
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of metallosupramolecular polymer concentration on the synthesis of poly[n]catenanes†
Marissa M.
Tranquilli
a,
Qiong
Wu
 b and
Stuart J.
Rowan
*abc
b and
Stuart J.
Rowan
*abc
aDepartment of Chemistry, University of Chicago, Chicago, IL, USA
bPritzker School of Molecular Engineering, University of Chicago, Chicago, IL, USA. E-mail: stuartrowan@uchicago.edu
cChemical and Engineering Sciences, Argonne National Laboratory, Lemont, IL, USA
First published on 26th May 2021
Abstract
Poly[n]catenanes are a class of polymers that are composed entirely of interlocked rings. One synthetic route to these polymers involves the formation of a metallosupramolecular polymer (MSP) that consists of alternating units of macrocyclic and linear thread components. Ring closure of the thread components has been shown to yield a mixture of cyclic, linear, and branched poly[n]catenanes. Reported herein are investigations into this synthetic methodology, with a focus on a more detailed understanding of the crude product distribution and how the concentration of the MSP during the ring closing reaction impacts the resulting poly[n]catenanes. In addition to a better understanding of the molecular products obtained in these reactions, the results show that the concentration of the reaction can be used to tune the size and type of poly[n]catenanes accessed. At low concentrations the interlocked product distribution is limited to primarily oligomeric and small cyclic catenanes 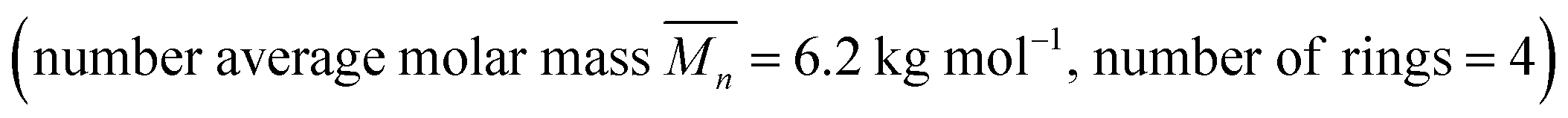 . However, the same reaction at increased concentration can yield branched poly[n]catenanes with an
. However, the same reaction at increased concentration can yield branched poly[n]catenanes with an 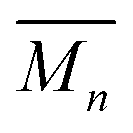 ca. 21 kg mol−1, with evidence of structures containing as many as 640 interlocked rings (1000 kg mol−1).
ca. 21 kg mol−1, with evidence of structures containing as many as 640 interlocked rings (1000 kg mol−1).
Introduction
Catenated compounds—molecules composed of interlocked rings—have been of interest since the mid 20th century when they were first synthesized.1,2 The interlocked rings are held together by what is termed the mechanical bond, which is a bonding motif that offers a unique combination of flexibility and strength.3–5 A covalent bond must be broken in order to separate the two components; however, the mechanically bonded nature of the rings allow the components to twist, rotate, and expand/contract relative to each other, offering modes of motion that are not possible with molecules comprised of only covalent bonds (Fig. 1).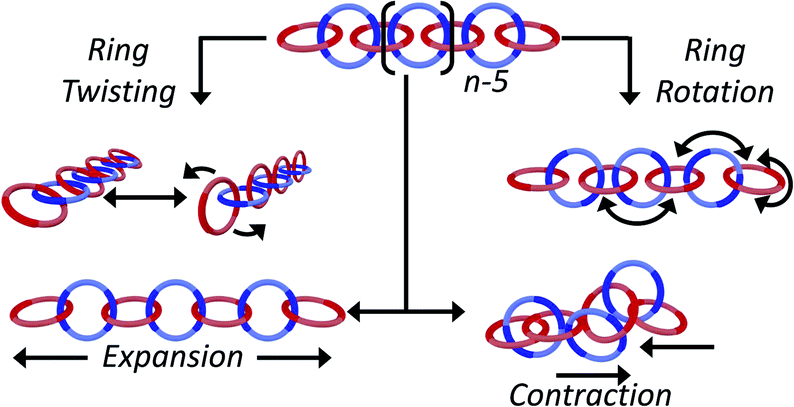 | ||
| Fig. 1 A poly[n]catenane consists of n interlocked rings held together by the mechanical bond which imparts unique mobility elements to the polymer, such as ring twisting and rotation and chain collapse/expansion. | ||
It is therefore perhaps not surprising that the incorporation of the mobile mechanical bond into polymer architectures has been an active area of research since the early days of the field.6 As more efficient synthetic methods have been developed to access interlocked molecules, there has been a corresponding growth of activity into mechanically interlocked polymers (MIPs).7–10 These new polymeric materials exhibit unusual property profiles,11 that include increased stress dissipation and high extensibility.12–14 One of the more intriguing MIP architectures is the poly[n]catenane—a polymer chain that is entirely comprised of interlocked macrocycles where the number of mechanical bonds is optimized (Fig. 1). Molecular simulations of these poly[n]catenanes have begun to outline the unusal dynamic and rheological properties they possess.15 In solution, modelling shows that a single poly[n]catenane chain demonstrates dynamics qualitatively similar to those found in entangled polymer melts, however these effects are observed at shorter time scales, suggesting these polymers will demonstrate unique rheological and dynamical responses.16 Similarly, modelling of these polymers in the melt has shown several phenomena including fast stress relaxation and unusual viscosity trends.17
While networks of catenanes (so-called Olympic networks) occur naturally in the DNA of some protozoic organisms,18–20 the laboratory synthesis of polymeric catenanes composed of only interlocked rings has long been a challenge.7 Early examples of (oligo)[n]catenanes include the step-wise synthesis of linear [5]catenanes (Olympiadane) and branched [7]catenanes.21–23 Other promising methodologies, such as ring merging24 or ring-opening of a [2]catenane25 have been investigated. While both are attractive synthetic routes, high molecular weight poly[n]catenanes structures have yet to be reported with such approaches. However, it is worth mentioning that the formation of Olympic networks has been proposed as a by-product in the polymerization of 1,2-dithianes.26,27
The synthesis of high molecular weight poly[n]catenanes7,28,29 is challenging, as catenane synthesis requires a ring closing step that is notoriously low yielding.7 A common approach to low molecular weight catenanes, pioneered by Sauvage, involves the threading of a linear component (or thread) through a pre-made ring driven by metal–ligand coordination,30–33 followed by ring closing of the thread under dilute conditions.34 Recently, this metal-templated approach has been expanded35 to allow access to poly[n]catenanes via the self-assembly of ditopic macrocyclic and thread components with a metal ion to form a metallosupramolecular polymer (MSP), followed by the ring closing reaction of the thread component and demetallation (Fig. 2a). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of macrocycle (1) and thread (2), where both components contain two 2,6-bisbenzimidazolylpyridine (Bip) ligands (Fig. 2b), were mixed with two equivalents of Zn2+ ions to yield the pseudopolyrotaxane MSP (3) (Fig. 3). Ring closing of 2 was achieved through olefin metathesis of its alkene tails at a concentration of 2.5 mM (w.r.t. 2) resulting in a mixture that contains the metallated catenane (4). Ideally, the alkene tails of 2 react intramolecularly to undergo a ring closing metathesis reaction, yielding the desired poly[n]catenanes (5a) after demetallation. However, studies on the demetallated products showed that in addition to the targeted linear poly[n]catenane (5a) the reaction mixture also contained cyclic and branched catenated products (5b/c) as well as the starting materials (1, 2). In addition, non-interlocked by-products derived from 2, namely macrocycle 7 (formed by ring closing of 2) and oligomer 6, formed via the Acyclic Diene Metathesis (ADMET) of 2 (which can be formed either via incorrect intramolecular MSP reactions, as in 4d, or intermolecular MSP reactions, as in 4e). Through a combination of NMR and GPC-MALS (gel permeation chromatography coupled to a multi-angle light scattering detector) studies, it was shown that the overall yield of the interlocked compounds was ca. 75% and, after some purification to remove the non-interlocked products (along with a small amount of low molecular weight interlocked oligomers), the poly[n]catenanes were obtained with a number average molar mass
:1 solution of macrocycle (1) and thread (2), where both components contain two 2,6-bisbenzimidazolylpyridine (Bip) ligands (Fig. 2b), were mixed with two equivalents of Zn2+ ions to yield the pseudopolyrotaxane MSP (3) (Fig. 3). Ring closing of 2 was achieved through olefin metathesis of its alkene tails at a concentration of 2.5 mM (w.r.t. 2) resulting in a mixture that contains the metallated catenane (4). Ideally, the alkene tails of 2 react intramolecularly to undergo a ring closing metathesis reaction, yielding the desired poly[n]catenanes (5a) after demetallation. However, studies on the demetallated products showed that in addition to the targeted linear poly[n]catenane (5a) the reaction mixture also contained cyclic and branched catenated products (5b/c) as well as the starting materials (1, 2). In addition, non-interlocked by-products derived from 2, namely macrocycle 7 (formed by ring closing of 2) and oligomer 6, formed via the Acyclic Diene Metathesis (ADMET) of 2 (which can be formed either via incorrect intramolecular MSP reactions, as in 4d, or intermolecular MSP reactions, as in 4e). Through a combination of NMR and GPC-MALS (gel permeation chromatography coupled to a multi-angle light scattering detector) studies, it was shown that the overall yield of the interlocked compounds was ca. 75% and, after some purification to remove the non-interlocked products (along with a small amount of low molecular weight interlocked oligomers), the poly[n]catenanes were obtained with a number average molar mass 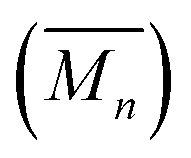 of 21.4 kDa
of 21.4 kDa 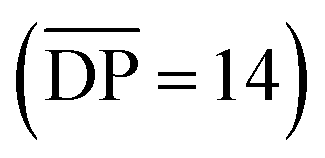 (note
(note  as it is calculated here, eqn (S1),† is the average number of rings in the polymer). Fractionation of this product confirmed that it consisted of a mixture of linear (5a), cyclic (5b), and branched (5c) poly[n]catenanes (Fig. 3). It was suggested that the cyclic poly[n]catenanes are formed from a cyclic MSP template (3b) while the branched poly[n]catenanes result from inter-MSP reactions.
as it is calculated here, eqn (S1),† is the average number of rings in the polymer). Fractionation of this product confirmed that it consisted of a mixture of linear (5a), cyclic (5b), and branched (5c) poly[n]catenanes (Fig. 3). It was suggested that the cyclic poly[n]catenanes are formed from a cyclic MSP template (3b) while the branched poly[n]catenanes result from inter-MSP reactions.
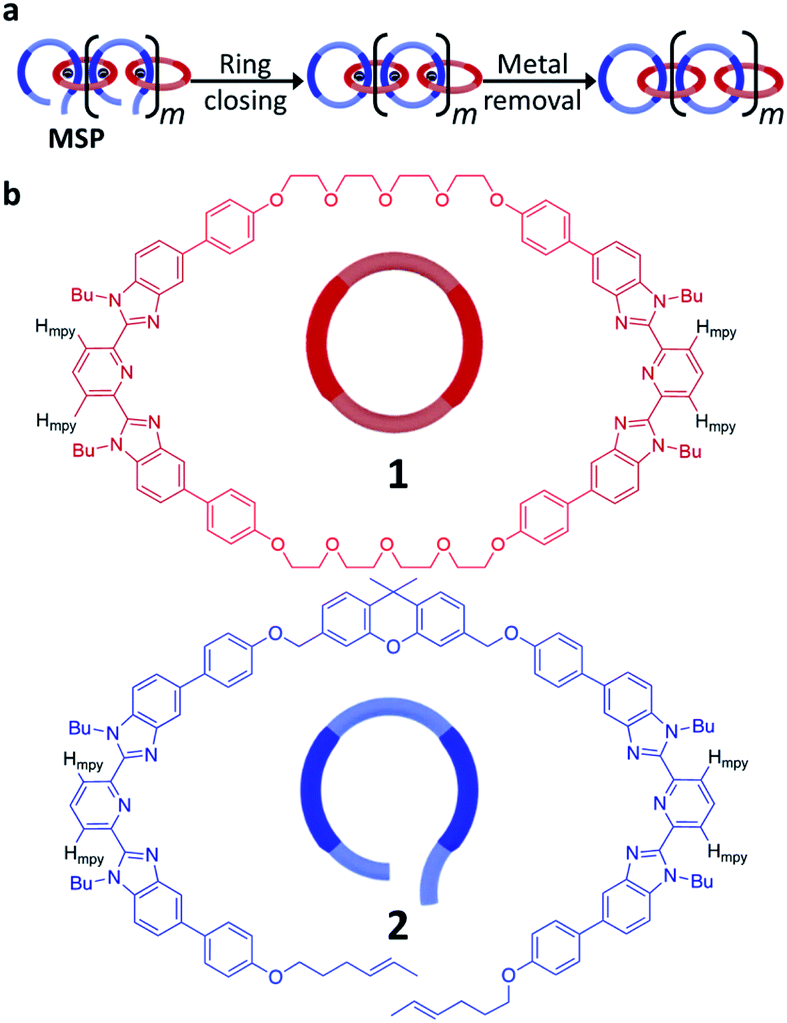 | ||
| Fig. 2 (a) Poly[n]catenanes can be synthesized by assembly of a ditopic thread-like component (blue) and a ditopic macrocycle (red) via metal-templating to yield a metallosupramolecular polymer (MSP). Ring closure of the blue component within the MSP followed by demetallation yields the poly[n]catenane. (b) Chemical structures and cartoon representations of the macrocyclic (1) and thread (2) components used in the synthesize the poly[n]catenane. The 2,6-bisbenzimidazolylpyridine (Bip) component is darkened for identification and the m-pyridyl protons (Hmpy) are indicated in black. | ||
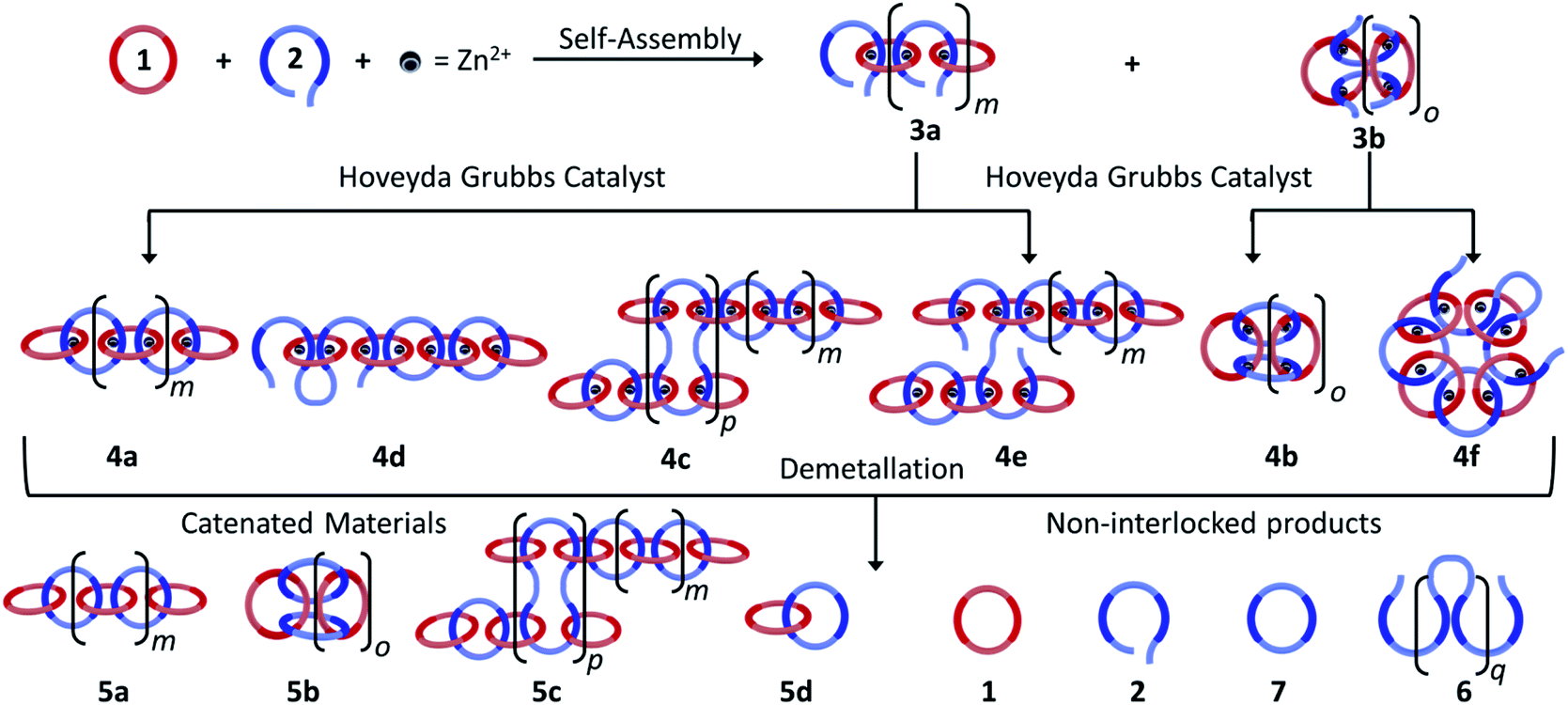 | ||
| Fig. 3 Mixing macrocycle (1) and thread (2) in a 1:1:2 ratio with zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(Tf2N)2) results in the self-assembly of linear (3a) and/or cyclic (3b) metallosupramolecular polymers (MSP). Subsequent reaction of the MSP with Hoveyda–Grubbs catalyst can yield a mixture of metallated products (some of which are shown as 4a–f) whose distribution depends on the nature of the MSP and concentration of the reaction. Demetallation of these reaction products yields poly/oligo[n]catenanes (e.g., 5a–d) along with recovery of some starting material and some non-interlocked byproducts. | ||
This prior study focused primarily on confirming the synthesis of poly[n]catenanes. As such, the reaction was primarily studied at a single concentration (2.5 mM w.r.t. 2) and no detailed characterization of the crude product distribution was undertaken. It is reasonable to expect that both the MSP assembly and the ring closing olefin metathesis reaction will be sensitive to monomer concentration. Thus, the goal of this work is to build on these prior observations by carrying out the ring closing reactions across a range of monomer concentrations (0.25–10 mM) and analyzing the crude reaction mixtures (after demetallation) to obtain a better understanding of how different reaction conditions impact this synthetic route to poly[n]catenanes.
Results and discussion
Analysis of MSP
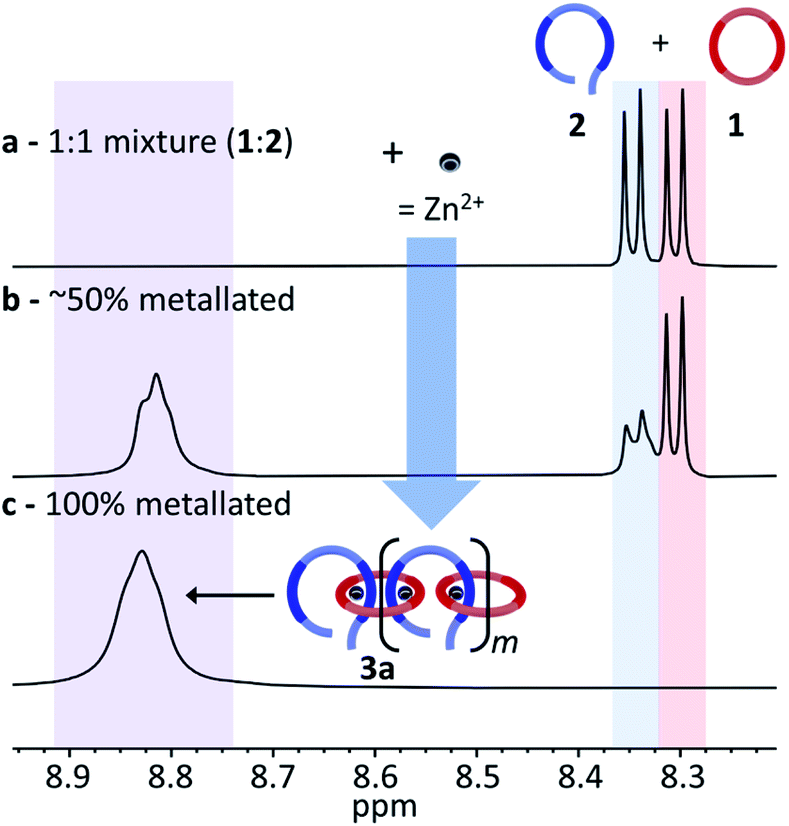
As the nature of the assembled MSP template is critical to the final product distribution, initial studies focused on obtaining a better understanding of the MSP assembly. A 1:1 mixture of macrocycle 1 and thread 2 in CDCl3 was prepared and titrated with zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(Tf2N)2). 2D NMR studies35,36 have shown that the most downfield shifted proton corresponds to the meta-proton of the pyridine moiety of the Bip ligands (Hmpy, Fig. 2b), which appear at 8.30 ppm and 8.35 ppm for 1 and 2, respectively (Fig. 4a). Coordination of the Zn2+ ions with the Bip ligand can be followed by 1H NMR as the peaks corresponding to Hmpy shift to ca. 8.82 ppm (Fig. 4b). It is worthwhile noting that the metal–ligand complex and the uncomplexed ligand are in slow exchange on the NMR timescale, allowing for the amount of free species present to be monitored during the titration. The Zn2+ ions are added into the reaction mixture until all the Hmpy peaks around 8.30–8.35 disappear (Fig. 4c). As both Bip moieties in macrocycle 1 cannot bind to the same Zn2+ ion,36 the only way for all the Bip ligands to bind a Zn2+ ion is the self-assembly of MSP (3) that has alternating 1 and 2 units along its backbone.
 | ||
| Fig. 4 The region of the 1H NMR (500 MHz, 298 K) corresponding to the Hmpy protons peaks of (a) a 1:1 mixture of macrocycle 1 and thread 2 (in CDCl3), (b) partial metalation of the mixture by titration of Zn(NTF2)2 (in CD3CN) and (c) the MSP 3a (in 1:5 CD3CN:CDCl3, 20 mM w.r.t. 2). | ||
In order to better understand if/how the MSP changes with concentration, it was dried and then redissolved in a mixture of acetonitrile-d3 and chloroform-d (1:5) to obtain a series of samples at different concentrations, 0.25 mM, 1.0 mM, 2.5 mM, and 10.0 mM with respect to 2 (Fig. 5a and S1†). The solutions were then characterized using a combination of 1H NMR and diffusion ordered spectroscopy (DOSY). At the highest concentration (10.0 mM), the Hmpy appears almost entirely as a one broad peak at around 8.8 ppm (Fig. 5a, Region i) which was assigned to the formation of MSP 3a (and/or very large cyclic 3b). This peak appears similar to what is seen at 20 mM (Fig. 4c) during sample preparation. There is a slight shift upfield of this peak at the lower concentration (Fig. 5a), but no free ligand is observed. As the concentration decreases, a second set of peaks appear upfield (around 8.67, Region ii in Fig. 5a) which grow in intensity at lower concentrations. The dynamic nature of the Zn2+/Bip complex results in a ring-chain equilibria existing in these MSPs and, as rings are more likely to be formed at lower concentrations,37 this suggests that the more upfield shifted Region ii peaks correspond to the cyclic MSP 3b.
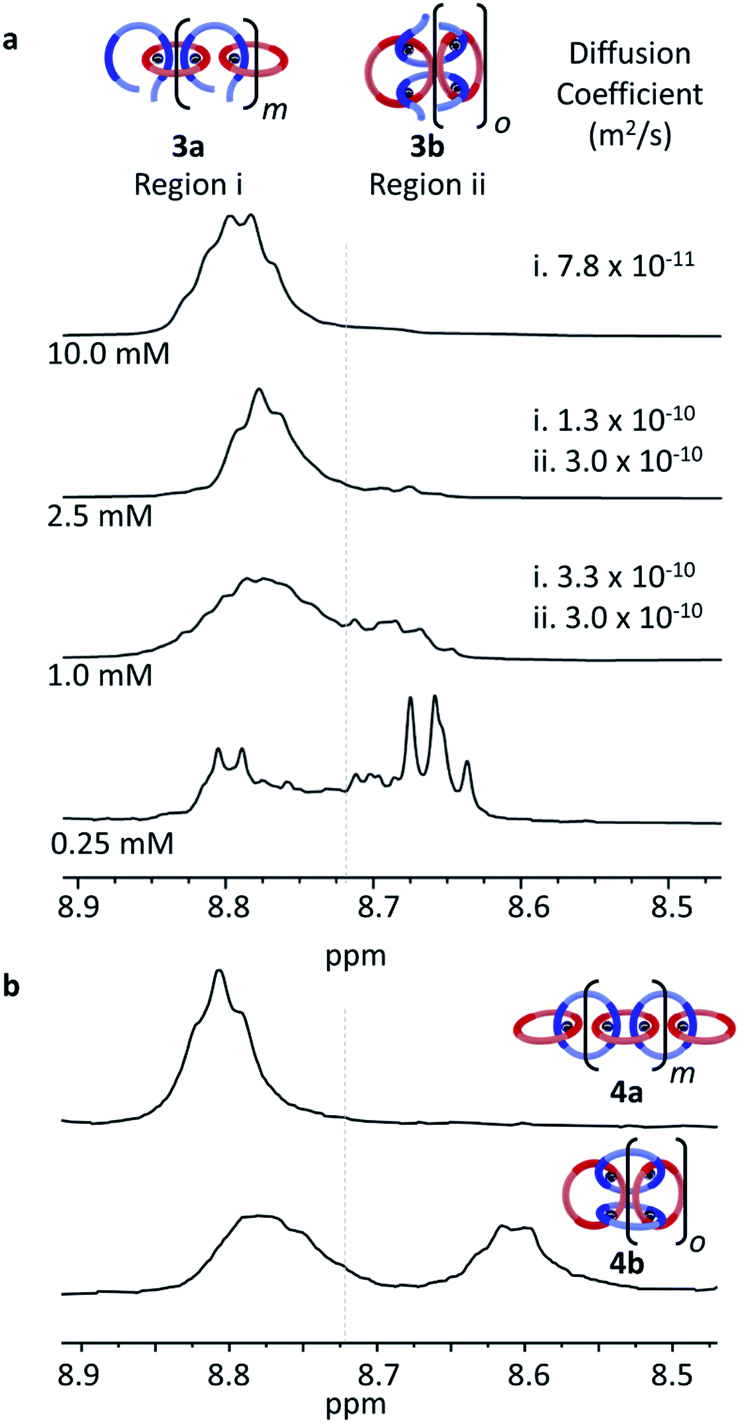 | ||
| Fig. 5 Region of the 1H NMR spectra (500 MHz, 1:5 CD3CN:CDCl3, 298 K) corresponding to the Hmpy protons for (a) the MSP (3a/3b) at 0.25 mM, 1.0 mM, 2.5 mM, and 10.0 mM (w.r.t. 2) along with the diffusion coefficients associated with Region i and ii at the different concentrations and (b) for poly[n]catenanes 4a and 4b. | ||
DOSY NMR of the MSP at the different concentrations provided information on the size of the compounds that correspond to the two regions in the NMR. Note: the 0.25 mM sample was too dilute to obtain reliable DOSY data. The diffusion coefficients (Fig. S2–S4†) for Region i decrease from 3.3 × 10−10 m2 s−1 to 7.8 × 10−11 m2 s−1 as the concentration increases from 1 to 10 mM, consistent with an increase in assembly size/MSP molecular weight with concentration, as would be expected for the linear MSP 3a. Region ii, on the other hand, exhibits similar diffusion coefficients for the concentrations tested. In addition, the diffusion coefficient of the Region ii protons (3.0 × 10−10 m2 s−1) is similar to the diffusion coefficient of the individual thread (3.0 × 10−10) or macrocycle (3.8 × 10−10), suggesting that the MSP in this region is a more compact structure. Taken together, this data is consistent with Region ii corresponding to smaller cyclic MSP assemblies (3b).
If these two regions in the NMR are representative of cyclic and linear MSPs, then it could be expected that a similar difference in the NMR would be observed in the metallated catenanes. Thus, using literature procedures35 samples that primarily consist of cyclic polycatenane (5b) (via Fe2+ templated synthesis35) or linear polycatenane (5a) (obtained by SEC purification of the polycatenane synthesis35) were re-metallated with Zn(Tf2N)2 to yield 4a and 4b. As can be seen in Fig. 5b, the 1H NMR of cyclic 4b resembles that of the 0.25 mM MSP, with two broad peaks between 8.60–8.90 ppm. This provides further support to the upfield shifted peaks observed in the more dilute MSPs corresponding to the cyclic assemblies. Likewise, the 1H NMR of 4a only shows a broad peak in Region i (around 8.8 ppm) consistent with the MSP at 10.0 mM (Fig. 5a, top) being predominantly linear 3a.
NMR analysis of poly[n]catenane
To carry out the olefin metathesis ring closing reaction, the MSPs were dried and dissolved in dry dichloromethane to yield a series of samples at 0.25, 0.5, 1.0, 2.5, 5.0, and 10.0 mM, with respect to 2. The MSP in dichloromethane-d (Fig. S5†) shows a similar 1H NMR Region i and Region ii distribution to that in Fig. 5a, suggesting that the linear/cyclic distribution of MSP observed is maintained during sythesis. The Hoveyda–Grubbs catalyst (0.32 mM) was added to each reaction before it was heated to reflux for 48 h, with a second addition of the same amount of catalyst after the first 24 hours elapsed. After deactivation of the catalyst, the crude material is demetallated using diethylenetriamine.Fig. 6a shows the m-pyridyl proton (Hmpy) region of the 1H NMR spectra for each of the demetallated crude reaction mixtures (the full NMR can be found in Fig. S6†). The non-interlocked byproducts are easily identifiable in the NMR, with the macrocycle 1 at 8.31 ppm and the thread 2 and thread-based byproduct 6 around 8.35 ppm. For the interlocked compounds, the Hmpy's shift upfield to between 8.05–8.27 ppm.35 The integration of these peaks allows determination of the average interlocked yield of each reaction (Fig. 6b and Table S1†). Within this region, a triplet from the 2/6 byproducts appears centered at 8.07 ppm which is excluded from the integration to more accurately calculate the yield of interlocked products (Fig. S7†). These integrations also allow for a determination of the residual macrocycle (1) (Fig. 6b) and residual thread (2)/ADMET (6) yields (Table S1†). The crude NMRs can also provide preliminary data on the yield of cyclic catenane (5b) via the previously reported35 calculation method (eqn (S2) and Fig. S7†). This cyclic yield (Fig. S8†) was found to follow a similar trend to the amount of cyclic MSP (3b) given for each concentration (Fig. S9†). The 2.5 mM reaction has an average interlocked yield of ca. 73 ± 5% (n = 5), consistent with the previously reported value.35 The average interlocked yield remains above 70% for all concentrations except those at the extremes (0.25 mM and 10.0 mM). This is in part a consequence of a lower average alkene conversion (87% for both 0.25 and 10.0 mM, cf. 0.5–2.5 mM show ca. 95% conversion). Presumably at 0.25 mM, the relatively high dilution leads to a decrease in reaction rate and a slightly lower yield of interlocked materials (69 ± 5%). At the higher concentrations (5 and 10 mM), a significant amount of insoluble product is obtained during the reaction. This product was still insoluble after demetallation and was only observed to swell in a range of organic solvents, such as chloroform and tetrahydrofuran. These products are assumed to be interlocked networks and are included in the overall interlocked yield in Fig. 6b. None-the-less, at 10 mM, the interlocked yield was only 65 ± 10%. This lower value may result from partial insolubility of the both the MSP and the partially reacted species at these higher reaction concentrations. Additionally, the higher concentration will likely promote inter-MSP reactions (4e), resulting in an increased formation of ADMET polymer 6. Preliminary data from DOSY NMR analysis demonstrated a consistent decrease in diffusion coefficient as the reaction concentration increased (Fig. S10 and Table S1†), suggesting that the higher reaction concentrations yielded larger products.
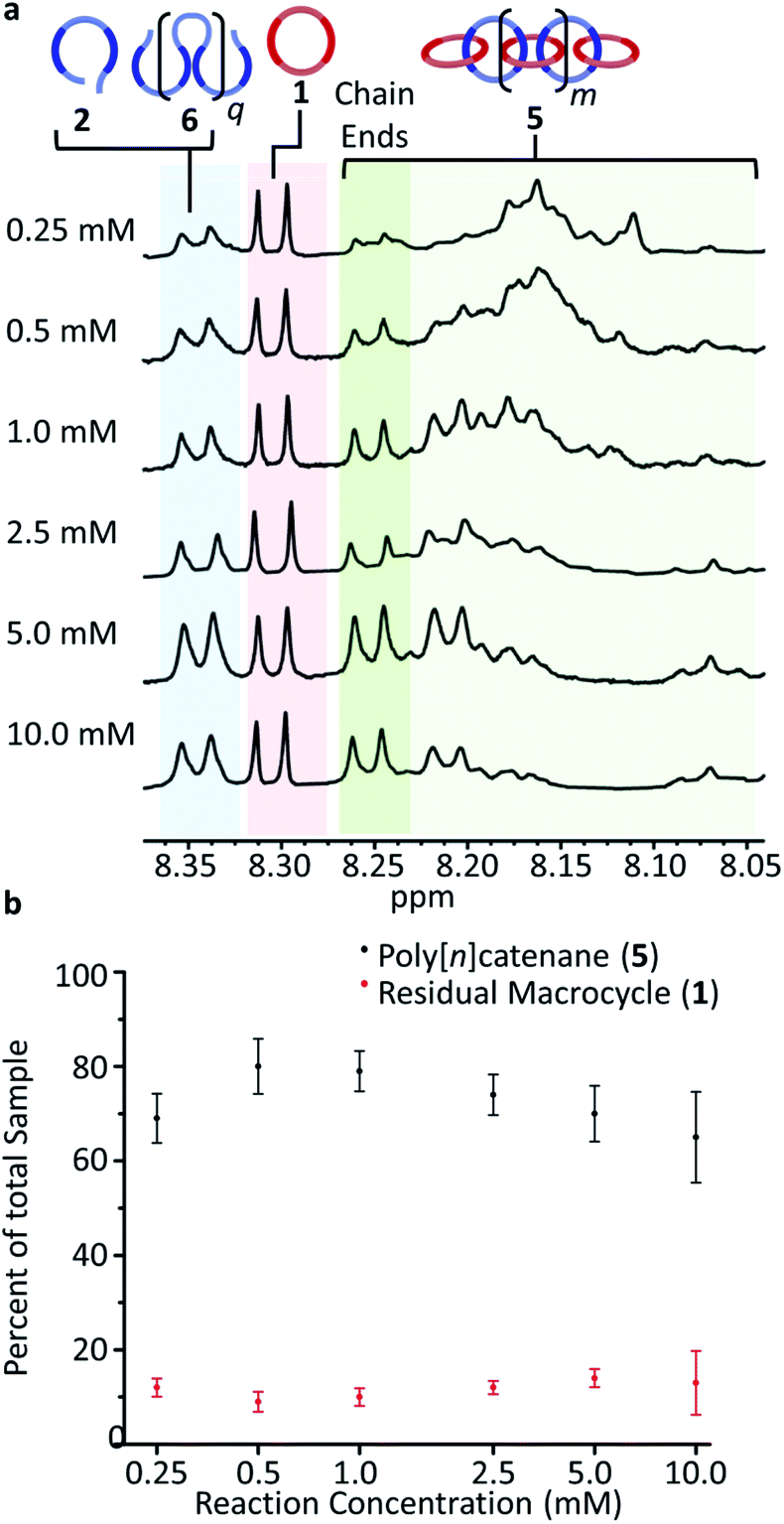 | ||
| Fig. 6 (a) Region of the 1H NMR spectra (500 MHz, CDCl3, 298 K) corresponding to the Hmpy protons for the demetallated crude reaction mixtures at different concentrations. (b) Average poly[n]catenane (5) (black) yields and amount of residual macrocycle (1) at reaction error bars for data sets taken at 95% confidence interval (n = 5). | ||
GPC analysis of poly[n]catenane architecture
To fully determine the extent of the size variations between reaction concentrations, the crude products were then analyzed via GPC-MALS. While initial studies of the poly[n]catenanes via GPC studies were conducted in THF,35 it was found that a 1:3 mixture of DMF:THF was a better mobile phase for these polymers. In addition, a GPC column with better low molecular weight resolution (relative to the prior studies)35 was employed to allow better resolution of such species. While the resulting GPC traces for the poly[n]catenane samples (Fig. 7a and S11–S16†) clearly show that a complicated range of products are formed during these reactions, it is possible to draw some conclusions from this data. Perhaps the most obvious data trend is a move to faster elution times with the reactions carried out at the higher concentrations, confirming the formation of higher molecular weight species in these more concentrated reactions. It is also possible to clearly see the presence of the residual macrocycle 1 in all the GPC spectra, which is consistent with what is observed in the NMR. In an attempt to obtain more information on the reaction products, deconvolution of the GPC traces was explored using a Gaussian algorithm38–40 to fit the data to the minimum number of peaks (see ESI† for more details). As can be seen in Fig. 7 (Fig. S12, S13 and S15†) excellent fits to the experimental GPC trace can be obtained using 6–9 Gaussian peaks (depending on the concentration of the reaction). Pure samples of macrocycle 1 and thread 2 were run using the same conditions and eluted at 17.2 minutes and 16.5 minutes, respectively (Fig. S17†), allowing those deconvoluted peaks (macrocycle (A, red) and thread (C, blue)) to be assigned to these starting materials. In addition, macrocycle 7, which can be formed by ring closing of thread 2, elutes ca. 17.3 minutes and as such, if present, would also be present as part of deconvoluted Peak A (red).
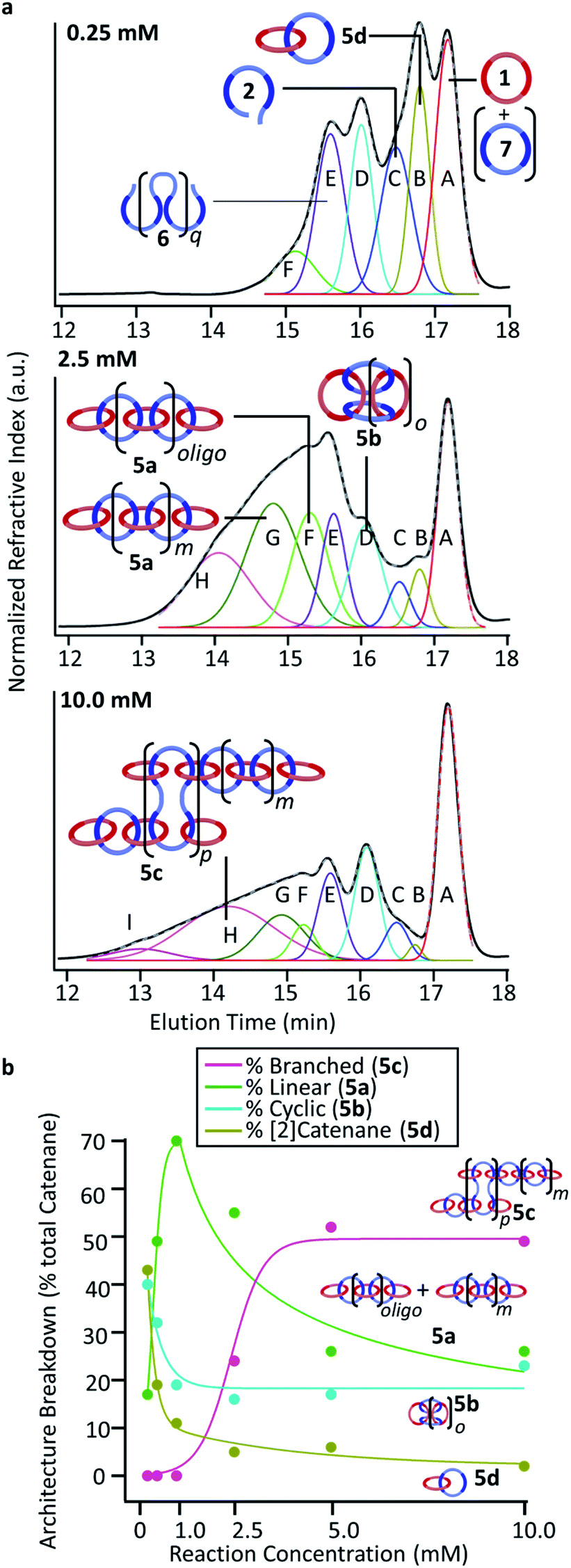 | ||
| Fig. 7 (a) GPC refractive index traces (black) for 0.25, 2.5, and 10.0 mM (mobile phase – 25% DMF in THF). Each trace shows deconvoluted peaks using a Gaussian fit (cummulative peak fit, grey dash) with each peak showing its (tentative) assignment. (b) Poly/oligo[n]catenane (5a, 5b, 5c (+insoluble fraction), and 5d) distribution versus reaction concentration (byproducts are excluded). | ||
Between the macrocycle peak (A, red) and the linear thread peak (C, blue) there is a peak that elutes at approximately 16.8 minutes (B, gold in Fig. 7). Based on the elution time of this peak, this compound is more compact than the thread (2) but bigger than macrocycles 1 and 7. MALS data corresponding to this peak (Fig. S11–S16†) shows that the molecular weight of this compound is approximately double that of macrocycle Peak A. This suggests that Peak B either corresponds to a [2]catenane (5d) and/or possibly the non-interlocked cyclic dimer of thread 2. The fact that this peak is more prominent at the lowest concentration (0.25 mM, Fig. 5, top), is consistent with either of these assignments.
Peak D (cyan in Fig. 7a) elutes at 16.0 minutes and is assigned as the cyclic catenane 5b, as it corresponds to the elution time of a purified cyclic sample, with an average of 5 rings, based on MALS data (Fig. S17†).35 The next identified peak elutes at approximately 15.6 minutes (Peak E, purple, Fig. 7a). While this peak does not correspond to any isolated samples, it is tentatively assigned to the ADMET (6) product in each of the samples.
Based on the previous poly[n]catenane characterization,35 it is proposed that Peaks F, G, H, and I correspond to different populations of linear (5a) and branched (5c) poly[n]catenanes.
There are a number of different routes that linear poly/oligo[n]catenanes can be accessed via during the synthesis. Fig. 8 highlights two possible routes to these structures: namely incomplete ring closing of the linear (3a) or cyclic (3b) MSPs. It can be expected that at low concentrations (when most of the MSP is the cyclic 3b, see above) the majority of the linear catenane will be formed via3b while at higher concentrations the linear catenane will accessed predominantly via3a. If both of these routes occur simultaneously, it can then be expected that there will be two molecular weight populations of linear 5a. These two populations are tentatively assigned to Peaks F and G (light and dark green) based on the GPC elution time of a sample of purified primarily linear (5a) poly[n]catenane (Fig. S17†) that spans both peaks.
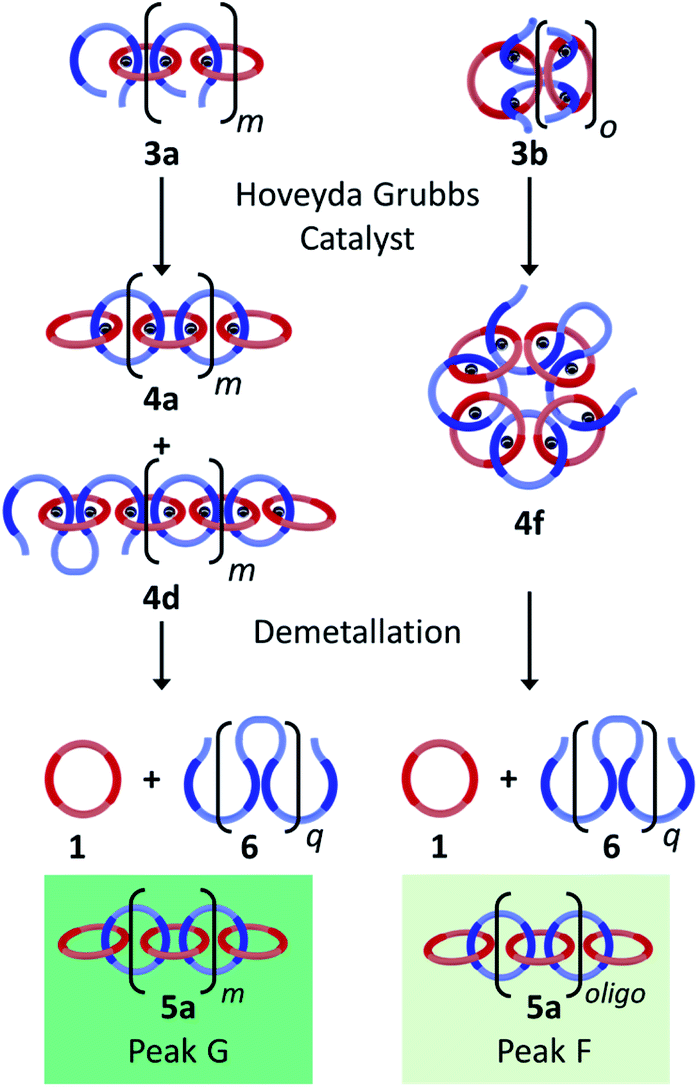 | ||
| Fig. 8 Proposed routes to linear poly/oligo[n]catenanes (5a) with different molecular weight populations (byproducts are excluded). | ||
For the 0.25 mM sample, where the MSP is almost exclusively cyclic 3b, only Peak F is present (15.2 minutes, light green). Based on MALS analysis of this peak, it corresponds to relatively low molecular weight oligomers/catenanes (average molecular weight Mnca. 8 kg mol−1). For this reaction, the 1H NMR of the sample shows evidence of catenane chain ends (Fig. 6a), indicating a mixture of linear 5a and cyclic 5b poly[n]catenanes. As such, it is proposed that Peak F corresponds to the linear oligo[n]catenanes 5a formed from via the cyclic MSP (Fig. 8). This hypothesis is supported by the fact that Peak F corresponds to a smaller percentage of the overall product distribution at higher concentrations (Table S2†). NMR of the MSP shows that, as the reaction concentration increases, the MSP shifts from being predominantly cyclic (3b) to being predominately linear (3a), resulting in a decrease in the yield of this oligomeric linear catenane (5a) at higher concentrations. As the relative size of Peak F decreases with reaction concentration there is a concomitant appearance and increase in the size of Peak G. It is proposed that Peak G corresponds to the higher molecular weight linear catenanes that result from the successful ring-closing of the linear MSP 3a (Fig. 8).
Peaks H and I correspond to the largest molecular weights observed in the catenanes. Based on chain end analysis of fractionated samples in prior work and comparison to an isolated branched poly[n]catenane sample (Fig. S17†), these peaks are tentatively assigned to branched 5c poly[n]catenanes. Peak H (light pink) at 14.1, begins to appear at 2.5 mM (Fig. 7a, middle), where the presence of branched catenanes has already been reported.35 As the concentration increases further however, a second peak (Peak I, dark pink) at 13.0 minutes is required for the Gaussian fit (Fig. 7a, bottom and Table S2†), suggesting the formation of a new population of highly branched polymers (MALS, Mn = 60 kg mol−1, 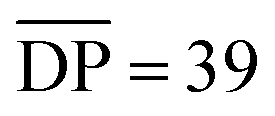 (eqn (S1))†).
(eqn (S1))†).
Based on these peak assignments, some general trends can be elucidated (Fig. 7b and Table S2†) regarding the impact of reaction concentration on product distribution. As may be expected, the [2]catenane (5d) (and/or cyclic dimer of 2) and the cyclic (5b) catenanes are the dominant products at the lowest concentration (0.25 mM). As the reaction concentration increases, the yields of 5d/2 and 5b drop and eventually plateau for concentrations between 2.5–10 mM.41
The linear (5a) catenanes (polymer + oligomer) show a significant increase in yield from 0.25 mM to 1.0 mM (Fig. 7b, green) and become the dominant product (70% 5a at 1.0 mM). However, the linear 5a formed at 1.0 mM appears to be primarily oligomeric in nature (Peak F), presumably a consequence of being formed predominantly from the cyclic MSP precursor (3b). While increasing the reaction concentration to 2.5 mM results in a slight drop in the overall yield of linear 5a (Fig. 8), the resulting 5a now is predominately the higher molecular 5a fraction (Peak G). As a result, carrying out this reaction at 2.5 mM optimizes both the yield and molecular weight of the linear 5a products. Between 2.5–5.0 mM the overall amount of linear polycatenane decreases as branched poly[n]catenanes 5c (Fig. 7a, pink) start to be formed. It is worthwhile also noting that at the higher concentrations, the larger molecular weight fraction (Fig. 7a, Peak G) of linear polycatenane 5a becomes the dominant linear species. However, the yield of the branched polycatenanes (5c plus networks as these numbers also include insoluble polymers) quickly dominates the reaction products, becoming the major poly[n]catenane formed above 5.0 mM (52% 5c) and 10.0 mM (48% 5c).
Chain end analysis via GPC/NMR techniques
One way to further elucidate the trends in architecture (and to back up the assignment of the deconvoluted peaks in the GPC) is to determine the number of average numbers of chain ends (Nc) in each sample. For these samples, cyclic poly[n]catenanes have no chain ends (Nc = 0), linear poly[n]catenanes have Nc = 2 and branched poly[n]catenanes have Nc ≥ 3, therefore determination of the average number of chain-ends in the sample can give an idea of the architecture distribution. Using a qualitative assessment of the 1H NMR data in Fig. 6a, the chain end region from 8.24–8.27 reflects the observed GPC trends: products formed at low reaction concentrations show diminutive peaks in this region, reflecting fewer chain ends and the primarily cyclic nature of the materials. However, at the higher reaction concentrations the products show increased prevalence of these chain end peaks consistent with the transition into linear and highly branched materials.To quantify this analysis, a new set of poly[n]catenanes were synthesized and were partially purified before analysis using literature procedures.35 This purification was performed in an attempt to remove as much of the byproducts as possible, particularly ADMET 6, to minimize the influence of these byproducts on the reported poly[n]catenane molecular weight and calculated Nc.
For this purification, the poly[n]catenanes were partially remetallated with Zn2+ ions and the mixture was dried and washed with 2:1 chloroform:hexanes to remove the soluble unmetallated (by)products (see ESI† and previous work35 for more details on the purification procedure). These samples were then demetallated and analyzed by GPC MALS (using 25 vol% DMF in THF as the mobile phase) and 1H NMR. Fig. 9a shows the 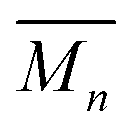 and calculated average number of chain ends (Nc), determined by a combination of the NMR and the GPC-MALS data using previously published procedures (eqn (S3)†).35 For example, based on this combination of NMR and GPC-MALS data, the poly[n]catenanes prepared at 2.5 mM have an average number of chain-ends of ca. 2.7, consistent with the product architecture being predominantly linear (5a) with some branched. In fact, deconvolution of the GPC trace (Fig. S18†) shows the dominance of the linear poly[n]catenane assigned Peak G (50% of the total catenated material) in this sample.
and calculated average number of chain ends (Nc), determined by a combination of the NMR and the GPC-MALS data using previously published procedures (eqn (S3)†).35 For example, based on this combination of NMR and GPC-MALS data, the poly[n]catenanes prepared at 2.5 mM have an average number of chain-ends of ca. 2.7, consistent with the product architecture being predominantly linear (5a) with some branched. In fact, deconvolution of the GPC trace (Fig. S18†) shows the dominance of the linear poly[n]catenane assigned Peak G (50% of the total catenated material) in this sample.
 | ||
| Fig. 9 (a) Number average molar mass (Mn) determined by GPC-MALS plotted for the reaction concentrations to observe the trends. Number of chain ends (Nc) determined by a combination of GPC-MALS and NMR analysis.29 (b) GPC RI trace from the 10.0 mM (black) and 0.25 mM (grey, dash) purified sample. Absolute molecular weight (determined by MALS) for the 10.0 mM purified sample (red, dots) and the 0.25 mM purified sample (blue, dashes). | ||
The data obtained from the partially purified samples also allows for a more complete analysis of the molecular weight trends throughout the various concentrations with the reduced presence of ADMET 6 (Fig. S19†). Consistent with the prior data, lower molecular weights result from the samples where the primary starting material was the cyclic MSP 3b. For these, at 0.25 mM and 0.5 mM, the average molecular weight appeared as 9.8 and 11.6 kg mol−1 respectively. These would correspond to catenanes with approximately 6 and 8 interlocked macrocycles. For both of these samples, chain end analysis35 revealed that the average poly[n]catenane contained fewer than 2 chain ends (0.8, 1.5, Fig. 9a) demonstrating that the material was likely a combination of the [2]catenane (5d), oligomeric 5a, and cyclic 5b poly[n]catenanes.
At higher concentrations the MSP precursor shifts to a higher percentage of the linear (3a) and as such the resulting products now consist of more linear (5a) and branched (5c) poly[n]catenanes (Nc > 2). This is concomitant with an increase of the 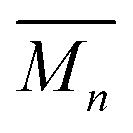 for the samples: at 2.5 mM the
for the samples: at 2.5 mM the 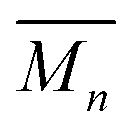 is 19.5 kg mol−1,
is 19.5 kg mol−1, 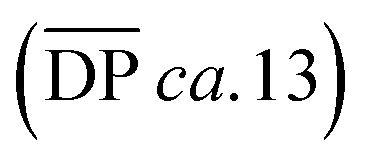 and Nc is 2.7. At this reaction concentration, the GPC MALS trace also shows evidence of larger catenanes, with molecular weights up to 100 kg mol−1.
and Nc is 2.7. At this reaction concentration, the GPC MALS trace also shows evidence of larger catenanes, with molecular weights up to 100 kg mol−1.
The highest reaction concentration studied (10.0 mM) produces the highest molecular weight species, 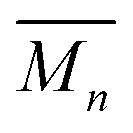 of 21 kg mol−1
of 21 kg mol−1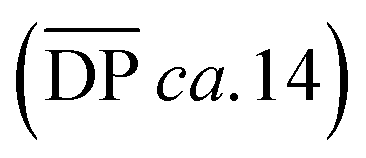 . The poly[n]catenanes synthesized here exhibit the largest Nc (5.6), indicative of the presence of a significant amount of highly branched poly[n]catenanes. It is important to note that this molecular weight analysis includes the lower molecular weight products (ADMET (6), cyclic (5b), and [2]catenane (5d)) that were not completely removed during the partial purification step, suggesting that the average size of the linear 5a and branched 5c materials is larger than this value.
. The poly[n]catenanes synthesized here exhibit the largest Nc (5.6), indicative of the presence of a significant amount of highly branched poly[n]catenanes. It is important to note that this molecular weight analysis includes the lower molecular weight products (ADMET (6), cyclic (5b), and [2]catenane (5d)) that were not completely removed during the partial purification step, suggesting that the average size of the linear 5a and branched 5c materials is larger than this value.
For comparison Fig. 9b shows the GPC-MALS data for these partially purified reactions mixtures that were obtained a 0.25 and 10 mM. The highest molecular weight products obtained at 0.25 mM are ca. 33 kg mol−1. However, the MALS data of products obtained from the 10 mM show evidence of soluble (branched) catenanes up to 1000 kg mol−1 (DP > 640), which are the largest reported poly[n]catenane compounds to date.
Conclusions
This study has been able to clarify several aspects of synthesis of poly[n]catenanes from an MSP template. The combination of carrying out the reaction at different concentrations along with analysis of the crude reaction mixture has shown how the architecture of the resulting poly[n]catenane can be altered. At low concentrations, cyclic and oligomeric linear poly[n]catenanes will predominate, while at the highest reaction concentrations, high molecular weight branched poly[n]catenanes are the major product, with poly[n]catenanes of up to 1000 kg mol−1 being observed. The formation of insoluble but swellable materials at these higher reaction concentrations may suggest the formation of interlocked networks and current research is underway to better characterize and understand these materials.Author contributions
SJR and QW proposed the study. QW conducted preliminary studies and provided a set of comparative poly[n]catenanes. All further synthesis and analysis were conducted by MMT. SJR supervised the work. MMT and SJR wrote the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by National Science Foundation (NSF) grant number CHE-1903603. Parts of this work were carried out at the Soft Matter Characterization Facility of the University of Chicago. We would like to thank the director of this facility, Dr Phillip Griffin, for assistance with GPC characterization. We would like to thank the University of Chicago Chemistry NMR Facility and the facility mangers past and present, Dr Antoni Jurkiewwicz and Dr Josh Kurutz. Finally, we would like to acknowledge Dr Benjamin Rawe for his intellectual contributions to this work.References
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- A. Harada, Nature, 1992, 356, 325–327 CrossRef CAS.
- Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17–29 RSC.
- M. Arunachalam and H. W. Gibson, Prog. Polym. Sci., 2014, 39, 1043–1073 CrossRef CAS.
- T. Takata, N. Kihara and Y. Furusho, Adv. Polym. Sci., 2004, 171, 1–75 CrossRef CAS.
- L. F. Hart, J. E. Hertzog, P. M. Rauscher, B. W. Rawe, M. M. Tranquilli and S. J. Rowan, Nat. Rev. Mater., 2021 DOI:10.1038/s41578-021-00278-z.
- T. Takata, Polym. J., 2006, 38, 1–20 CrossRef CAS.
- H. Xing, Z. Li, Z. L. Wu and F. Huang, Macromol. Rapid Commun., 2018, 39, 1700361 CrossRef PubMed.
- W. Wang and H. Xing, Polym. Chem., 2018, 9, 2087–2091 RSC.
- P. M. Rauscher, K. S. Schweizer, S. J. Rowan and J. J. de Pablo, Macromolecules, 2020, 53, 3390–3408 CrossRef CAS.
- P. M. Rauscher, S. J. Rowan and J. J. de Pablo, ACS Macro Lett., 2018, 7, 938–943 CrossRef CAS.
- P. M. Rauscher, K. S. Schweizer, S. J. Rowan and J. J. de Pablo, J. Chem. Phys., 2020, 152, 214901 CrossRef CAS PubMed.
- H. C. Renger and D. R. Wolstenholme, J. Cell Biol., 1972, 54, 346–364 CrossRef CAS PubMed.
- G. Riou and E. Delain, Proc. Natl. Acad. Sci. U. S. A., 1969, 62, 210–217 CrossRef CAS PubMed.
- P. Borst and J. H. J. Hoeijmakers, Plasmid, 1979, 2, 20–40 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1994, 33, 1286–1290 CrossRef.
- C. H. Lu, X. J. Qi, A. Cecconello, S. S. Jester, M. Famulok and I. Willner, Angew. Chem., Int. Ed., 2014, 53, 7499–7503 CrossRef CAS PubMed.
- H. Iwamoto, S. Tafuku, Y. Sato, W. Takizawa, W. Katagiri, E. Tayama, E. Hasegawa, Y. Fukazawa and T. Haino, Chem. Commun., 2016, 52, 319–322 RSC.
- N. Watanabe, Y. Ikari, N. Kihara and T. Takata, Macromolecules, 2004, 37, 6663–6666 CrossRef CAS.
- J. A. Berrocal, L. M. Pitet, M. M. L. Nieuwenhuizen, L. Mandolini, E. W. Meijer and S. Di Stefano, Macromolecules, 2015, 48, 1358–1363 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143–3150 CrossRef CAS.
- K. Endo and T. Yamanaka, Macromolecules, 2006, 39, 4038–4043 CrossRef CAS.
- H. Xing, Z. Li, W. Wang, P. Liu, J. Liu, Y. Song, Z. L. Wu, W. Zhang and F. Huang, CCS Chem., 2019, 1, 513–523 Search PubMed.
- B. Nisar Ahamed, R. Duchêne, K. Robeyns and C.-A. Fustin, Chem. Commun., 2016, 52, 2149–2152 RSC.
- N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- J. P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1985, 107, 6108–6110 CrossRef CAS PubMed.
- Modern Supramolecular Chemistry, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley, 2008 Search PubMed.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, J. J. de Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- R. J. Wojtecki, Q. Wu, J. C. Johnson, D. G. Ray, L. S. T. J. Korley and S. J. Rowan, Chem. Sci., 2013, 4, 4440–4448 RSC.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- S. A. Mirmohammadi, M. Imani, H. Uyama, M. Atai, M. B. Teimouri and N. Bahri-Lale, Polym. Int., 2014, 63, 479–485 CrossRef CAS.
- N. Cozza, W. Bonani, A. Motta and C. Migliaresi, Int. J. Biol. Macromol., 2016, 87, 504–513 CrossRef CAS PubMed.
- Y. Zheng, Y. Huang and B. C. Benicewicz, Macromol. Rapid Commun., 2017, 38, 1700300 CrossRef PubMed.
- J. A. Semlyen, in Mechanisms of Polyreactions-Polymer Characterization, Springer Berlin Heidelberg, Berlin, Heidelberg, 1976, pp. 41–75 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Novel experimental procedures, 1H NMR spectra, DOSY NMR analysis, NMR analysis calculations, GPC MALS deconvolution and MALS analysis, chain end analysis calculation. See DOI: 10.1039/d1sc02450g |
| This journal is © The Royal Society of Chemistry 2021 |