
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of stereochemistry on the activity of rapadocin, an isoform-specific inhibitor of the nucleoside transporter ENT1†
Yuefan
Wang‡
 ab,
Hanjing
Peng‡
ab,
Zufeng
Guo‡
abc,
Brett R.
Ullman
d,
Kana
Yamamoto
d,
Sam Y.
Hong
d and
Jun O.
Liu
*abe
ab,
Hanjing
Peng‡
ab,
Zufeng
Guo‡
abc,
Brett R.
Ullman
d,
Kana
Yamamoto
d,
Sam Y.
Hong
d and
Jun O.
Liu
*abe
aDepartment of Pharmacology, Johns Hopkins School of Medicine, 725 North Wolfe Street, Baltimore, MD 21205, USA. E-mail: joliu@jhu.edu
bSJ Yan and HJ Mao Laboratory of Chemical Biology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
cCenter for Novel Target and Therapeutic Intervention, Institute of Life Sciences, Chongqing Medical University, Chongqing, 400016, China
dRapafusyn Pharmaceuticals Inc., Baltimore, MD 21205, USA
eSidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
First published on 21st July 2021
Abstract
Rapadocin is a novel rapamycin-inspired polyketide–tetrapeptide hybrid macrocycle that possesses highly potent and isoform-specific inhibitory activity against the human equilibrative nucleoside transporter 1 (hENT1). Rapadocin contains an epimerizable chiral center in phenylglycine and an olefin group, and can thus exist as a mixture of four stereoisomers. Herein, we report the first total synthesis of the four stereoisomers of rapadocin using two different synthetic strategies and the assignment of their structures. The inhibitory activity of each of the four synthetic isomers on both hENT1 and hENT2 was determined. It was found that the stereochemistry of phenylglycine played a more dominant role than the configuration of the olefin in the activity of rapadocin. These findings will guide the future design and development of rapadocin analogs as new modulators of adenosine signaling.
Introduction
Rapamycin and FK506 are macrocyclic natural products with potent immunosuppressive activity, making them powerful drugs for preventing graft rejection and treating a variety of autoimmune diseases. These drugs work by inducing the formation of ternary complexes between FK506-binding protein (FKBP) and their targets, mTOR and calcineurin, respectively.1,2 The ability of these drugs to bind FKBP confers a number of advantages, including superior bioavailability and distribution in vivo.3 Inspired by the unique structure and mode of action of these drugs, we recently developed rapamycin-like hybrid macrocycles by replacing the effector domain of rapamycin with tetrapeptides and named them rapafucins.4 High-throughput screening led to the discovery of rapadocin, a highly potent, isoform-specific, and FKBP-dependent inhibitor of the human equilibrative nucleoside transporter 1 (hENT1) with an IC50 of 5 nM for adenosine uptake.4 ENTs are an important family of nucleoside transporters, which play a major role in the transport of nucleoside-based small molecular drugs for cancer and several viral diseases in addition to their physiological substrate adenosine.5,6 Rapadocin has been shown to protect against ischemia-reperfusion kidney injury by potentiating adenosine signaling through blocking adenosine uptake, making it a promising lead to develop drugs for treating kidney ischemia/reperfusion injury and other diseases.7–9Rapadocin shares the two-domain architecture of rapamycin and FK506: an FKBP-binding domain (FKBD) and an effector domain consisting of tetrapeptides.10–12 In our previous synthesis of the rapafucin library,4 the effector domain in rapadocin is generated using solid-phase peptide synthesis before it is coupled to a preassembled FKBD. The resulting FKBD-peptide conjugate is cyclized and released from the solid support via ring-closing metathesis (RCM). Under those conditions, we found that the phenylglycine residue can undergo epimerization during solid-phase synthesis13,14 and the olefin formed during the RCM reaction can assume either Z- or E-configuration (Fig. 1).15 Due to the complexity of the rapadocin structure, it was difficult to separate and detect each of the four possible stereoisomers for further characterization, leaving open the question of whether the four stereoisomers differ in their inhibitory activities towards hENT1. To address those uncertainties, we embarked on the total synthesis of each of the four possible stereoisomers of rapadocin to determine their biological activity.
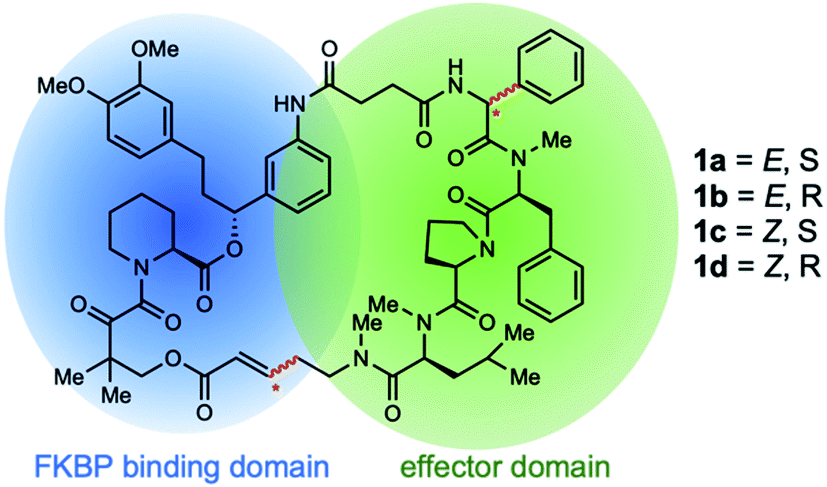 | ||
| Fig. 1 Possible stereoisomers of rapadocin. | ||
Results and discussion
Design of solution phase synthesis of rapadocin stereoisomers
When we did the solid phase synthesis of rapadocin, we found a significant racemization of the phenylglycine moiety. The configuration of the RCM reaction generated double bond was also difficult to confirm using NMR. We designed two different routes for the total synthesis of rapadocin 1 (Scheme 1). The retrosynthetic planning of macrolactamization began with the disconnection of the macrocycle at the FKBD-mLeu amide bond (2) and then on the either side of the D-proline residue, leading to an olefin-containing FKBD and a dipeptide (4) precursor. This synthetic route required the development of an efficient synthetic approach for the stereoselective synthesis of 4 bearing a highly racemization-prone phenylglycine moiety.16–20 The second route is based on RCM for macrocyclization from linearized precursor 3, which can be broken down on either side of the D-proline residue to the precursor dipeptide 4. Both synthetic routes required the development of an efficient synthesis of dipeptide 4. The late-stage solution-phase RCM reaction is similar to that used in the solid phase synthesis,4 while the macrolactamization strategy could confirm the double bond configuration.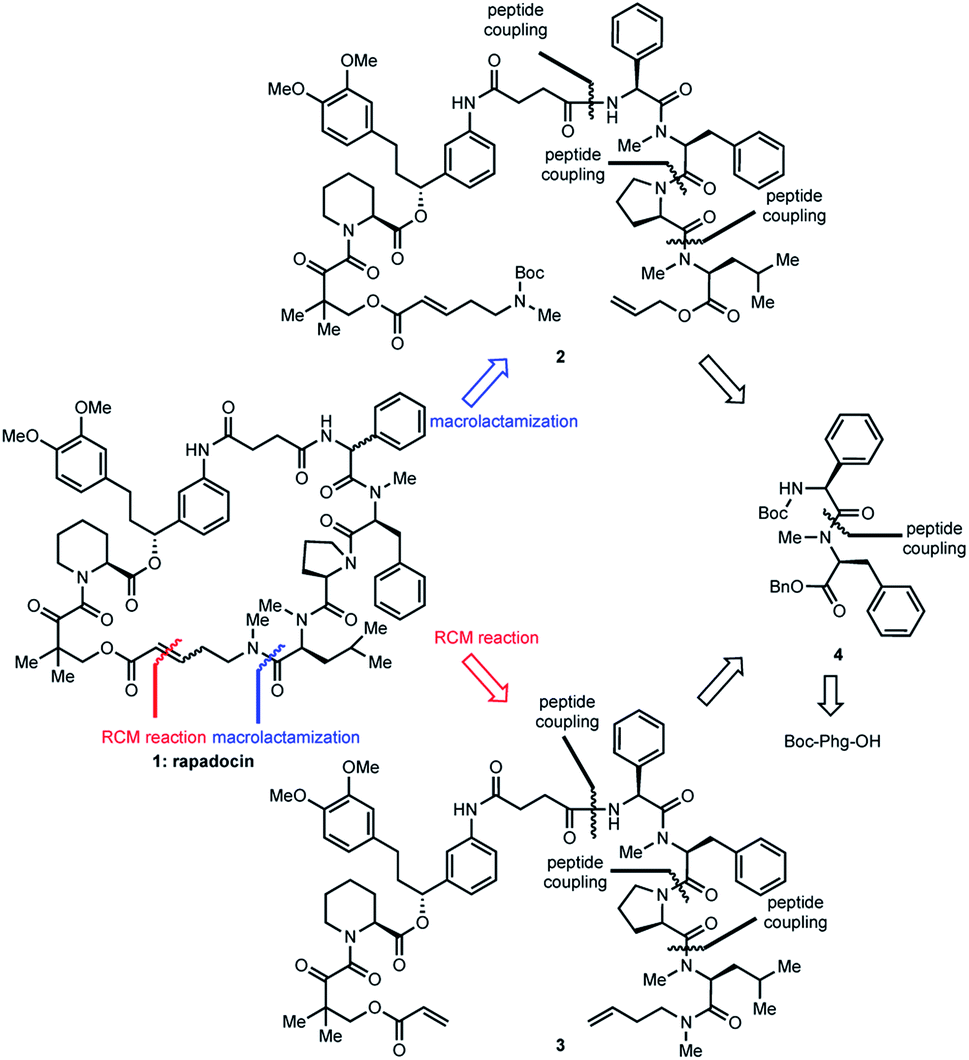 | ||
| Scheme 1 Retrosynthetic analyses of rapadocin. | ||
Two synthetic routes of rapadocin isomers
The synthesis of dipeptide 4 commenced with diastereoselective amidation from commercially available Boc-L-α-phenylglycine (Boc-Phg-OH). Boc-Phg-OH was activated as a mixed acid anhydride and coupled with N-methyl phenylalanine 5 to give the highly diastereoselective dipeptide 4 with an excellent d.r. ratio (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1),20,21 as determined by 1H-NMR (Table 1).
:1),20,21 as determined by 1H-NMR (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent (equiv.) | Base (equiv.) | Temperature | Solvent | Ratio (4:6)a |
| a The ratio of the phenylglycine epimers in the dipeptide was determined by 1H-NMR. b This is the most optimal value attained. | |||||
| 1 | HATU (1.5) | DIPEA (3) | 0 °C to r.t. | CH2Cl2 | 2:3 |
| 2 | EDCI (1.6) | DMAP (2.0) | 0 °C to r.t. | CH2Cl2 | 1:1 |
| 3 | EDCI/HOAT (1.5/1.5) | NaHCO3 (1.5) | 0 °C to r.t. | THF | 3:2 |
| 4 | BOPCI (1.0) | NMM (2.0) | −15 °C to r.t. | CH2Cl2 | 5:1 |
| 5 | DEPBT (1.6) | NaHCO3 (1.6) | −15 °C to r.t. | THF | 7:1 |
| 6 | ClCO2iBu (1.0) | NMM (1.5) | −15 °C to 0 °C | CH2Cl2 | >20:1b |
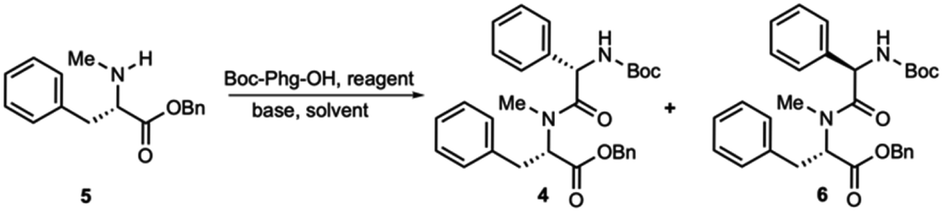
The hydrogenolysis of benzyl ester 4 with 10% Pd/C gave the corresponding carboxylic acid 7 in 88% yield (Scheme 2). The amidation of 7 with allyl ester of D-proline in the presence EDCI and DMAP in CH2Cl2 afforded tripeptide 8 in 73% yield. Further allyl deprotection of 8 under Tsuji–Trost allylation conditions in the presence of a catalytic amount of Pd(PPh3)4 afforded carboxylic acid 9 in 93% yield.22 The final methyl-leucine residue was attached using coupling agents EDCI and DMAP in CH2Cl2 to afford the tetrapeptide, which was then reacted with hydrogen chloride dioxane solution (4 M) to expose the N-terminus of tetrapeptide 10.
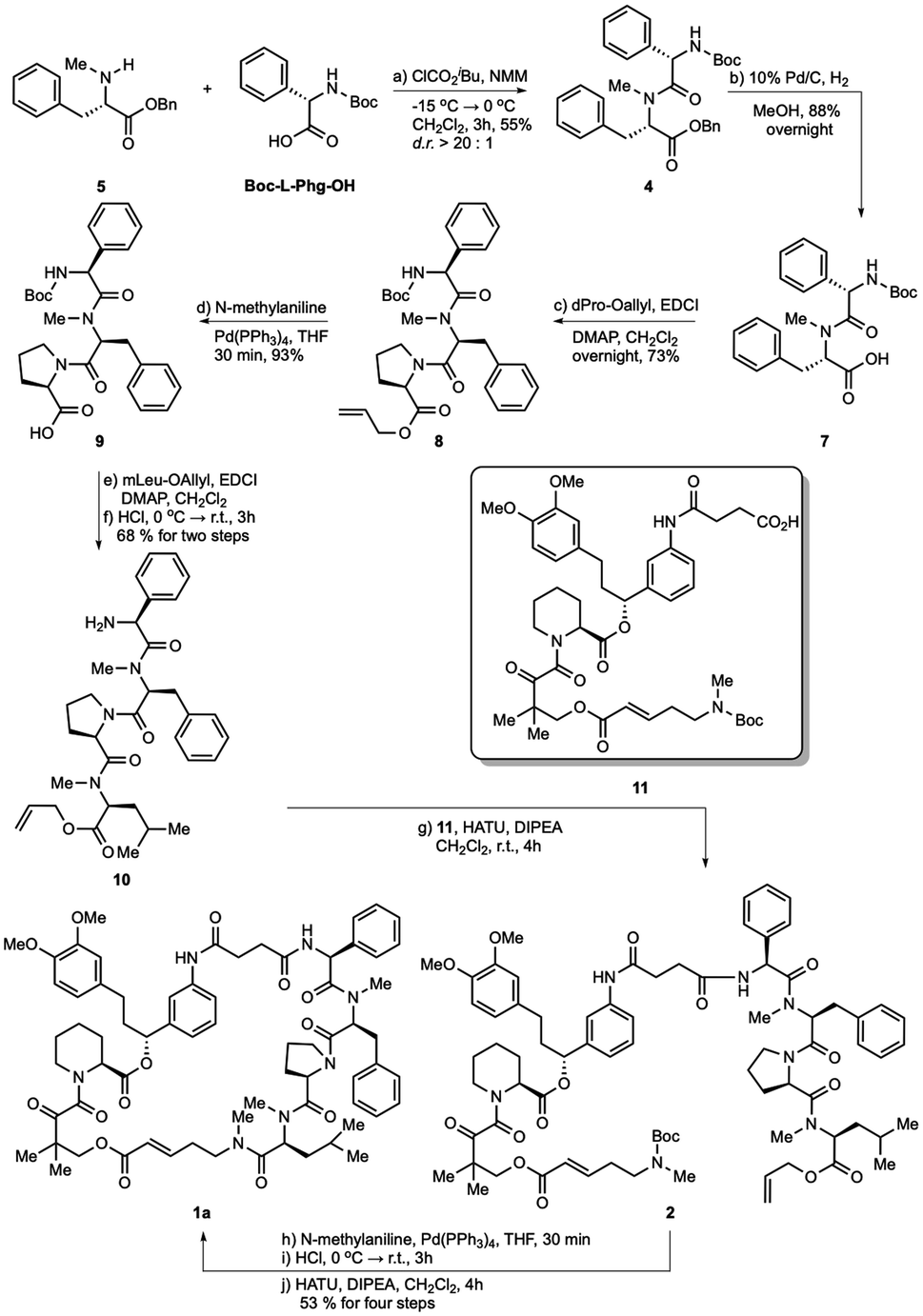 | ||
| Scheme 2 Total synthesis of rapadocin via macrolactamization strategy. | ||
With tetrapeptide fragment 10 in hand, we explored the pathway for the completion of the total synthesis. Tetrapeptide 10 was coupled with Boc-E-FKBD fragment 11 upon treatment with HATU and Hünig's base in CH2Cl2 to obtain a linear product 2. The fragment 11 was provided by Affinity Research Chemicals with an established E- or Z-configuration of the olefin moiety. The deprotection of the allyl ester of 2 with Pd(PPh3)4 was followed by the removal of the Boc group with dioxane HCl solution (4 M). Finally, the lactamization of the acyclic rapadocin with HATU and Hünig's base in CH2Cl2 gave rapadocin 1a in 53% yield over four steps.
With the successful synthesis of the first rapadocin isomer 1a with a L(S)-Phg-(mPhe) moiety and E configuration of the double bond, we proceeded to prepare 1b and other isomers. As shown in Scheme 3, 1b was readily made using the same synthetic procedure as that for 1a with the dipeptide 6 from Boc-D-Phg-OH instead of Boc-L-Phg-OH. We found that when DEPBT and NaHCO3 were used as coupling reagents for the synthesis of 6, the yield was higher than that for the formation of dipeptide 4 with a 70% yield and the d.r. ratio of the product was greater than 20:1, as determined by 1H-NMR. Compounds 1c and 1d were synthesized with the same synthetic strategy using the Z-FKBD fragment.
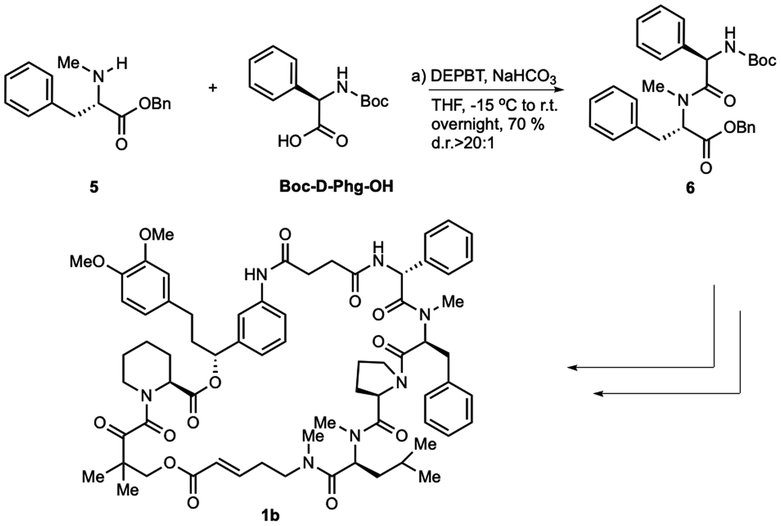 | ||
| Scheme 3 Synthesis of rapadocin isomers. | ||
For polypeptides and cyclic peptides to which rapafucin belong, 1H- and 13C-NMR are no longer suited to judge the purity of those compounds, as each amide bond may exist in two rotamer conformations and different rotamer conformations could show distinct spectra, complicating the 1H- and 13C-NMR spectra. This situation is made worse in the case of rapadocin due to the different rotamer conformations arising from the pipecolic ester of FKBD that has been seen in FK506 and rapamycin. We thus resorted to using a combination of NMR, HPLC and high-resolution MS to confirm the purity of the different stereoisomers of rapadocin.23–25 Based on the HSQC result of 1a (see the ESI†), we confirmed that the double bond proton is in the range of ppm 6.0–5.5 in the 1H-NMR spectrum. To provide further support for the successful synthesis of all four isomers 1a–1d, we compared the four 1H-NMR spectra for olefin protons. As shown in Fig. 2, the four isomers showed different 1H-NMR, suggestion that four isomers are not the same compound.
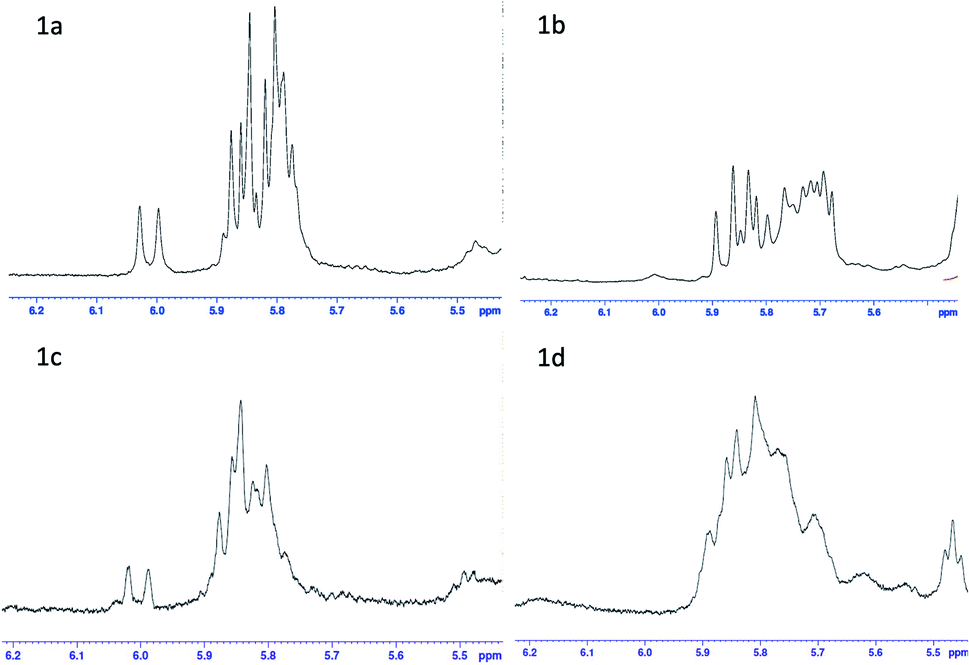 | ||
| Fig. 2 1H-NMR comparison of rapadocin four isomers. | ||
After the successful syntheses of the rapadocin isomers via the macrolactamization strategy, we investigated the RCM strategy that mimics the solid-phase synthesis to verify the configuration of the double bond formed during the RCM reaction and to explore more efficient synthetic routes to pure rapadocin stereoisomers.
As shown in Scheme 4, tripeptide 9 was coupled with methyl-leucine derivative 12 upon treatment with HATU and Hünig's base in CH2Cl2 to obtain tetrapeptide 13 in 80% yield. Intermediate 13 was reacted with hydrogen chloride dioxane solution (4 M) to expose the N-terminus of tetrapeptide 14, which was then condensed with FKBD 15 to give linear product 3 in 54% yield over two steps. FKBD 15 was made using a reported method.4 To complete the total synthesis, 3 was treated with the Zhan-1b catalyst to give the final product,26,27 which could be the isomer 1a, 1c or a mixture of both. By the comparison of the 1H-and 13C-NMR spectra and optical rotations of the product from the RCM reaction with those obtained using the macrolactamization strategy, the RCM product was assigned as 1a ([α]23D = −36.5 and [α]23D = −35.9). Thus, the double bond configuration of rapadocin from the RCM reaction used in previous study was confirmed to have the E configuration.
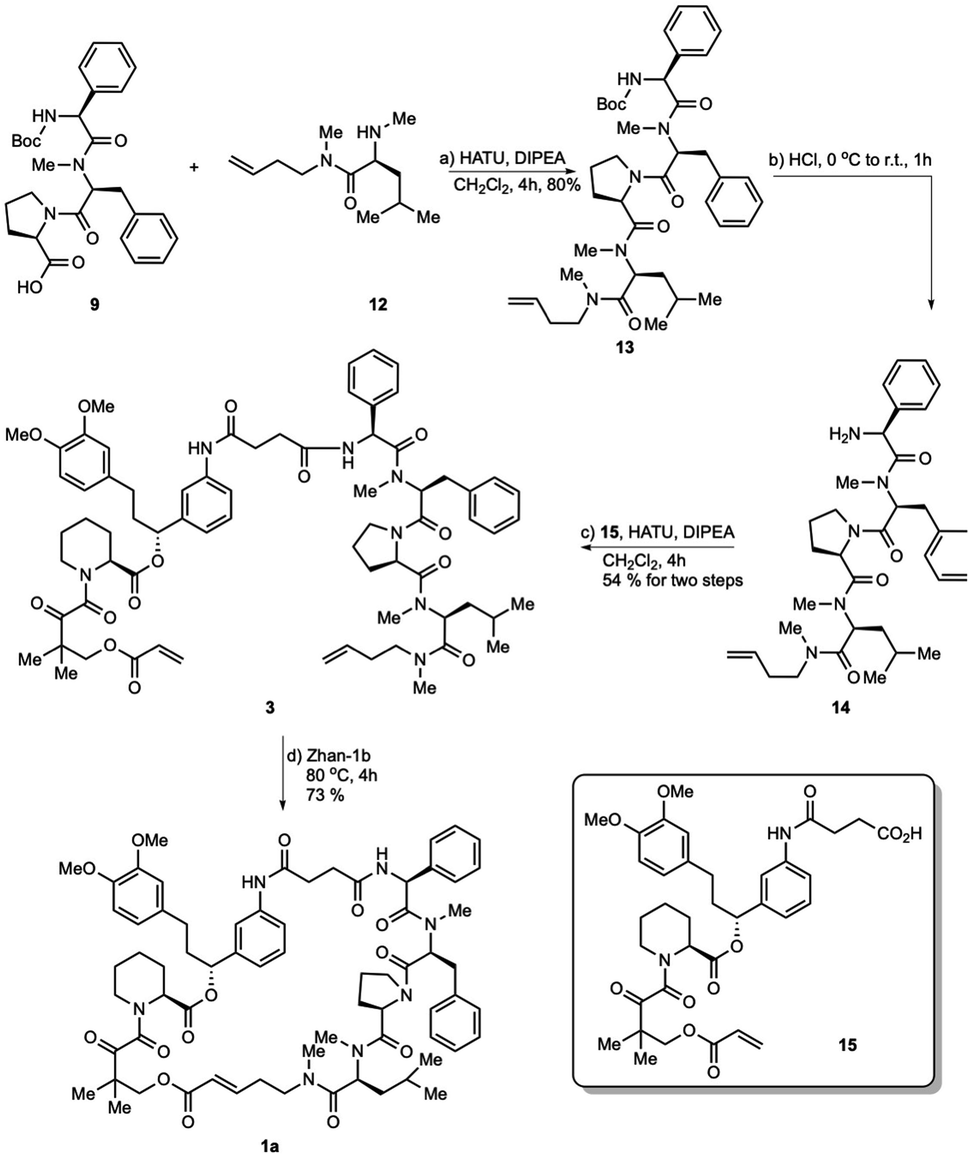 | ||
| Scheme 4 Total synthesis of rapadocin via the RCM strategy. | ||
The configuration of the olefin has a major impact on the stability of the stereoisomers. In contrast to 1a and 1b, 1c and 1d with the Z olefin configuration are unstable in solution. At room temperature, Z isomers in DMSO changed to a new undefined compound with a molecular weight of 1270 in one week. At −20 °C, Z-isomers underwent the same change in one month. In contrast, E-isomers are stable in solution at −20 °C for over a year. This result suggests that the E-isomers of rapafucins with the current backbone structures are thermodynamically favored by the RCM reaction.
Biological evaluations of the four isomers
With each of the four isomers of rapadocin in hand, we took advantage of a window of time before the Z-isomers of rapadocin underwent appreciable degradation and determined their inhibitory activity toward hENT1-mediated nucleoside uptake using [3H]-thymidine uptake assay in porcine kidney (PK15) cell lines exclusively expressing hENT1.28 Interestingly, rapadocin isomer 1a, with a L-Phg moiety and E-double bond, was the most potent (IC50 = 3.3 nM) among the four stereoisomers. In contrast, the other three isomers 1b, 1c, and 1d exhibited lower potency against hENT1 with IC50 values of 589.2 nM, 14.5 nM, and 469.3 nM, respectively (Fig. 3a). These results suggested that isomer 1a is the most active component of rapadocin from the solid-phase synthesis. As the potency of the RCM reaction product (rapadocin) is comparable to that of isomer 1a, it is likely that this stereoisomer is the predominant product from the RCM reaction. The fact that isomers 1a and 1c are significantly more active than 1b and 1d also suggests that the stereochemistry of the phenylglycine residue has a much greater influence on the activity of rapadocin than the configuration of the olefin moiety.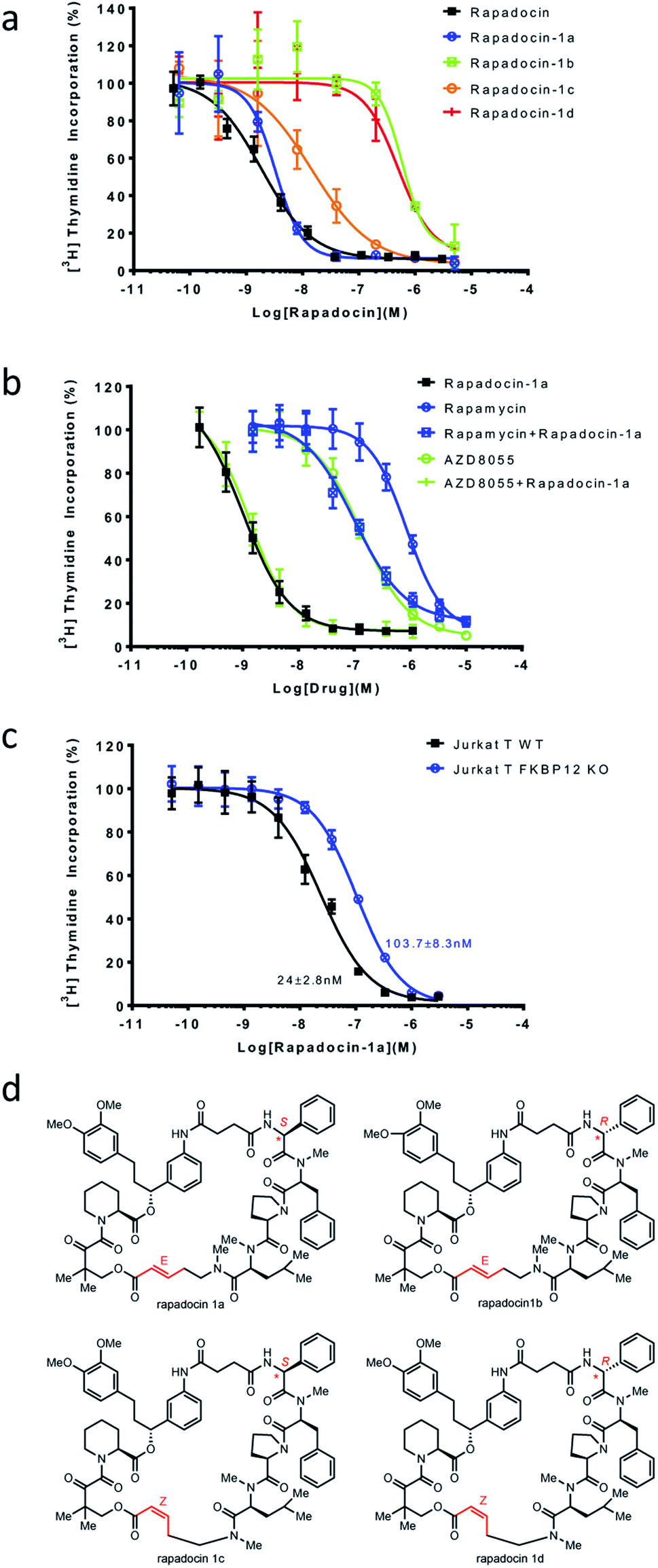 | ||
| Fig. 3 Inhibition of hENT1 by different stereoisomers of rapadocin. (a) Inhibition of [3H]-thymidine uptake in PK15 cell lines exclusively expressing hENT1 by the four stereoisomers of rapadocin. (b) Rapamycin, but not AZD8055, antagonizes isomer 1a inhibition of hENT1-mediated thymidine uptake. The dose–response curves were obtained using [3H]-thymidine uptake in PK15-ENT1 cells. (c) Knockout of FKBP12 imparts resistance to isomer 1a. Dose response curves for the inhibition of [3H]-thymidine up-take by isomer 1a in wild type (WT) and FKBP12 knockout Jurkat T cells. Error bars represent s.d. Data are presented as mean ± s.d.; n = 3 independent experiments. (d) Structures of the four rapadocin isomers. | ||
To further assess the dependence of isomer 1a on endogenous FKBPs, we determined mutual antagonism between two mTOR inhibitors, rapamycin and AZD8055, and rapadocin isomer 1a as previously described.4 As expected, combining rapamycin (5 μM) with isomer 1a significantly decreased its potency (Fig. 3b). However, the high concentration of AZD8055 (5 μM), an FKBP-independent mTOR inhibitor, had a negligible effect on the activity of isomer 1a. We also determined the inhibitory activity of isomer 1a in either a wild type Jurkat T or an FKBP12 knockout cell line using the [3H]-thymidine incorporation assay. The FKBP12 knockout Jurkat T cells also gained resistance to 1a (Fig. 3c). These results suggested that, like rapadocin, the inhibitory activities of the four isomers, especially isomer 1a, are also dependent on endogenous FKBP12.
In addition, we also investigated the inhibition of [3H]-thymidine uptake by 5 μM of rapadocin isomers in PK15 cell lines exclusively expressing either hENT1 or hENT2. Similar to rapadocin, isomers 1a–1d had little effect on [3H]-thymidine uptake in the PK15-ENT2 cell line, suggesting that the four isomers retained high specificity for hENT1 (Fig. 4).
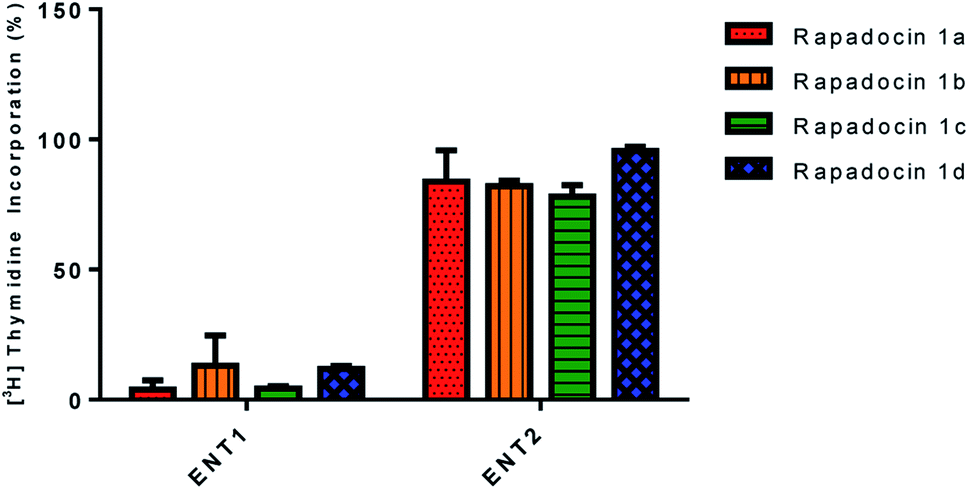 | ||
| Fig. 4 Effects of rapadocin isomers on hENT1 and hENT2. | ||
Conclusion
In summary, we designed two synthetic strategies to synthesize the four stereoisomers of rapadocin. We confirmed that the structure of the most active isomer of rapadocin contains L-Phg (S stereochemistry) with an E configuration of the double bond (1a). We also developed an RCM strategy to confirm our structural elucidation result and efficient synthesis of the most active isomer 1a. We found that isomers with an E-configuration of the double bond are much more stable than those with a Z-configuration. We observed that the stereochemistry of phenylglycine played a significant role in the observed FKBP12-dependent and isoform-selective inhibition of hENT1. The complete assignment of the phenylglycine stereochemistry and the double bond configuration of the most potent stereoisomer of rapadocin with efficient scalable solution-phase synthetic routes will greatly facilitate the future development of rapadocin as a new molecular probe for hENT1 and as a lead for developing drugs to treat ischemic kidney reperfusion injury among other diseases through the modulation of adenosine signaling.Data availability
All additional experimental data and procedures are included in the ESI.†Author contributions
J. L. conceived the project, oversaw the execution of the project and edited the manuscript. Y. W. designed the synthetic routes, completed the total synthesis and wrote the first manuscript. H. P. helped to finish the synthesis of 1c and 1d. Z. G. and H. P. performed the biological assays and analysed the data. B. R. U., K. Y. and S. Y. H. assisted the chemical synthesis.Conflicts of interest
The authors declare conflict of interest. Patent applications covering rapadocin have been filed by Johns Hopkins University and licensed to Rapafusyn Pharmaceuticals, Inc. J. O. L. is a co-founder of, as well as a Scientific Advisory Board Member for, Rapafusyn Pharmaceuticals, Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.Acknowledgements
This work was made possible by the NIH Director's Pioneer Award, the Flight Attendant Medical Research Institute, a generous gift from Mr Shengjun Yan and Ms. Hongju Mao and NCI (P30CA006973) (J. O. L.) and a Damon Runyon Postdoctoral Fellowship (H. P.). We thank Dr Yixin Han in Professor E. J. Corey's lab (Harvard) for help with the optical rotation analysis.References
- J. Heitman, N. R. Movva and M. N. Hall, Science, 1991, 253, 905–909 CrossRef CAS PubMed
.
- J. Liu, J. D. Farmer, Jr, W. S. Lane, J. Friedman, I. Weissman and S. L. Schreiber, Cell, 1991, 66, 807–815 CrossRef CAS
- P. S. Marinec, L. Chen, K. J. Barr, M. W. Mutz, G. R. Crabtree and J. E. Gestiwicki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1336–1341 CrossRef CAS
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C. M. Tse, Z. Tarmakova, C. Schiene-Fischer, G. Fischer, I. Coe, V. O. Paavilainen, Z. Sun and J. O. Liu, Nat. Chem., 2019, 11, 254–263 CrossRef CAS
- J. D. Young, S. Y. Yao, J. M. Baldwin, C. E. Cass and S. A. Baldwin, Mol. Aspects Med., 2013, 34, 529–547 CrossRef CAS
- R. C. Boswell-Casteel and F. A. Hays, Nucleosides, Nucleotides Nucleic Acids, 2017, 36, 7–30 CrossRef CAS PubMed
- Y. J. Day, L. Huang, H. Ye, J. Linden and M. D. Okusa, Am. J. Physiol. Ren. Physiol., 2005, 288, F722–F731 CrossRef CAS PubMed
- C. M. Lappas, Y. J. Day, M. A. Marshall, V. H. Engelhard and J. Linden, J. Exp. Med., 2006, 203, 2639–2648 CrossRef CAS PubMed
- A. Grenz, H. Osswald, T. Eckle, D. Yang, H. Zhang, Z. V. Tran, K. Klingel, K. Ravid and H. K. Eltzschig, PLoS Med., 2008, 5, e137 CrossRef PubMed
- J. P. Griffith, J. L. Kim, E. E. Kim, M. D. Sintchak, J. A. Thomson, M. J. Fitzgibbon, M. A. Fleming, P. R. Caron, K. Hsiao and M. A. Navia, Cell, 1995, 82, 507–522 CrossRef CAS PubMed
- C. R. Kissinger,
et al.
, Nature, 1995, 378, 641–644 CrossRef CAS PubMed
- J. Choi, J. Chen, S. L. Schreiber and J. Clardy, Science, 1996, 273, 239–242 CrossRef CAS PubMed
- C. Brieke and M. J. Cryle, Org. Lett., 2014, 16, 2454–2457 CrossRef CAS PubMed
- C. Liang, M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Tetrahedron Lett., 2017, 58, 2325–2329 CrossRef CAS
- A. H. Hoveyda, J. Org. Chem., 2014, 79, 4763–4792 CrossRef CAS PubMed
- G. G. Smith and T. Sivakua, J. Org. Chem., 1983, 48, 627–634 CrossRef CAS
- M. Bodanszky and A. Bodanszky, Chem. Commun., 1967, 12, 591–593 RSC
- K. C. Nicolaou,
et al.
, Chem.–Eur. J., 1999, 5, 2602–2621 CrossRef CAS
- D. A. Evans, J. L. Katz, G. S. Peterson and T. Hintermann, J. Am. Chem. Soc., 2001, 123, 12411–12413 CrossRef CAS PubMed
- L. Tan and D. Ma, Angew. Chem., Int. Ed., 2008, 47, 3614–3617 (
Angew. Chem.
, 2008
, 120
, 3670–3673
) CrossRef CAS PubMed
- T. Noguchi, S. Jung and N. Imai, Tetrahedron Lett., 2014, 55, 394–396 CrossRef CAS
- F. Guibé, Tetrahedron, 1998, 54, 2967–3042 CrossRef
- P. Karuso, H. Kessler and D. F. Mierke, J. Am. Chem. Soc., 1990, 112, 9434–9436 CrossRef CAS
- D. F. Mierke, P. Schmieder, P. Karuso and H. Kessler, Helv. Chim. Acta, 1991, 74, 1027–1047 CrossRef CAS
- H. Kessler, R. Haessner and W. Schüler, Helv. Chim. Acta, 1993, 76, 117–130 CrossRef CAS
- A. Kajetanowic and K. Grela, Angew. Chem., Int. Ed., 2021, 60, 13738–13756 CrossRef PubMed
- T. K. Olszewski, M. Bieniek, K. Skowerski and K. Grela, Synlett, 2013, 24, 903–919 CrossRef CAS
- J. L. Ward, A. Sherali, Z. P. Mo and C. M. Tse, J. Biol. Chem., 2000, 275, 8375–8381 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02295d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |