
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning of solvent effects on molecular spectra and reactions†
Michael
Gastegger
 *a,
Kristof T.
Schütt
ab and
Klaus-Robert
Müller
abcd
*a,
Kristof T.
Schütt
ab and
Klaus-Robert
Müller
abcd
aMachine Learning Group, Technische Universität Berlin, 10587 Berlin, Germany. E-mail: michael.gastegger@tu-berlin.de
bBerlin Institute for the Foundations of Learning and Data, 10587 Berlin, Germany
cDepartment of Artificial Intelligence, Korea University, Anam-dong, Seongbuk-gu, Seoul 02841, Korea
dMax-Planck-Institut für Informatik, 66123 Saarbrücken, Germany
First published on 23rd July 2021
Abstract
Fast and accurate simulation of complex chemical systems in environments such as solutions is a long standing challenge in theoretical chemistry. In recent years, machine learning has extended the boundaries of quantum chemistry by providing highly accurate and efficient surrogate models of electronic structure theory, which previously have been out of reach for conventional approaches. Those models have long been restricted to closed molecular systems without accounting for environmental influences, such as external electric and magnetic fields or solvent effects. Here, we introduce the deep neural network FieldSchNet for modeling the interaction of molecules with arbitrary external fields. FieldSchNet offers access to a wealth of molecular response properties, enabling it to simulate a wide range of molecular spectra, such as infrared, Raman and nuclear magnetic resonance. Beyond that, it is able to describe implicit and explicit molecular environments, operating as a polarizable continuum model for solvation or in a quantum mechanics/molecular mechanics setup. We employ FieldSchNet to study the influence of solvent effects on molecular spectra and a Claisen rearrangement reaction. Based on these results, we use FieldSchNet to design an external environment capable of lowering the activation barrier of the rearrangement reaction significantly, demonstrating promising venues for inverse chemical design.
1 Introduction
The presence of a solvent can dramatically change molecular properties as well as the outcome of reactions.1,2 Hence, a profound understanding of the interactions between molecules and their environments is tantamount not only for rationalizing experimental results, but also guiding the way towards controlling, or even designing, reactions and properties.1,3,4 While computational chemistry has described such phenomena with reasonable success, obtaining accurate predictions which can be related to experiment is a highly non-trivial task. Due to the large number of species involved, treating a system and its environment entirely with electronic structure theory is prohibitively expensive, especially for accurate high-level methods. Approximate schemes, on the other hand, are often unable to capture important physical aspects, such as structural features of the environment in the case of continuum models or chemical reactions in the case of classical force-fields. To overcome these issues, we propose a deep neural network potential that includes the influence of external fields, to capture the interactions of the chemical system with the environment.Recently, machine learning (ML) methods have emerged as a powerful strategy to overcome this trade-off between accuracy and efficiency inherent to computational chemistry approaches.5–8 Highly efficient ML models now provide access not only to interatomic potential energy surfaces,9–17 but also to a growing range of molecular properties.18–24 Specialized ML architectures have been developed for the prediction of vectorial and tensorial quantities,25–27 such as dipole moments,28 polarizabilities29 and non-adiabatic coupling vectors.30 Such advances pave the way for using ML models in practical applications, with the simulation of infrared,28 Raman,31,32 ultraviolet33 and nuclear magnetic resonance (NMR) spectra34 being only a few examples. At the same time, there is an ongoing effort to incorporate more physical knowledge into ML algorithms, giving rise to semi-empirical ML schemes35,36 and even models based on electron densities37–39 and wavefunctions.40–42
Most of these approaches operate on closed systems, where the molecule is not subjected to any external environment. Christensen et al.43 proposed a general framework for modeling response properties with kernel methods. In this context, and in order to model electric field dependent properties, the FCHL representation44 was extended by an electric field model based on rough estimates of atomic partial charges. This makes it possible to predict various response properties beyond atomic forces, such as dipole moments across compositional space as well as static infrared spectra. Note that this approach inherently relies on molecular representations which are able to capture the required perturbations of the energy.
In this work, we propose the FieldSchNet deep learning framework, which models the interactions with arbitrary environments in the form of vector fields. As a consequence, our model is able to describe various implicit and explicit interactions with the molecular environment using external fields as a physically motivated interface. Moreover, the field-based formalism automatically grants access to first and higher order response functions of the potential energy, with polarizabilities and nuclear magnetic shielding tensors being only a few examples. This enables the prediction of a wide range of molecular spectra (e.g. infrared, Raman and NMR) without the need for additional, specialized ML models.
FieldSchNet can be used as a polarizable continuum model for solvation45 or to interact with an electrostatic field generated by explicit external charges in an QM/MM-like setup46 – an approach we refer to as ML/MM. As the model offers speed-ups of up to four orders of magnitude compared to the original electronic structure reference, we can employ FieldSchNet to investigate the influence of solvent effects on the Claisen rearrangement reaction of allyl-p-tolyl ether – a study out of reach for conventional high-level electronic structure or ML approaches. While these simulations would take 18 years with the original electronic structure reference, they could be performed within 5 hours with FieldSchNet.
FieldSchNet goes well beyond the scope of previous ML approaches by providing an analytic description of a chemical system in its environment. We exploit this feature by designing an external field in order to minimize the height of the reaction barrier in the above Claisen reaction. Hence, FieldSchNet constitutes a unified framework that enables not only the prediction of spectroscopic properties of molecules in solution, but even the inverse chemical design of catalytic environments.
2 FieldSchNet
2.1 Field-dependent representation
FieldSchNet allows to model interactions between atomistic systems and external fields εext(R) in a natural way. It predicts molecular energies and properties based on local representations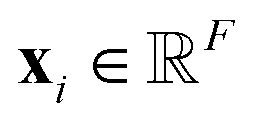 of atomic environments embedded in external vector fields, where F is the number of features. These representations are constructed iteratively, where the representation xli of atom i in each layer l is refined via interactions with the neighboring atoms (see Fig. 1a)
of atomic environments embedded in external vector fields, where F is the number of features. These representations are constructed iteratively, where the representation xli of atom i in each layer l is refined via interactions with the neighboring atoms (see Fig. 1a)| xl+1i = xli + wli + uli + vli, | (1) |
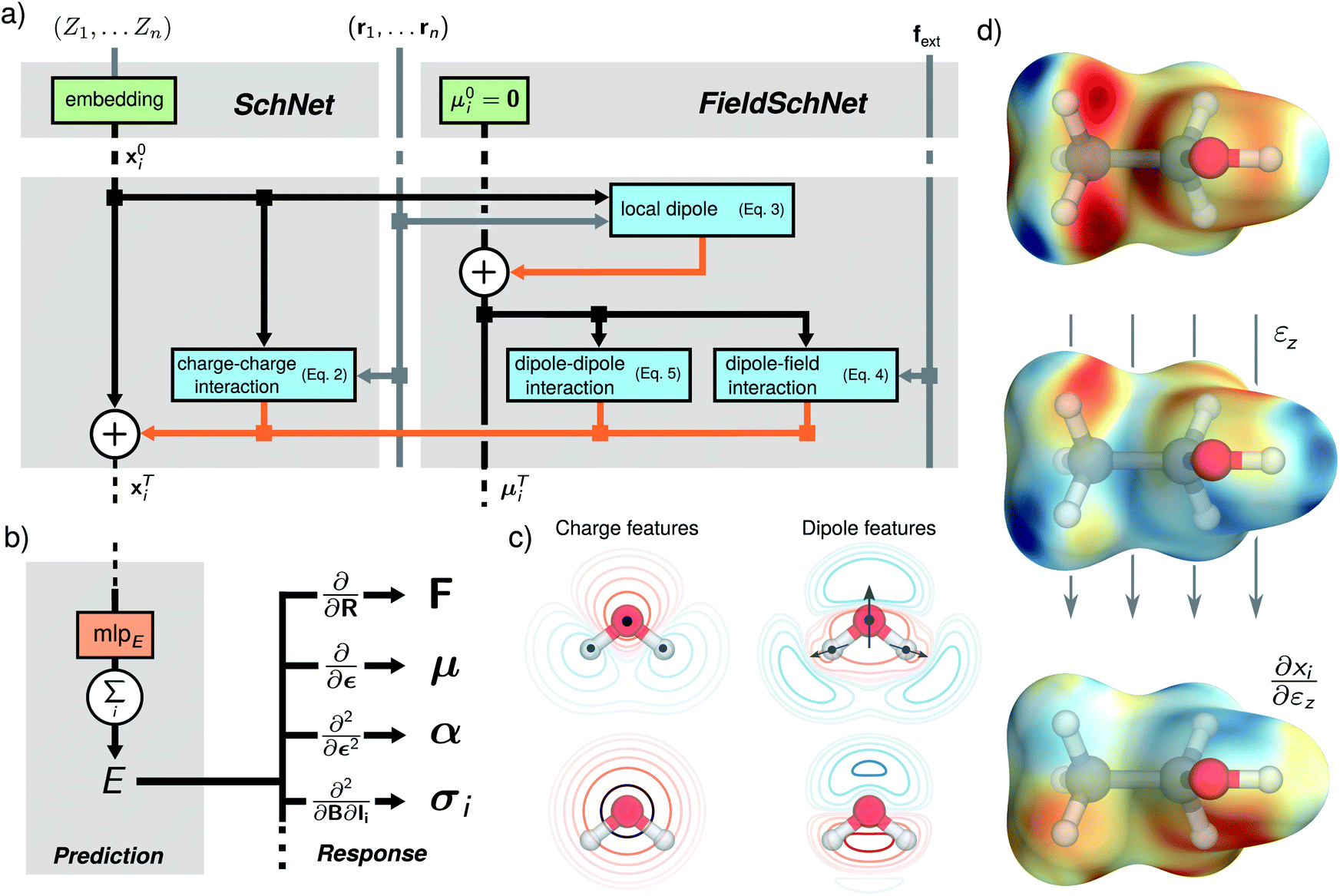 | ||
| Fig. 1 Network architecture and learned representations. (a) Scheme of the network blocks for generating the field-dependent representation. Starting from an initial representation of atom types and zero vectors for the dipole features, the conventional SchNet features xi are augmented by a set of local dipole features μi. Interactions between the dipole features and the dipole features with the external field are used to update the representation xTi in each layer. (b) The energies are predicted from the final set of features based on a sum of atomic contributions. By applying the appropriate derivative operations to the energy, different response properties can be computed, such as molecular forces F, dipole moments μ, polarizabilities α and shielding tensors σi. (c) Schematic comparison of charge and dipole based interactions in the water molecule shown for the whole molecule (top) and the oxygen atom (bottom). The introduction of dipoles allows for a more fine grained spatial resolution of the environment which is particularly clear to see in the case of the atomic oxygen contributions where charge like features only capture radial dependencies. (d) Representations for a hydrogen probe at position R projected onto a ∑i|Ri − R|2 isosurface, shown for the ethanol molecule. From top to bottom: FieldSchNet descriptor without an external field, FieldSchNet descriptor in the presence of an external field applied to the z-plane of the molecule and response of the descriptor with respect to the z-component of an external field. | ||
Starting from an initial embedding depending only on the respective atom type. Here, wli is the standard SchNet interaction (Fig. 1a, left block), while the added terms uli and vli correspond to dipole-field and dipole–dipole interactions (Fig. 1a, right block). The SchNet interaction update then takes the form
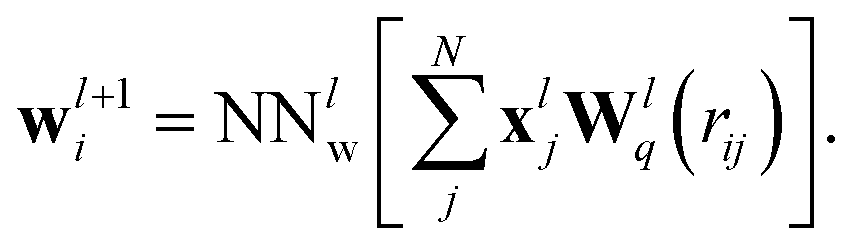 | (2) |
The radial interaction functions Wlq depend on the interatomic distance rij and are learned from reference data. A fully-connected neural network is applied afterwards performing a non-linear transformation. From here on, we will represent networks of this type as NNlw, where the superscript l indicates the current interaction layer and the subscript different sets of parameters.
In terms of rotational symmetry, the SchNet feature refinements can be interpreted as charge–charge interactions, as the features of xi are scalars. Hence, the internal structure of SchNet, as well as the generated representation, is invariant with respect to rotations and translations of the molecule. This is an important requirement for machine learning potentials, as the energy of an atomic system exhibits the same symmetry.47 However, this invariance breaks down in the presence of external fields. In this case, models also need to be constructed such that they are able to resolve rotations and translations relative to an external frame of reference.
FieldSchNet achieves this requirement by introducing an additional vector valued representation based on atomic dipole moments 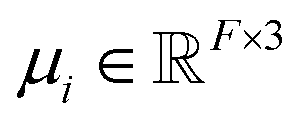 (left side of Fig. 1a), which is refined analogous to eqn (1):
(left side of Fig. 1a), which is refined analogous to eqn (1):
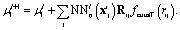 | (3) |
Based on these features, FieldSchNet models the interaction between molecule and external fields uli with the term
| uli = NNlε[(μli)Tεext(Ri)], | (4) |
In addition to the dipole-field interaction, FieldSchNet introduces an update vli based on the interaction between neighboring dipoles
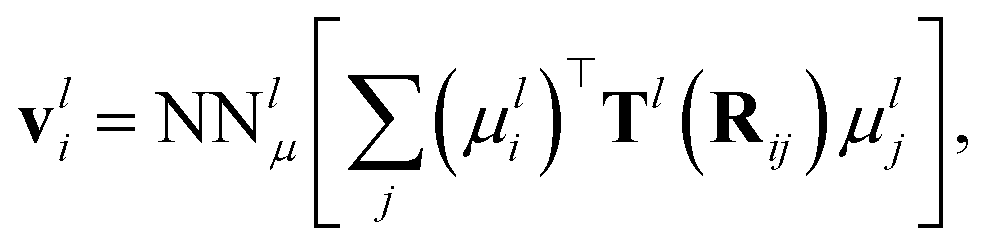 | (5) |
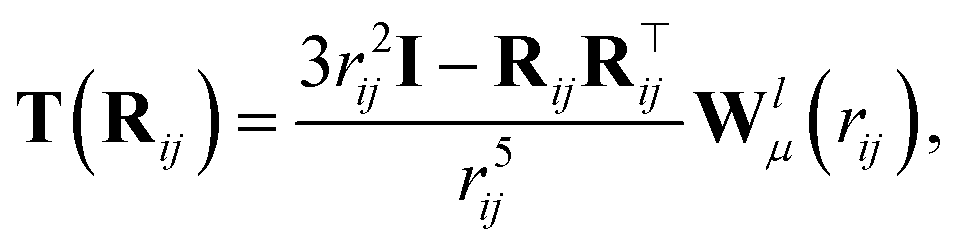 | (6) |
For each external field, FieldSchNet adds a set of dipole features and expands the update in eqn (1) by the corresponding dipole-field and dipole–dipole terms. As the scalar features xi of the next layer depend on the dipole and field interactions of the current layer, those in turn are coupled with the preceding scalar features via the dipoles (eqn (3)). This allows FieldSchNet to capture interactions with external fields far beyond the linear regime.
As visualized in Fig. 1d at the example of an ethanol molecule, the FieldSchNet descriptor exhibits the same symmetry as the molecule (top). Upon introducing an electric field in the z-axis of the molecule, the descriptor adapts to the changed environment and the original symmetry is broken (middle). This effect can also be observed in the response of the descriptor with respect to the field (bottom). At the same time, multiple successive SchNet and dipole–dipole updates enable the efficient construction of higher-order features of the molecular structure (see Fig. 1c), going beyond the purely radial dependence of charge-like features. Although inspired by electric dipoles μ, the dipole-like representations in FieldSchNet are auxiliary constructs and may also represent other quantities, e.g. the nuclear magnetic moments Ii when modeling magnetic fields. An appropriate form of the dipoles corresponding to each field is learned purely data-driven.
Once the FieldSchNet representation xi has been constructed, the potential energy is predicted via the atomic energy contributions typical for atomistic networks (Fig. 1b)
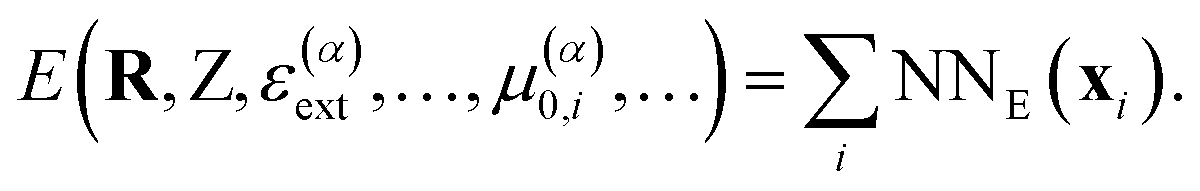 | (7) |
Since the features xi depend on the interactions between dipoles and fields of each previous layer and the dipoles are in turn constructed from the features, the potential energy is now a function of the atomic positions, nuclear charges and all external fields as well as initial atomic dipoles. This makes it possible to obtain response properties from the energy model by taking the corresponding derivatives.
2.2 Modeling continuum solvation
A FieldSchNet-based machine learning potential for continuum solvent effects can be derived by adapting the Onsager expression for the reactive field.48 Modeling the external electric field εsolv experienced by each atom i due to a continuum solvent with dielectric constant ε as | (8) |
| aWli = NNla(xli), | (9) |
2.3 Hybrid machine learning/molecular mechanics (ML/MM)
We adopt a QM/MM-like approach for FieldSchNet by coupling the classical and quantum region with electrostatic embedding, resulting in a ML/MM approach. In this case, the quantum region is polarized by the charges of the classical region, while the electrostatic energy of the classical region is in turn modified by the point charges computed for the quantum region. The influence of the external charges on the molecule can be modeled via the electrostatic field exerted by a collection of point charges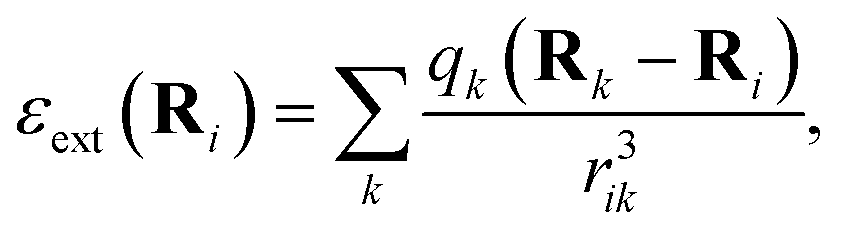 | (10) |
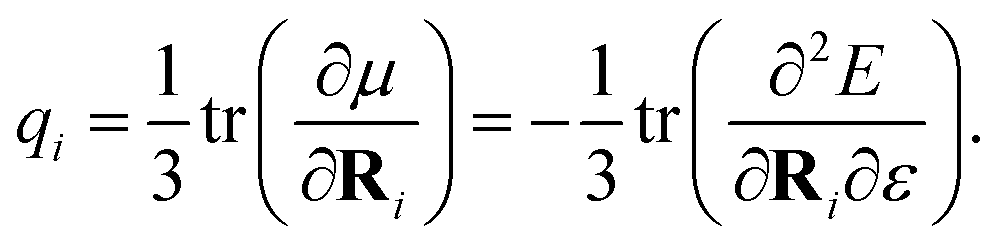 | (11) |
These charges are fully polarized charges, depending not only on the molecular structure but also the charge distribution of the environment.
3 Results and discussion
3.1 Molecular spectroscopy
FieldSchNet is particular promising for the prediction of molecular spectra, as a single model provides access to a wide range of response properties. A variety of spectra can be simulated in this manner, ranging from vibrational spectra such as infrared and Raman to nuclear magnetic resonance spectra. The availability of molecular forces makes it possible to go beyond static approximations and even derive vibrational spectra from molecular dynamics simulations.50 Moreover, the high computational efficiency of the machine learning model renders otherwise costly path integral molecular dynamics (PIMD) simulations feasible, which are able to account for nuclear quantum effects51 and yield predicted spectra close to experiment.52We train a single FieldSchNet model on reference data generated with the PBE0 functional for an ethanol molecule in vacuum. The dataset is based on the MD17 ethanol data53 and contains a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures (see ESI† for more details). A combined loss function incorporates the energy (E), atomic forces (F), dipole moment (μ), polarizability (α) and nuclear shielding tensors (σ) as target properties. Details on reference data generation and training are provided in the ESI.† Excellent fits were obtained for all quantities, as can be seen based on the test accuracy reported in ESI Table S3.† Energies and force predictions fall well within chemical accuracy, with mean absolute errors (MAEs) of 0.017 kcal mol−1 and 0.128 kcal mol−1 Å−1. The response properties of the electric field exhibit MAEs as low as 0.04 D (μ) and 0.008 bohr3 (α) compared to value ranges of 4.56 D and 13.58 bohr3 present in the reference data. In a similar manner, FieldSchNet yields low MAEs for the shielding tensor σ, exhibiting an error of 0.123 ppm for hydrogen (range of 29.43 ppm) and 0.194 ppm for carbon atoms (range of 153.94 ppm).
000 structures (see ESI† for more details). A combined loss function incorporates the energy (E), atomic forces (F), dipole moment (μ), polarizability (α) and nuclear shielding tensors (σ) as target properties. Details on reference data generation and training are provided in the ESI.† Excellent fits were obtained for all quantities, as can be seen based on the test accuracy reported in ESI Table S3.† Energies and force predictions fall well within chemical accuracy, with mean absolute errors (MAEs) of 0.017 kcal mol−1 and 0.128 kcal mol−1 Å−1. The response properties of the electric field exhibit MAEs as low as 0.04 D (μ) and 0.008 bohr3 (α) compared to value ranges of 4.56 D and 13.58 bohr3 present in the reference data. In a similar manner, FieldSchNet yields low MAEs for the shielding tensor σ, exhibiting an error of 0.123 ppm for hydrogen (range of 29.43 ppm) and 0.194 ppm for carbon atoms (range of 153.94 ppm).
In the following, we simulate a range of spectra using the multi-property FieldSchNet model (see ESI† for computational details). Fig. 2a shows infrared spectra obtained from static calculations and the dipole time-autocorrelation functions simulated by molecular dynamics and ring-polymer PIMD. In addition, a static spectrum obtained with the reference method, as well as the gas-phase experimental spectrum are provided.54 While the FieldSchNet model is able to reproduce the static reference almost exactly, comparison of both spectra to experiment demonstrates the drawbacks of relying on purely static calculations for the prediction of vibrational spectra in general.
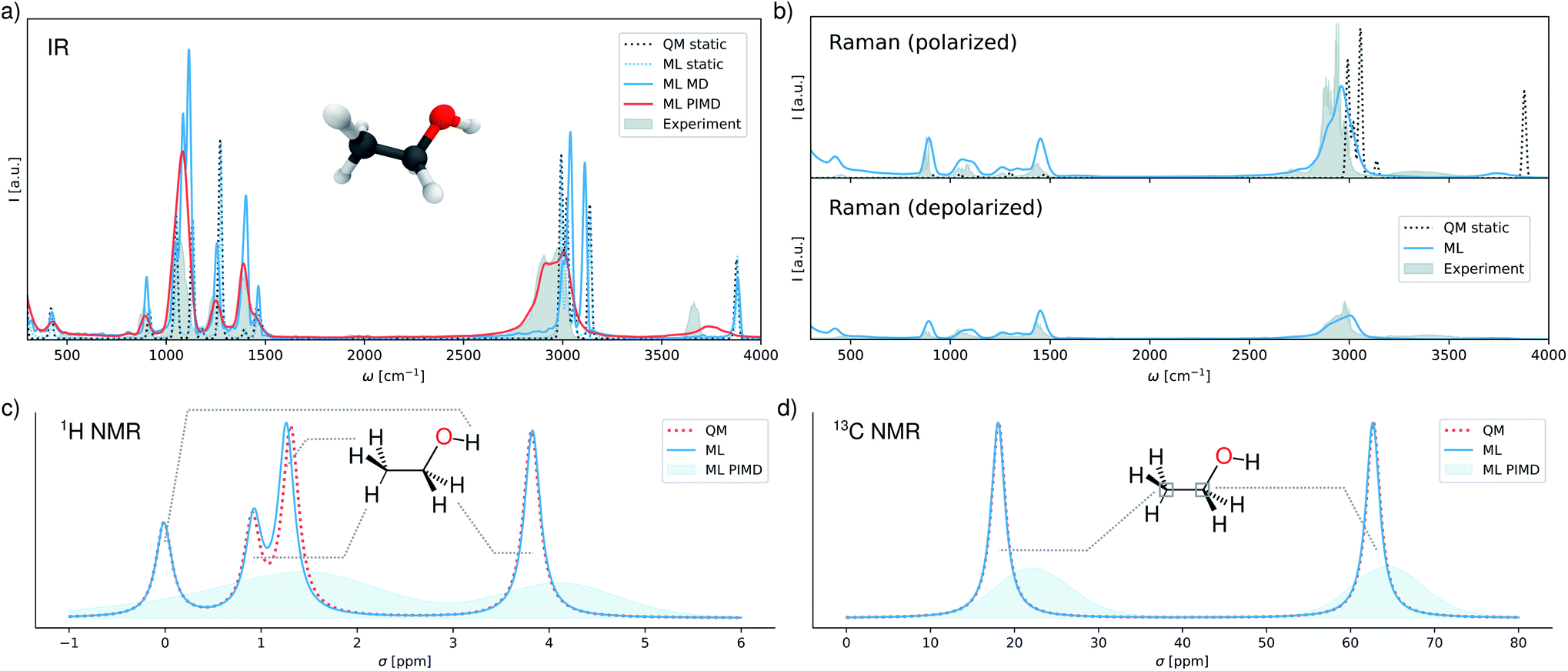 | ||
| Fig. 2 Predicted spectra (a) infrared spectra for ethanol in vacuum predicted by the different methods compared to an experimental spectrum. (b) Polarized and depolarized Raman spectra simulated with FieldSchNet. The experimental spectra are shown in gray. (c and d) 1H and 13C chemical shifts predicted by the model and reference method, as well as the shift distributions sampled during ring-polymer molecular dynamics. | ||
The potential of the machine learning model becomes apparent when going beyond the static picture. In order to predict vibrational spectra from molecular dynamics simulations, a large number of successive computations are necessary, which quickly become prohibitive when relying on electronic structure methods. However, due to the computational efficiency of FieldSchNet, these simulations can be carried out with little effort. A single evaluation of all response properties takes 220 ms on a Nvidia Tesla P100 GPU compared to a computation time of 207 s with the original electronic structure method on a Intel Xeon E5-2690 CPU, a speedup by almost three orders of magnitude. As a consequence, a simulation which would take 240 days with conventional approaches can now be performed in approximately 6 hours. This huge step in efficiency grows even stronger for larger systems.
The benefits of performing molecular dynamics simulations can be observed in the spectrum recovered in this manner, as it exhibits a much better agreement with experiment in the low frequency regions. Nevertheless, this approach still fails to reproduce positions and intensities of the bands associated with the stretching vibrations of the C–H bonds (∼3000 cm−1) and the O–H bond (∼3600 cm−1). These differences are primarily due to the neglect of anharmonic and nuclear quantum effects. One way to include these effects is via PIMD, where multiple coupled replicas of the molecule are simulated. While this approach is computationally more demanding than classical molecular dynamics due to the additional replicas, the simulations can still be carried out efficiently with FieldSchNet. As can be seen in Fig. 2a, accounting for anharmonic effects does indeed shift the C–H stretching vibrations to the experimental wavelengths and improves the position the O–H band, yielding a predicted spectrum close to experiment.
In addition to infrared spectra, FieldSchNet enables the simulation of Raman spectra, which rely on molecular polarizabilities α. Since the full polarizability tensor is predicted by the model, it is possible to compute polarized as well as depolarized Raman spectra. Fig. 2b depicts both types of spectra as obtained with FieldSchNet via path integral molecular dynamics, as well as their experimental counterparts.55 In both cases, very good agreement with experiment is observed, with the most prominent difference being once again the vibrations in the O–H stretching regions. The quality of the predicted spectra is particularly striking, when comparing to the statically computed polarized Raman spectrum, which fails to reproduce the shapes and magnitudes of several peaks.
Beyond vibrational spectra, FieldSchNet can be used to obtain NMR chemical shifts via the nuclear shielding tensors σi. In this manner, chemical shifts can be obtained for all NMR active isotopes, which in the case of ethanol are 1H, 13C and 17O. The predicted and reference chemical shifts for the equilibrium configuration of ethanol are provided in Fig. 2c and d. In addition, the distribution of shifts sampled during PIMD simulations are shown. 17O shifts are omitted from the analysis, as only one oxygen nucleus is present. However, the shifts are still reproduced accurately and the associated error is given in ESI Table S3.† FieldSchNet predictions agree closely with the reference method for the 1H and 13C isotopes. The 1H chemical shifts of the hydrogens in the CH2 and CH3 groups are close to their expected experimental values of 1.2 ppm and 3.8 ppm, respectively. The peak shifted to 1 ppm is associated with the hydrogen atom in plane with the O–H bond. The resulting band structure vanishes in the molecular dynamics simulation due to rotations of the methyl group. A major disagreement with experiment is the shift of the hydrogen in the O–H group which shows uncharacteristically low values of 1 ppm. This can be attributed to a shortcoming of the reference method, which exhibits the same behavior. The 13C shifts of the CH2 and CH3 carbon atoms agree almost perfectly with their experimental values of ∼60 ppm and ∼20 ppm.
3.2 Implicit environments with polarizable continuum models
Accounting for effects of the molecular environment and solvent effects in particular is crucial for a wide range of chemical applications. These effects can critically influence the properties of compounds and the outcome of chemical reactions. Due to the large number of species involved, treating a molecule and surrounding environment entirely with electronic structure methods is impractical. In the case of solutions, approximating the solvent by a polarizable continuum model (PCM) with specific dielectric constant ε has proven as a powerful tool.45By using an expression for the external field adapted from the reaction field approach of Onsager,48 FieldSchNet can operate as a machine learning model for polarizable continuum solvents with an explicit dependency on ε (see Section 2.2). To study this mode of operation, we train such a polarizable continuum FieldSchNet (pc-FieldSchNet) on a reference data set composed of computations for a ethanol molecule in the gas phase, as well as ethanol (ε = 24.3) and water (ε = 80.4) continuum solvents. To this end, the 10000 ethanol structures in vacuum were recomputed for each environment (ethanol and water) and the combined dataset was then screened for numerical instabilities due to the cavity generation process in the PCM computations, yielding a total of 27990 reference structures (see ESI† for details). Again, the model is trained to predict the potential energy, the atomic forces and the response properties for the molecular spectra. pc-FieldSchNet reproduces all quantities with high accuracy, comparable with that of the model for response properties in vacuum (see ESI Table S3†). This demonstrates that pc-FieldSchNet is able to learn the correct dependence on the specific dielectric constant ε.
ESI Table S3† furthermore shows results for two benchmark datasets of methanol (ε = 32.63) and toluene (ε = 10.3), solvents that have not been included in training. We find that pc-FieldSchNet generalizes well to these solvents with errors comparable to the prediction of solutions used for training. The accuracy for toluene is slightly lower with the polarizability showing particularly high mean absolute errors of 0.243 bohr3. However, this is can be attributed to an insufficient sampling of the regions of low polarity, as the most similar solvent included in training is vacuum. The methanol dataset is reproduced with high accuracy, demonstrating that the model is able to generalize across unseen continuum solvents.
3.3 Explicit environments with ML/MM
Although continuum models are powerful tools for the description of solutions, they break down in situations where direct interactions between molecule and environment need to be considered, e.g. solute–solvent hydrogen bonds. In these cases, the solvent has to be treated explicitly, e.g. using QM/MM schemes.46,56 These retain the full atomistic structure of the environment but instead treat it with more affordable classical force fields, while the molecule itself is described with electronic structure methods. By expressing the external electric field as the field generated by the point charges of the MM region, FieldSchNet can operate in a similar manner and replace the quantum region in such a simulation, essentially yielding an ML/MM approach. We use generalized atomic polar tensor charges49 computed with FieldSchNet to model the electrostatic interactions between both regions (see Section 2.3 for details). In the following, we study the impact of using an ML/MM solvent model on the simulated infrared spectrum of liquid ethanol.We train a FieldSchNet model on a set of PBE0 reference data of 30000 ethanol configurations polarized by external charge distributions that have been sampled from a classical force field (three classical ethanol environments for each vacuum structure, more details on reference data generation and model training can be found in the ESI†). The test set performance of the resulting model is provided in ESI Table S3.† We observe slightly increased errors for the ML/MM model in energy and forces compared to the vacuum and continuum models due to the more complex environment. Still, the model is able to reach high accuracy.
ML/MM molecular dynamics simulations are carried out for a single machine learning ethanol surrounded by a solvent box of 1250 ethanol molecules treated at force field level (see ESI† for computational details.). The resulting infrared spectrum is depicted in Fig. 3, alongside spectra computed in the same manner using the vacuum model (Section 3.1) and continuum model (ε = 24.3), as well as an experimental spectrum of liquid ethanol.57 Comparing the gas phase and continuum models, we find that in this case implicit solvent effects lead to no major improvements with respect to experiment. The spectrum simulated via the ML/MM approach on the other hand, yields significantly better predictions. The low wavelength regions in particular show excellent agreement with experiment. The high wavelength regions are shifted to higher wavelengths since anharmonic effects are neglected in the classical ML/MM simulation. However, we still observe the broadening and red-shift of the O–H stretching vibration present in the experimental spectrum. This effect is caused by hydrogen bonding between the O–H groups in the machine learning region and the surrounding ethanols (see inset Fig. 3). Continuum models fail to account for these kind of interactions, as they neglect the structure of the solvent (see ML/PCM Fig. 3).
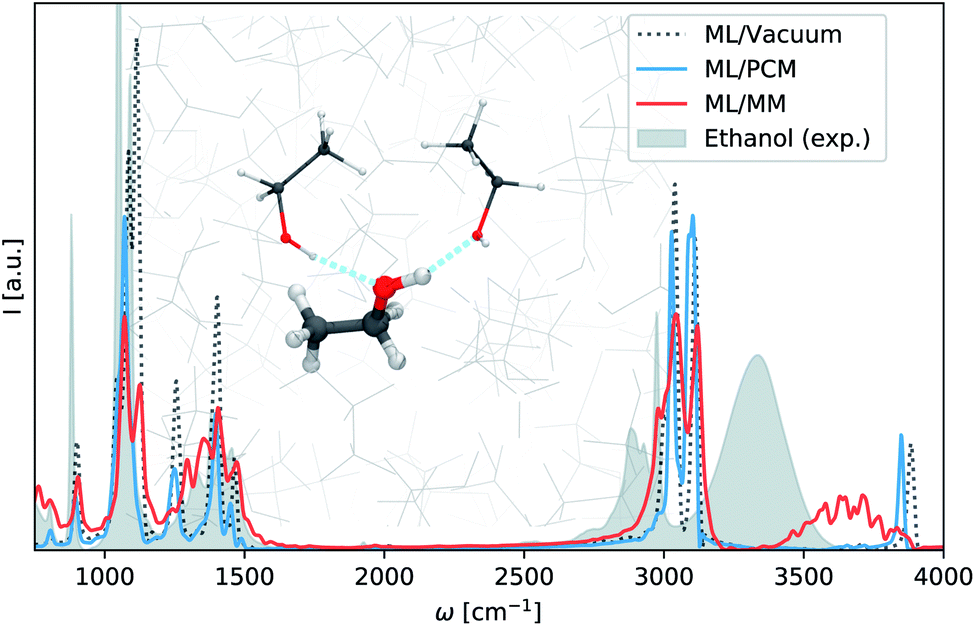 | ||
| Fig. 3 Solvent effects on the infrared spectrum of ethanol. Infrared spectra of liquid ethanol predicted without solvent (ML/Vacuum), with continuum solvent (ML/PCM) and explicit solvent molecules (ML/MM). The experimental spectrum is shown in gray. The inset depict hydrogen bonds between the hydroxyl group and the environment, responsible for the broadening and red-shift of the O–H stretching vibration. | ||
While we have trained the FieldSchNet ML/MM model using a rather large training set, adaptive sampling schemes can drastically reduce the amount of required reference calculations in practice.28,58–60 To demonstrate this, we select a representative set of configurations from the ML/MM ethanol reference data (see ESI† for details). An initial ensemble of models is first trained on a small subset (100 configurations) randomly selected from the original data. We then use variance of the predictions of this ensemble to estimate the uncertainty associated with the other samples in the ethanol reference data. The 100 configurations with the highest uncertainty are added to the initial dataset, the FieldSchNet ensemble is retrained, and the procedure is repeated until the desired accuracy is obtained. The final data set contains only 2000 configurations in contrast to the initial 30000 while still yielding an accurate IR spectrum (see ESI Fig. S4†).
Beyond that, the FieldSchNet ML/MM model can be extended in a systematic manner using adaptive sampling to include other solutes or solvents. We demonstrate this by constructing an ML/MM model for liquid methanol based on the FieldSchNet ensemble for ethanol from above. The system consists of a single ML-modeled methanol suspended in a box of 1859 methanol molecules treated at force field level. The predictive uncertainty of the pretrained ensemble is monitored and methanol ML/MM configurations with high uncertainty are selected. We recompute these geometries with the electronic structure reference and retrain our models on the expanded data set.
As can be seen in Fig. 4 even a single adaptive sampling iteration where the model has been trained 100 additional methanol configuration is enough to yield a qualitatively accurate infrared spectrum of liquid methanol in a ML/MM simulation. The spectrum accurately reproduces the difference between ethanol and methanol in the angular deformation vibrations at 1500 cm−1 and the C–C–O asymmetric (1080 cm−1) and symmetric (900 cm−1) stretching vibrations observed in ethanol are now absent.
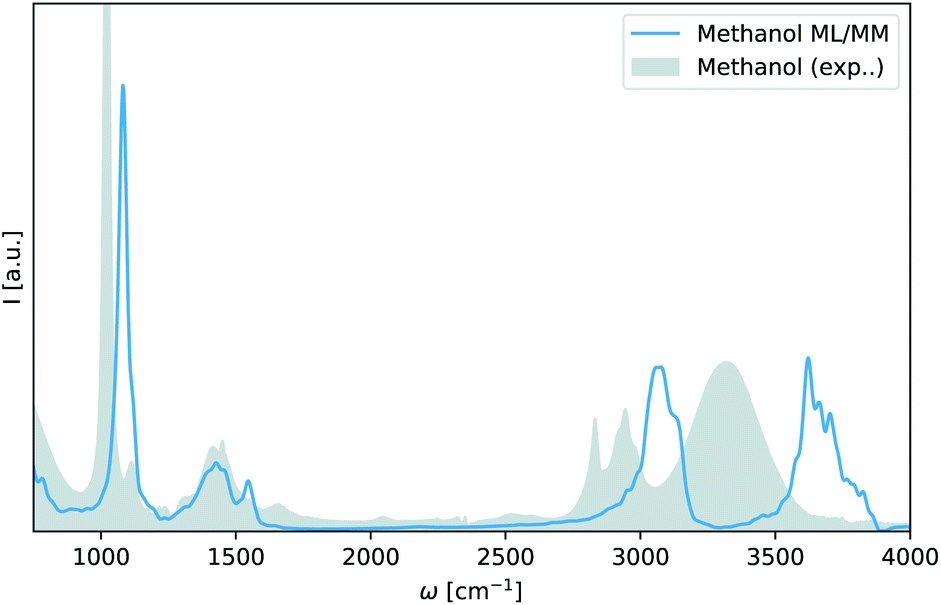 | ||
| Fig. 4 Infrared spectrum of methanol obtained via adaptive sampling. ML/MM spectrum of liquid methanol as obtained with a FieldSchNet model trained on 100 additional methanol configurations selected with adaptive sampling (blue). An experimental spectrum for liquid methanol is shown in gray.57 | ||
3.4 Modeling organic reactions
Effects of the environment and solvents play a central role in many molecular reactions. Certain arrangements and combinations of molecules in the environment can promote or inhibit reactions in a dramatic fashion. As such, a good understanding of these effects is crucial for the development of new catalysts and drugs. However, accurate computational simulations of such systems are hard to obtain since intense sampling procedures are required in order to obtain reliable free energy profiles of the studied reaction. This is further complicated by the need to account for the large number of molecules in the environment, which in many cases cannot be described by more affordable continuum models of solvation due to explicit interactions between solute and environment.An example for such a reaction is the Claisen rearrangement of allyl-p-tolyl ether (Fig. 5a). The presence of water as a solvent accelerates this reaction by a factor of 300 compared to reaction rates in the gas-phase.61 Computational and experimental studies have determined that the main reason for this acceleration is explicit hydrogen bonding between the transition state and the water molecules of the solvent. These lead to a lowered barrier and promote the reaction.62,63 The combination of computational efficiency and accuracy with the ability to perform ML/MM simulations makes FieldSchNet well suited for modeling such reactions. Beyond that, the model provides access to a range of properties which can be used to characterize the different species formed during reaction.
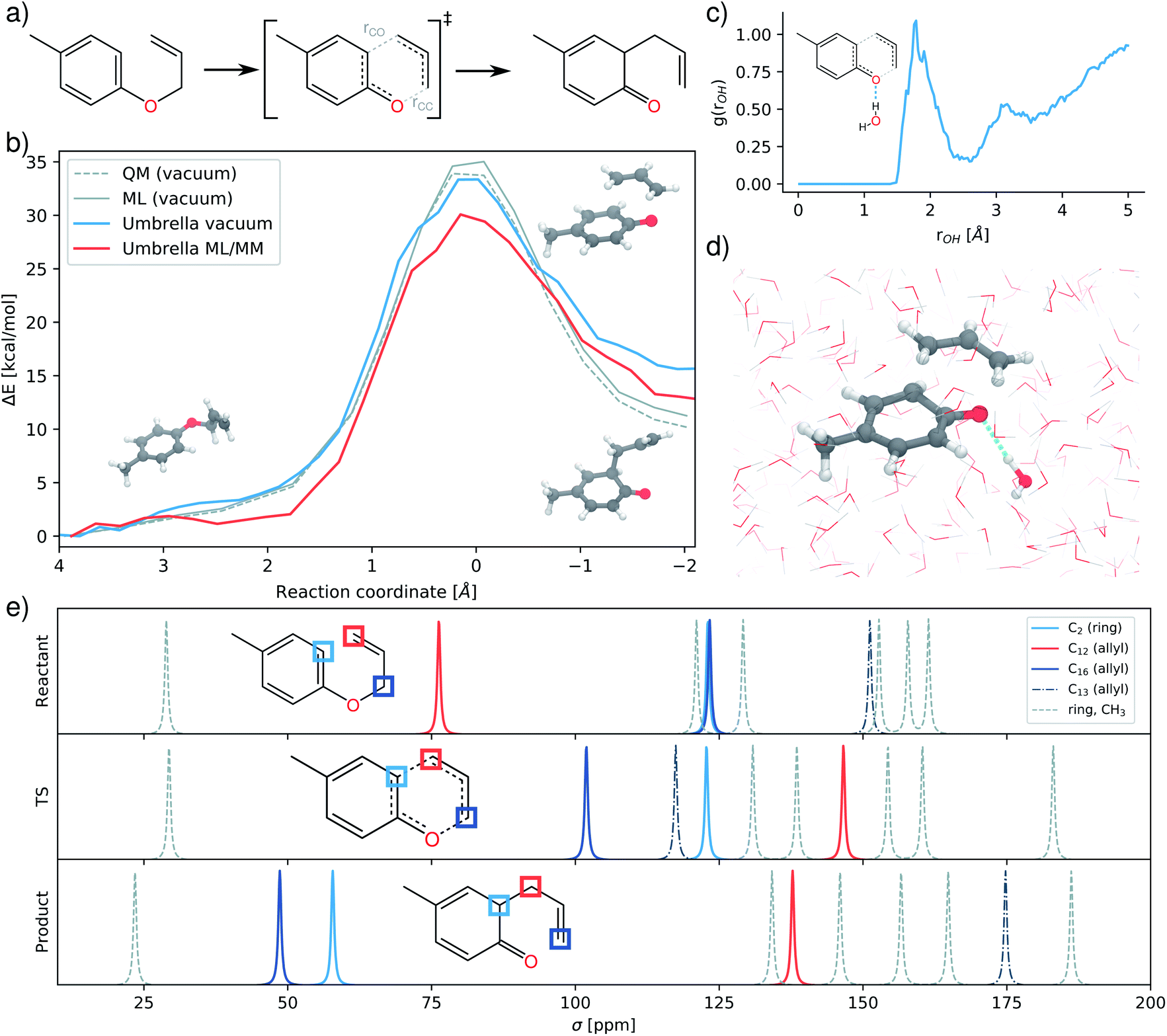 | ||
| Fig. 5 Study of the allyl-p-tolyl ether Claisen rearrangement. (a) Scheme of the reaction, with the two bonds used to determine the reaction coordinate marked in gray. (b) Reaction free energy barriers as computed in gas-phase via nudged elastic band optimization (gray) and via umbrella sampling in vacuum (blue) and water (red). QM refers to the energy profile computed with the PBE0 reference. (c) Radial distribution function of the distance between ether oxygen and water hydrogens in the transition state. (d) Transition state configuration with stabilizing hydrogen bond. (e) NMR chemical shifts predicted for the different stages of the simulation. | ||
We train two FieldSchNet models to simulate the first step in the Claisen rearrangement of allyl-p-tolyl ether. The first model is trained on 61000 PBE0 reference configurations sampled from a metadynamics trajectory of the reaction simulated at a lower level of theory (PBE, see ESI† for details). A second model is a ML/MM model based on the reference configurations determined above augmented by different MM charge distributions sampled via the TIP3P force field for water64 (three charge configurations for every third structure of the metadynamics simulation for a total of 61002 structures, see ESI† for details). Errors for both models are reported in ESI Table S4.†
We perform umbrella sampling65 in vacuum as well as a solvent box containing ∼7000 TIP3P water molecules using the respective FieldSchNet models (see ESI†). The difference between the two bonds broken and formed during rearrangement is chosen as reaction coordinate (indicated as rCO and rCC in Fig. 5a). The speedup offered by FieldSchNet for this system is even more pronounced than for ethanol. A single computation which takes 1.6 hours with the reference method can now be performed in 180 ms, corresponding to an acceleration by a factor of over ∼30000.
The resulting free energy barriers are depicted in Fig. 5b, along with potential energy barriers computed in vacuum using the reference method and vacuum model. The FieldSchNet ML/MM model correctly predicts a lower activation barrier (30.08 kcal mol−1) for the aqueous environment compared to the gas phase reaction (33.35 kcal mol−1). The overall difference in the barrier height ΔΔG = 3.28 kcal mol−1 is close to the experimental value of ΔΔG = 4 kcal mol−1.61 Analyzing the configurations sampled during the ML/MM simulation, we observe the hydrogen bonding between the ether oxygen and water molecules in the solvent responsible for the acceleration of the reaction.62,63Fig. 5c shows the radial distribution function between hydrogens in the solvent and the oxygen of the transition state. A pronounced peak at a distance of 2 Å indicates that hydrogen bonds between solvent and solute form frequently at this stage.
The NMR chemical shifts provided by FieldSchNet can be used to trace structural changes occurring during the rearrangement and allow to connect theoretical predictions to experiment. Fig. 5d depicts the 13C chemical shifts predicted for different stages of the reaction. For example, it shows how the first (C12) and third (C16) carbon atom in the allyl ether exchange chemical environments during reaction. The former moves from typical shifts of allyl ether groups (∼68 ppm) to shifts associated with terminal carbons in conventional allyl groups (∼130 ppm). At the same time, the C16 undergoes this process in reverse, ending at shifts of ∼50 ppm characteristic for this position in allyl substituents. Another change of interest are the shifts of the carbon C2 in the aromatic ring where a new bond forms. This atom starts at typical values for aromatic carbons (∼120 ppm), staying there during the transitions state. Upon formation of the product, the aromaticity of the ring is lost and the shift moves to regions more indicative for carbon atoms in cyclic ketones (∼25–50 ppm).
3.5 Designing molecular environments
The analytic nature of neural networks allows to establish a direct relation between molecular structure and properties which can be exploited in inverse chemical design applications.40,66 FieldSchNet is well suited for such tasks, as it provides access to a wide range of response properties as a function not only of molecular structure but also the external environment. This offers the possibility to manipulate external fields and molecular environments in order to optimize certain properties or to control reaction rates. In the following, we apply FieldSchNet to design a chemical environment promoting the Claisen rearrangement reaction studied above.The external field in the ML/MM FieldSchNet model used to describe the reaction depends on the charge distribution of the environment. Thus, the external charges can be optimized to lower the reaction barrier by minimizing
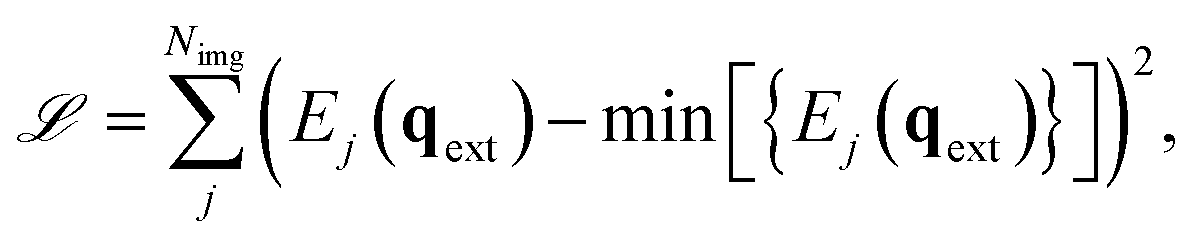 | (12) |
Fig. 6a shows the evolution of the barrier during various stages of the optimization procedure. By designing an optimal environment, the activation barrier can be reduced by ∼25 kcal mol−1, lowering it from an initial 35 kcal mol−1 to 10 kcal mol−1. These findings correspond to a rate acceleration by a factor of ∼2 × 1018 at 300 K. Fig. 6b and c show the optimized environment in presence of the transition state, visualizing regions of negative (b) and positive (c) charge. Motifs such as the strong negative density close to the oxygen atom and the neighboring carbons involved in the forming bond are in strong agreement with experimental studies, where it was found that electron donating groups in these regions promote the reaction.62,67
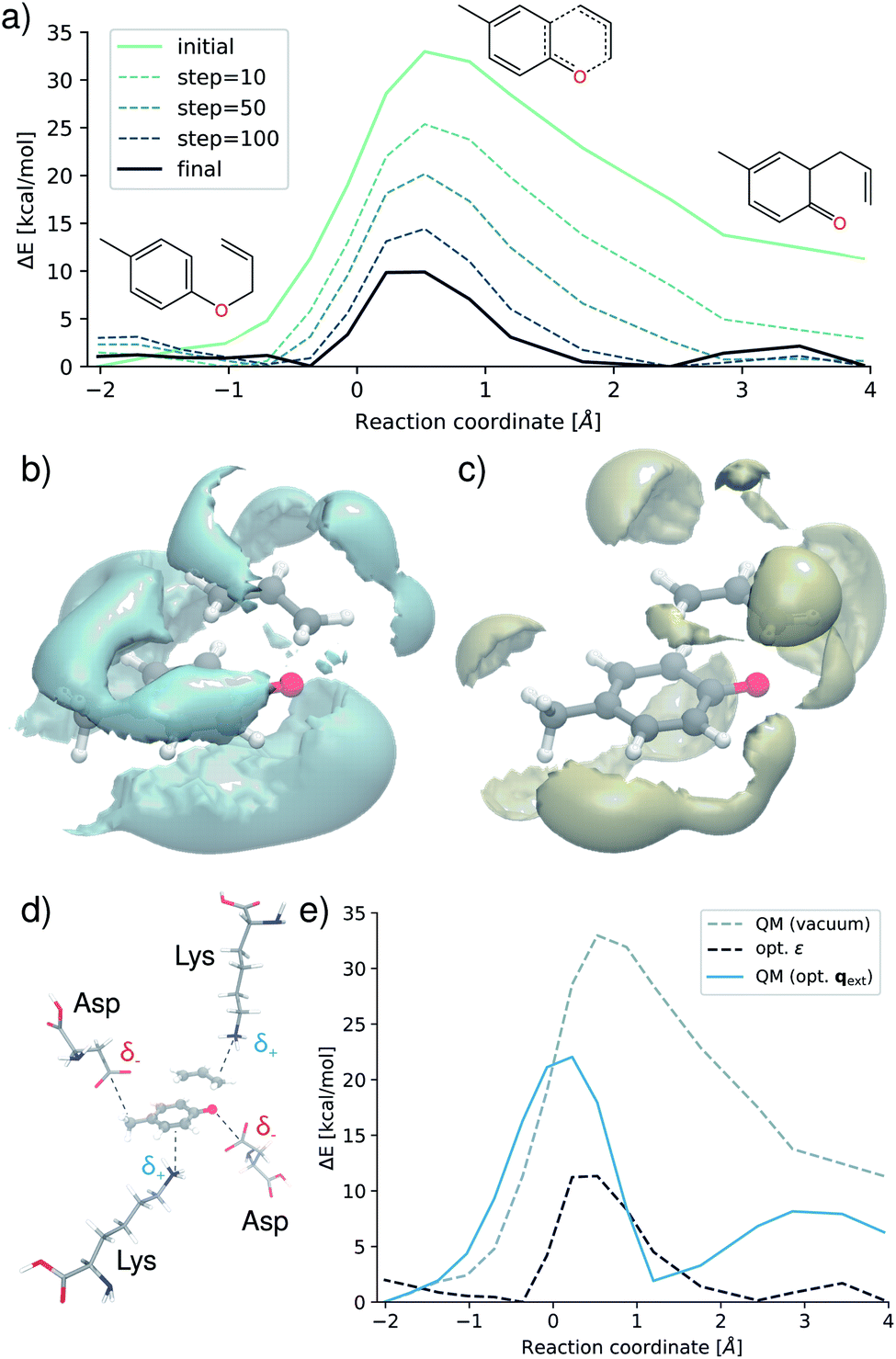 | ||
| Fig. 6 Design of reaction environments. (a) Evolution of the reaction barrier height during optimization of the environment. (b) Distribution of regions of negative charge around the transition state. (c) Regions of positive charge. (d) Transition state with optimal placement of charged amino acid residues (e) Reaction barrier recomputed for the optimal external charge arrangement generated by the amino acid residues (blue) compared to the original barrier (gray) and the barrier obtained for the optimized field (black). | ||
We outline how such an optimized field can guide design endeavors by translating these motifs into an atomic environment of amino acids as would be present in an enzymatic active site. Negatively charged aspartic acid residues (Asp) are placed close to regions exhibiting high negative density, while regions of high positive charge are populated with lysine (Lys) molecules. Using eqn (12), we optimize the placement of the amino acids based on the electrostatic field derived from their Hirshfeld atomic partial charges.68 The resulting environment is shown in Fig. 6d. Finally, we recompute the reaction barrier in presence of the amino acid charge distribution with the original electronic structure reference (Fig. 6e). Although the optimal environment cannot be reconstructed perfectly due to the constraint imposed by the structure of the molecules, this relatively straightforward approach leads to a significant reduction of the barrier. The activation energy is lowered from 35 kcal mol−1 to 22 kcal mol−1, which still corresponds to an acceleration by a factor of ∼3 × 109. This demonstrates that the theoretically optimized environment Fig. 6b and d can be mimicked retaining the same trend on the barrier and reaction speed.
4 Conclusions
FieldSchNet enables modelling the interactions of molecules with arbitrary external fields. This offers access to a wealth of molecular response properties such as polarizabilities and nuclear shielding tensors without the need for introducing specialized models. Leveraging external fields as an interface, the model can further operate as a polarizable continuum model for solvents, as well as a surrogate for the quantum mechanics region in quantum mechanics/molecular mechanics (QM/MM) schemes, yielding a ML/MM approach. As a consequence, FieldSchNet paves the way to many promising applications out of reach for previous techniques.Computational spectroscopy can benefit greatly from the FieldSchNet framework, as it provides efficient means for computing high quality spectra as we have demonstrated for IR and Raman spectra as well as NMR chemical shifts. Combining the latter with nuclear spin–spin coupling tensors, i.e. response properties of the magnetic moments, enables the accurate prediction of nuclear magnetic resonance spectra. In principle, all other spectroscopic quantities, derived via the response formalism, can be modeled by FieldSchNet as well.
The ability of our neural network to operate as a continuum model for solvation is not only highly attractive for applications in drug design, where efficient models of solvent effects are much sought after, but serves as a starting point for developing more powerful models, which could for example consider structural aspects of the solvent. When treating interactions with the environment explicitly, the introduced ML/MM procedure proves to be a powerful tool combining the accuracy of machine learning potentials with the even higher speed of force fields. While the presented study of solvent effects on the Claisen rearrangement reaction of allyl-p-tolyl ether would require 18 CPU years with the electronic structure reference, it was performed within 5 hours with FieldSchNet on a single GPU. This greatly expands the range of application of ML models and brings the simulation of large, biologically relevant systems, such as enzymatic reactions, within reach.
Moreover, the fully analytic nature of FieldSchNet enables inverse design as we have illustrated by minimizing the reaction barrier of the Claisen rearrangement through interactions with an optimized environment. Coupling this to a generative model of molecular structure,66 the charge distributions found using FieldSchNet may be populated in a fully automated fashion. Possible application of FieldSchNet to inverse design tasks include the targeted functionalization of compounds or the creation of enzyme cavities promoting reactions.
FieldSchNet constitutes a unified framework for describing reactions and spectroscopic properties in solution. Beyond that, it provides insights on how these quantities can be controlled via the molecular environment. As this opens up new avenues for designing workflows tightly integrated with experiment, we expect FieldSchNet to become a valuable tool for chemical research and discovery.
Data availability
The code for training and deploying the model can be found at https://github.com/atomistic-machine-learning/field_schnet data for this paper, including all datasets required for training the models are available at http://quantum-machine.org/datasets/.Author contributions
MG and KTS conceived the research. MG developed the method and carried out the reference computations and simulations. KTS, MG, KRM designed the experiments and analyses. MG and KTS wrote the paper. KTS, MG and KRM discussed results and contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding: This project has received funding from the European Unions Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 792572. KTS and KRM acknowledge support by the Federal Ministry of Education and Research (BMBF) for the Berlin Center for Machine Learning/BIFOLD (01IS18037A). KRM acknowledges financial support under the Grants 01IS14013A-E, 01GQ1115 and 01GQ0850; Deutsche Forschungsgemeinschaft (DFG) under Grant Math+, EXC 2046/1, Project ID 390685689 and KRM was partly supported by the Institute of Information & Communications Technology Planning & Evaluation (IITP) grants funded by the Korea Government (no. 2019-0-00079, Artificial Intelligence Graduate School Program, Korea University). Correspondence to MG.References
- J. J. Varghese and S. H. Mushrif, React. Chem. Eng., 2019, 4, 165–206 RSC.
- C. Reichardt and T. Welton, Solvents and solvent effects in organic chemistry, John Wiley & Sons, 2011 Search PubMed.
- A. Zunger, Nat. Rev. Chem., 2018, 2, 1–16 CrossRef.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- O. A. von Lilienfeld, K.-R. Müller and A. Tkatchenko, Nat. Rev. Chem., 2020, 4, 347–358 CrossRef.
- O. A. von Lilienfeld and K. Burke, Nat. Commun., 2020, 11, 1–4 Search PubMed.
- A. Tkatchenko, Nat. Commun., 2020, 11, 1–4 Search PubMed.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, A. Tkatchenko and K.-R. Müller, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c01111.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem., 2009, 28, 577–606 Search PubMed.
- A. P. Bartók, M. C. Payne, R. Kondor and G. Csányi, Phys. Rev. Lett., 2010, 104, 136403 CrossRef PubMed.
- A. P. Bartók, S. De, C. Poelking, N. Bernstein, J. R. Kermode, G. Csányi and M. Ceriotti, Sci. Adv., 2017, 3, e1701816 CrossRef PubMed.
- K. T. Schütt, F. Arbabzadah, S. Chmiela, K.-R. Müller and A. Tkatchenko, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- J. S. Smith, O. Isayev and A. E. Roitberg, Chem. Sci., 2017, 8, 3192–3203 RSC.
- S. Chmiela, H. E. Sauceda, K.-R. Müller and A. Tkatchenko, Nat. Commun., 2018, 9, 3887 CrossRef PubMed.
- E. V. Podryabinkin, E. V. Tikhonov, A. V. Shapeev and A. R. Oganov, Phys. Rev. B, 2019, 99, 064114 CrossRef CAS.
- O. T. Unke and M. Meuwly, J. Chem. Theory Comput., 2019, 15, 3678–3693 CrossRef CAS PubMed.
- M. Rupp, A. Tkatchenko, K.-R. Müller and O. A. Von Lilienfeld, Phys. Rev. Lett., 2012, 108, 058301 CrossRef PubMed.
- G. Montavon, M. Rupp, V. Gobre, A. Vazquez-Mayagoitia, K. Hansen, A. Tkatchenko, K.-R. Müller and O. A. Von Lilienfeld, New J. Phys., 2013, 15, 095003 CrossRef CAS.
- K. T. Schütt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko and K.-R. Müller, J. Chem. Phys., 2018, 148, 241722 CrossRef PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Proceedings of the 34th International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- F. A. Faber, L. Hutchison, B. Huang, J. Gilmer, S. S. Schoenholz, G. E. Dahl, O. Vinyals, S. Kearnes, P. F. Riley and O. A. Von Lilienfeld, J. Chem. Theory Comput., 2017, 13, 5255–5264 CrossRef CAS PubMed.
- A. V. Shapeev, Multiscale Model. Simul., 2016, 14, 1153–1173 CrossRef.
- B. Huang and O. A. von Lilienfeld, Nat. Chem., 2020, 12, 945–951 CrossRef CAS PubMed.
- N. Thomas, T. Smidt, S. Kearnes, L. Yang, L. Li, K. Kohlhoff and P. Riley, 2018, preprint at https://arxiv.org/abs/1802.08219.
- B. Anderson, T. S. Hy and R. Kondor, Advances in Neural Information Processing Systems, 2019, pp. 14510–14519 Search PubMed.
- A. Grisafi, D. M. Wilkins, G. Csányi and M. Ceriotti, Phys. Rev. Lett., 2018, 120, 036002 CrossRef CAS PubMed.
- M. Gastegger, J. Behler and P. Marquetand, Chem. Sci., 2017, 8, 6924–6935 RSC.
- D. M. Wilkins, A. Grisafi, Y. Yang, K. U. Lao, R. A. DiStasio and M. Ceriotti, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3401–3406 CrossRef CAS PubMed.
- J. Westermayr, M. Gastegger and P. Marquetand, J. Phys. Chem. Lett., 2020, 11, 3828–3834 CrossRef CAS PubMed.
- N. Raimbault, A. Grisafi, M. Ceriotti and M. Rossi, New J. Phys., 2019, 21, 105001 CrossRef CAS.
- G. M. Sommers, M. F. C. Andrade, L. Zhang, H. Wang and R. Car, Phys. Chem. Chem. Phys., 2020, 22, 10592–10602 RSC.
- Y. Zhang, S. Ye, J. Zhang, C. Hu, J. Jiang and B. Jiang, J. Phys. Chem. B, 2020, 124, 7284–7290 CrossRef CAS PubMed.
- F. M. Paruzzo, A. Hofstetter, F. Musil, S. De, M. Ceriotti and L. Emsley, Nat. Commun., 2018, 9, 1–10 CrossRef CAS PubMed.
- H. Li, C. Collins, M. Tanha, G. J. Gordon and D. J. Yaron, J. Chem. Theory Comput., 2018, 14, 5764–5776 CrossRef CAS PubMed.
- G. Hegde and R. C. Bowen, Sci. Rep., 2017, 7, 42669 CrossRef CAS PubMed.
- K. Ryczko, D. A. Strubbe and I. Tamblyn, Phys. Rev. A, 2019, 100, 022512 CrossRef CAS.
- F. Brockherde, L. Voigt, L. Li, M. E. Tuckerman, K. Burke and K.-R. Müller, Nat. Commun., 2017, 8, 872 CrossRef PubMed.
- M. Bogojeski, L. Vogt-Maranto, M. E. Tuckerman, K.-R. Müller and K. Burke, Nat. Commun., 2020, 11, 5223 CrossRef CAS PubMed.
- K. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller and R. J. Maurer, Nat. Commun., 2019, 10, 5024 CrossRef PubMed.
- G. Carleo and M. Troyer, Science, 2017, 355, 602–606 CrossRef CAS PubMed.
- J. Hermann, Z. Schätzle and F. Noé, Nat. Chem., 2020, 12, 891–897 CrossRef CAS PubMed.
- A. S. Christensen, F. A. Faber and O. A. von Lilienfeld, J. Chem. Phys., 2019, 150, 064105 CrossRef PubMed.
- F. A. Faber, A. S. Christensen, B. Huang and O. A. Von Lilienfeld, J. Chem. Phys., 2018, 148, 241717 CrossRef PubMed.
- B. Mennucci, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 CAS.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- K. T. Schütt, A. Tkatchenko and K.-R. Müller, Machine Learning Meets Quantum Physics, Springer, 2020, pp. 215–230 Search PubMed.
- L. Onsager, J. Am. Chem. Soc., 1936, 58, 1486–1493 CrossRef CAS.
- J. Cioslowski, Phys. Rev. Lett., 1989, 62, 1469 CrossRef PubMed.
- M. Thomas, M. Brehm, R. Fligg, P. Vöhringer and B. Kirchner, Phys. Chem. Chem. Phys., 2013, 15, 6608–6622 RSC.
- H. E. Sauceda, V. Vassilev-Galindo, S. Chmiela, K.-R. Müller and A. Tkatchenko, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- S. Habershon, D. E. Manolopoulos, T. E. Markland and T. F. Miller III, Annu. Rev. Phys. Chem., 2013, 64, 387–413 CrossRef CAS PubMed.
- S. Chmiela, A. Tkatchenko, H. E. Sauceda, I. Poltavsky, K. T. Schütt and K.-R. Müller, Sci. Adv., 2017, 3, e1603015 CrossRef PubMed.
- NIST Chemistry WebBook NIST Standard Reference Database Number 69, ed. P. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, retrieved September 24, 2020, DOI: DOI:10.18434/T4D303.
- J. Kiefer, Anal. Chem., 2017, 89, 5725–5728 CrossRef CAS PubMed.
- C. J. Cramer, Essentials of computational chemistry: theories and models, John Wiley & Sons, 2004 Search PubMed.
- I. Doroshenko, V. Pogorelov and V. Sablinskas, Dataset Pap. Chem., 2012, 2013, 329406 Search PubMed.
- G. Csányi, T. Albaret, M. Payne and A. De Vita, Phys. Rev. Lett., 2004, 93, 175503 CrossRef PubMed.
- J. Behler, Int. J. Quantum Chem., 2015, 115, 1032–1050 CrossRef CAS.
- A. Shapeev, K. Gubaev, E. Tsymbalov and E. Podryabinkin, Machine Learning Meets Quantum Physics, 2020, pp. 309–329 Search PubMed.
- W. N. White and E. F. Wolfarth, J. Org. Chem., 1970, 35, 2196–2199 CrossRef CAS.
- M. Irani, M. Haqgu, A. Talebi and M. Gholami, J. Mol. Struct.: THEOCHEM, 2009, 893, 73–76 CrossRef CAS.
- O. Acevedo and K. Armacost, J. Am. Chem. Soc., 2010, 132, 1966–1975 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, et al. , J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
- N. Gebauer, M. Gastegger and K. Schütt, Advances in Neural Information Processing Systems, 2019, pp. 7564–7576 Search PubMed.
- R. M. Coates, B. D. Rogers, S. J. Hobbs, D. P. Curran and D. R. Peck, J. Am. Chem. Soc., 1987, 109, 1160–1170 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chem. Acc., 1977, 44, 129–138 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02742e |
| This journal is © The Royal Society of Chemistry 2021 |