisition of D for compounds in mixtures can be used as a tool to identify new compounds. Recently developed tools such as SMART 2.0 (ref. 11) and MADByTE12 that use 2D NMR data derived from mixtures to dereplicate or predict structures in databases will significantly benefit from an orthogonal tool to correlate predicted D with experimentally derived D (a surrogate for MW). This DOSY methodology is highly applicable to areas outside of NP research and could be adapted more broadly in metabolomics and lipidomics research. We are currently investigating potential to apply the DOSY diffusion coefficient prediction tool to 3D NMR data such as DOSY-COSY and DOSY-HSQC for molecular network matching.Experimental
LC-MS analysis was carried out on an Agilent 6530 Q-TOF mass spectrometer with a 1200 Series autosampler and 1290 Infinity LC module using electrospray ionization with a mobile phase linear gradient of 100% H2O (10 mM ammonium acetate) to 100% MeOH on a Kinetex® 5 μm C18 100 Å (100 × 4.60 mm) column over 10 min. Each compound was injected individually, and retention times were determined in (+) or (−) mode and by UV. The total dwell time of the system was measured by UV and was found to be 1.2 min, this was then subtracted from the retention times of compounds and divided by the total number of mins to afford the % MeOH at which each compound eluted. HPLC separation was performed on a Merck Hitachi L-7100 pump equipped with a L-7455 diode array detector, and fractions were collected with a Gilson 215 liquid handler. The total dwell time of HPLC system was determined to be 3.7 min by UV measurement. The solvents used for HPLC separation were HPLC grade, and solvent used for LC-MS analysis were LC-MS grade. H2O was filtered using a Millipore Milli-Q PF.NMR data was recorded at 298 K on a Bruker Avance III HDX 800 MHz spectrometer with a triple resonance 5 mm cryoprobe, and a Bruker III 500 MHz spectrometer with a 5 mm probe. All compounds were prepared as individual samples in 3 mm NMR tubes with 200 μL of DMSO-d6 with 0.5% (v/v%) TTMS. The 1H DOSY data was acquired without sample spinning at 298 K. The pulse sequence used was LEDBP (ledbpgp2s in the Bruker library) with 32768 data points and 32 scans. The diffusion delay (Δ) was kept constant at 0.1 s and the diffusion pulse (δ) was adjusted to provide ∼90% signal attenuation for each compound, the spoil gradient was 0.6 ms, the gradient recovery delay was 0.2 ms and the eddy current delay was 5 ms. Each experiment was acquired with 24–32 diffusion gradients incremented linearly from 5 to 95% of the maximum gradient strength.
NMR data was processed in TopSpin 3.6.1, without zero-filling and with line broadening of 0.3–0.5 Hz. The 2D DOSY spectra were generated with Dynamics Center 2.5.2 using monoexponential curve fitting. The D for each compound was calculated as the average D of all signals that showed accurate D without any signal overlap. The average D for a compound was referenced to TTMS signal at 3.157 × 10−10 m2 s−1, which was determined from a mixture of only TTMS and DMSO in three different samples.
All calculations were carried out using MATLAB 2016a (The MathWorks, Inc.). The logP calculation and parameters used to establish model 4 were generated in OSIRIS DataWarrior Version 5.2.1 by adding substructure counts to the DEREP-NP database. Dpre and Dpre TFA were generated by applying the formula from model 4 using the “calculated values” function in DataWarrior.
The Tasmannia xerophila plants were purchased from Victorian Alps Nursery, Victoria, Australia. The dried and ground T. xerophila leaf material (80 g) was exhaustively extracted with CH2Cl2 and MeOH, collectively yielding a dark green gum (16 g). The combined extracts were dissolved in MeOH and adsorbed onto C18 silica and loaded to an HPLC cartridge (20 mm × 10 mm) and connected in series to a Betasil 5 μm 100 Å C18 HPLC column (21.2 × 150 mm). The column was eluted with a linear gradient from 100% H2O to 100% MeOH at a flow rate of 9 mL min−1 for 60 minutes with fractions collected every min. This provided mixtures of related compounds throughout all HPLC fractions, including 68 and 69 in fraction 47. Reverse-phase HPLC fractions 25–52 were then combined (189 mg) and adsorbed onto diol-bonded silica and loaded to an HPLC cartridge (20 mm × 10 mm) and connected in series to a YMC-pack diol 5 μm 120 Å HPLC column (21.2 × 150 mm). The column was eluted with a linear gradient from 85% hexane/15% CH2Cl2 to 100% CH2Cl2 over 55 min, then to 90% CH2Cl2/10% MeOH over 20 min at a flow rate of 9 mL min−1 for 60 minutes with fractions collected every min, providing a mixture of 68 and 69 in fraction 10 (0.6 mg).
Data availability
The ESI file contains data used to develop the MW and D prediction models. The DEREP-NP database containing predicted D can be downloaded at https://github.com/guykl/.Author contributions
Contributions to the preparation of this manuscript are as follows; GK: conceptualization, data curation, formal analysis, investigation, methodology, resources, validation, visualization, writing – original draft, writing – review & editing, DH: investigation, resources, writing – review & editing, JP: investigation, resources, AC: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Frederic Leusch for providing several compounds used to establish calibration curve. We thank Wendy Loa-Kum-Cheung for her assistance with NMR data acquisition.Notes and references
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141–2153 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. P. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrov, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. De Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- A. T. Aron, E. C. Gentry, K. L. McPhail, L. F. Nothias, M. Nothias-Esposito, A. Bouslimani, D. Petras, J. M. Gauglitz, N. Sikora, F. Vargas, J. J. J. van der Hooft, M. Ernst, K. Bin Kang, C. M. Aceves, A. M. Caraballo-Rodríguez, I. Koester, K. C. Weldon, S. Bertrand, C. Roullier, K. Sun, R. M. Tehan, C. A. Boya P, M. H. Christian, M. Gutiérrez, A. M. Ulloa, J. A. Tejeda Mora, R. Mojica-Flores, J. Lakey-Beitia, V. Vásquez-Chaves, Y. Zhang, A. I. Calderón, N. Tayler, R. A. Keyzers, F. Tugizimana, N. Ndlovu, A. A. Aksenov, A. K. Jarmusch, R. Schmid, A. W. Truman, N. Bandeira, M. Wang and P. C. Dorrestein, Nat. Protoc., 2020, 15, 1954–1991 CrossRef CAS PubMed.
- J. Hubert, J. M. Nuzillard and J. H. Renault, Phytochem. Rev., 2017, 16, 55–95 CrossRef CAS.
- K. Scheubert, F. Hufsky, D. Petras, M. Wang, L. Nothias, K. Dührkop, N. Bandeira, P. C. Dorrestein and S. Böcker, Nat. Commun., 2017, 8, 1494 CrossRef PubMed.
- O. Corcoran and M. Spraul, Drug Discovery Today, 2003, 8, 624–631 CrossRef CAS PubMed.
- O. Gökay and K. Albert, Anal. Bioanal. Chem., 2012, 402, 647–669 CrossRef PubMed.
- C. L. Zani and A. R. Carroll, J. Nat. Prod., 2017, 80, 1758–1766 CrossRef CAS PubMed.
- L. Buedenbender, L. J. Habener, T. Grkovic, D. İ. Kurtböke, S. Duffy, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2018, 81, 957–965 CrossRef CAS PubMed.
- R. Reher, H. W. Kim, C. Zhang, H. H. Mao, M. Wang, L.-F. Nothias, A. M. Caraballo-Rodriguez, E. Glukhov, B. Teke, T. Leao, K. L. Alexander, B. M. Duggan, E. L. Van Everbroeck, P. C. Dorrestein, G. W. Cottrell and W. H. Gerwick, J. Am. Chem. Soc., 2020, 142, 4114–4120 CrossRef CAS PubMed.
- J. M. Egan, J. A. van Santen, D. Y. Liu and R. G. Linington, J. Nat. Prod., 2021, 84, 1044–1055 CrossRef CAS PubMed.
- T. D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Elsevier, Boston, 3rd edn, 2016 Search PubMed.
- D. Li, G. Kagan, R. Hopson and P. G. Williard, J. Am. Chem. Soc., 2009, 131, 5627–5634 CrossRef CAS PubMed.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- M. Nilsson, M. A. Connell, A. L. Davis and G. A. Morris, Anal. Chem., 2006, 78, 3040–3045 CrossRef CAS PubMed.
- M. Nilsson and G. A. Morris, Magn. Reson. Chem., 2006, 44, 655–660 CrossRef CAS PubMed.
- M. Nilsson, A. M. Gil, I. Delgadillo and G. A. Morris, Chem. Commun., 2005, 1737–1739 RSC.
- G. Dal Poggetto, L. Castañar, M. Foroozandeh, P. Kiraly, R. W. Adams, G. A. Morris and M. Nilsson, Anal. Chem., 2018, 90, 13695–13701 CrossRef CAS PubMed.
- M. Foroozandeh, L. Castañar, L. G. Martins, D. Sinnaeve, G. D. Poggetto, C. F. Tormena, R. W. Adams, G. A. Morris and M. Nilsson, Angew. Chem., Int. Ed., 2016, 55, 15579–15582 CrossRef CAS PubMed.
- A. Chen, D. Wu and C. S. Johnson, J. Am. Chem. Soc., 1995, 117, 7965–7970 CrossRef CAS.
- W. Li, H. Chung, C. Daeffler, J. A. Johnson and R. H. Grubbs, Macromolecules, 2012, 45, 9595–9603 CrossRef CAS PubMed.
- D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2009, 42, 270–280 CrossRef CAS PubMed.
- R. T. Williamson, E. L. Chapin, A. W. Carr, J. R. Gilbert, P. R. Graupner, P. Lewer, P. McKamey, J. R. Carney and W. H. Gerwick, Org. Lett., 2000, 2, 289–292 CrossRef CAS PubMed.
- M. Tsuda, T. Yasuda, E. Fukushi, J. Kawabata, M. Sekiguchi, J. Fromont and J. Kobayashi, Org. Lett., 2006, 8, 4235–4238 CrossRef CAS PubMed.
- J. A. Jones, D. K. Wilkins, L. J. Smith and C. M. Dobson, J. Biomol. NMR, 1997, 10, 199–203 CrossRef CAS.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- S. Augé, P. Schmit, C. A. Crutchfield, M. T. Islam, D. J. Harris, E. Durand, M. Clemancey, A.-A. Quoineaud, J. Lancelin, Y. Prigent, F. Taulelle and M.-A. Delsuc, J. Phys. Chem. B, 2009, 113, 1914–1918 CrossRef PubMed.
- C. A. Crutchfield and D. J. Harris, J. Magn. Reson., 2007, 185, 179–182 CrossRef CAS PubMed.
- S. Bachmann, R. Neufeld, M. Dzemski and D. Stalke, Chem.–Eur. J., 2016, 22, 8462–8465 CrossRef CAS PubMed.
- E. J. Cabrita and S. Berger, Magn. Reson. Chem., 2001, 39, S142–S148 CrossRef CAS.
- G. S. Kapur, E. J. Cabrita and S. Berger, Tetrahedron Lett., 2000, 41, 7181–7185 CrossRef CAS.
- M. H. Abraham, P. L. Grellier, D. V Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 699 RSC.
- M. H. Abraham, Chem. Soc. Rev., 1993, 22, 73 RSC.
- M. H. Abraham, R. J. Abraham, J. Byrne and L. Griffiths, J. Org. Chem., 2006, 71, 3389–3394 CrossRef CAS PubMed.
- M. H. Abraham, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2867–2881 RSC.
- S. P. D. Senadeera, L. Lucantoni, S. Duffy, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2018, 81, 1588–1597 CrossRef CAS PubMed.
- A. K. Kreyenschmidt, S. Bachmann, T. Niklas and D. Stalke, ChemistrySelect, 2017, 2, 6957–6960 CrossRef CAS.
- S. P. D. Senadeera, S. Duffy, V. M. Avery and A. R. Carroll, Bioorg. Med. Chem. Lett., 2017, 27, 2602–2607 CrossRef CAS PubMed.
- P. S. Pregosin, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 261–288 CrossRef CAS.
- E. J. Cabrita and S. Berger, Magn. Reson. Chem., 2002, 40, S122–S127 CrossRef CAS.
- A. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- K. Valkó, C. Bevan and D. Reynolds, Anal. Chem., 1997, 69, 2022–2029 CrossRef PubMed.
- C. M. Du, K. Valko, C. Bevan, D. Reynolds and M. H. Abraham, J. Liq. Chromatogr. Relat. Technol., 2001, 24, 635–649 CrossRef CAS.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- R. C. Bowyer and P. R. Jefferies, Chem. Ind., 1963, 1245–1246 CAS.
- R. E. Corbett and R. N. Speden, J. Chem. Soc., 1958, 3710–3715 RSC.
- G. Kleks, D. C. Holland, E. K. Kennedy, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2020, 83, 3435–3444 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Structures of all compounds used in the analysis, tables of data used to generate molecular weight and diffusion co-efficient predictions and figures associated with models 1, 2, 3 and 4. See DOI: 10.1039/d1sc02940a |
| ‡ The DEREP-NP database in DataWarrior format (dwr) with predicted D can be downloaded at https://github.com/guykl/. |
| This journal is © The Royal Society of Chemistry 2021 |