
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of tryptophan-containing 2,5-diketopiperazines via sequential C–H activation: total syntheses of tryprostatin A, maremycins A and B†
Xue-Song
Yin‡
,
Wei-Yi
Qi‡
 and
Bing-Feng
Shi
*
and
Bing-Feng
Shi
*
Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: bfshi@zju.edu.cn
First published on 7th September 2021
Abstract
Indole 2,5-diketopiperazines (DKPs) are an important type of metabolic cyclic dipeptides containing a tryptophan (Trp) unit possessing a range of interesting biological activities. The intriguing structural features and divergent activities have stimulated tremendous efforts towards their efficient synthesis. Herein, we report the development of a unified strategy for the synthesis of three Trp-containing DKPs, namely tryprostatin A, and maremycins A and B, via a sequential C–H activation strategy. The key Trp skeletons were synthesized from the inexpensive, readily available alanine via a Pd(II)-catalyzed β-methyl C(sp3)–H monoarylation. A subsequent C2-selective prenylation of the resulting 6-OMe-Trp by Pd/norbornene-promoted C–H activation led to the total synthesis of tryprostatin A in 12 linear steps from alanine with 25% overall yield. Meanwhile, total syntheses of maremycins A and B were successfully accomplished using a sequential Pd-catalyzed methylene C(sp3)–H methylation as the key step in 15 linear steps from alanine.
Introduction
2,5-Diketopiperazines (DKPs), an important type of cyclic dipeptides derived from the condensation of two amino acids, represent a large family of biologically active molecules with a rigid conformation and high stability in vivo.1 Among them, indole DKP alkaloids that contain a tryptophan (Trp) unit are particularly interesting (Fig. 1). The intriguing structural features of these compounds enhance their binding affinity and specificity with biological targets, thus making them suitable for drug discovery with various biological activities such as antitumor, antibacterial, antiviral, antioxidant, and immunomodulatory.2 Representative members include tryprostatin A (1), maremycin A and B (2), cyclotryprostatin A and B (3), thaxtomin A (4) and norgeamide A and B (5) (Fig. 1). | ||
| Fig. 1 Representative Trp-containing DKP natural products. | ||
Tryprostatin A (1) was isolated as a secondary metabolite from a marine fungal strain Aspergillus fumigatus BM939.3a–c It was reported that tryprostatin A completely inhibited cell cycle progression of tsFT210 cells in the G2/M phase.3 The total synthesis of tryprostatin A has been achieved by several groups with the emphasis on the asymmetric preparation of the chiral 6-OMe-Trp moiety and a C2-selective prenylation of the indole ring.4,5 The Cook group first employed the Fischer indole/Schöllkopf protocol to furnish the chiral 6-OMe-Trp unit followed by bromination/lithiation/prenylation to install the C2 prenyl group, leading to the total synthesis in 13 steps with 14.7% overall yield.4a–d In 2010, the Fukuyama group elegantly applied radical-mediated indole synthesis/Pd-catalyzed prenylation to accomplish the total synthesis of tryprostatin in 11 linear steps with 30% yield.4e
Maremycin A and B (2a and 2b) are indole DKP alkaloids isolated from the culture broth of marine Streptomyces strain B 9173, featuring a 3-hydroxyindolin-2-one structure derived from (2S,3S)-β-methyltryptophan (β-MeTrp).6 The intriguing structure has stimulated tremendous efforts on the total synthesis.7 In 2008, Tamura and co-workers reported the first total synthesis of maremycin A using [3 + 2] cycloaddition as the key step in 9 linear steps with 10.6% yield.7a,e In 2011, the Melchiorre group reported the synthesis of the enantioenriched 3-substituted 3-hydroxyoxindoles by organocatalyzed addition of N–Me dioxindole to crotonaldehyde and accomplished the total synthesis of maremycin A in 7 linear steps with 15% yield.7b Shortly after, Scheidt and co-workers achieved a concise synthesis of maremycin B via a key N-heterocyclic carbene/Lewis acid-catalyzed formal [3 + 2] addition in 6 linear steps with 17% yield.7c Another strategic synthesis of maremycin A and B was accomplished by Jia and co-workers in 2012, featuring a chiral pool strategy starting from L-isoleucine followed by a Pd-catalyzed indole synthesis (14 steps, 4.8% yield for maremycin A; 13 steps, 8.2% yield for maremycin B).7d The biosynthesis of the key β-MeTrp unit has also been achieved by Lin,7f,g Buller and Arnold.7h
Although Trp is the least abundant of the 20 proteinogenic amino acids, its unique structure renders it to be easily derivatized both enzymatically and chemically. Thus, Trp has been found to be an important biosynthetic precursor to a variety of microbial natural products.8 Structurally, Trp-containing DKPs are characterized by a Trp or its derivatives, as exemplified in Fig. 1. For example, tryprostatin A is composed of a proline and an unnatural 2-prenyl-6-OMe-Trp unit; maremycins A and B are composed of an S-methylcysteine (S-Me-Cys) and an unnatural AA derived from β-MeTrp. Obviously, the concise synthesis of these Trp-containing DKP alkaloids highly relies on the development of a general and straightforward strategy to access various substituted Trps.
In recent years, C–H activation has revolutionized the retrosynthetic logic of natural products.9 Our group has long been interested in promoting the practical synthesis of natural products via C–H activation.10 Herein, we report the total synthesis of three Trp-containing DKP alkaloids, namely tryprostatin A, and maremycins A and B. The key Trp skeletons were synthesized from the inexpensive, readily available alanine via a Pd(II)-catalyzed β-methyl C(sp3)–H monoarylation.11,12 A subsequent C2-selective prenylation of the resulting 6-OMe-Trp by Pd/norbornene-promoted C–H activation led to the total synthesis of tryprostatin A. Meanwhile, total syntheses of maremycins A and B were successfully accomplished using a sequential Pd-catalyzed methylene C(sp3)–H methylation as the key step.
Results and discussion
Retrosynthetic analysis based on a sequential C–H activation strategy
Our retrosynthetic analysis is depicted in Fig. 2. The DKP moiety could be formed by the condensation of two amino acids [e.g. β-MeTrp (I) + S-Me-Cys (II) → maremycins; 2-prenyl-6-OMe-Trp (III) + Pro (IV) → tryprostatin A]. We reasoned that the syntheses of the two key Trp units, β-MeTrp (I) and 2-prenyl-6-OMe-Trp (III), were a good platform to verify the brevity and practicality of the sequential C–H activation strategy. Specifically, our recently developed Pd(II)-catalyzed β-methyl C(sp3)–H monoarylation of an alanine derivative (6)11 followed by methylene C(sp3)–H methylation13 directed by 8-aminoquinoline would enable the facile construction of β-MeTrp (I).14 2-Prenyl-6-OMe-Trp (III) could be assembled by the same β-methyl C(sp3)–H monoarylation strategy while the requisite prenyl moiety could be installed by a C2-selective C–H prenylation. The inspiration for this came from previous reports by Bach and co-workers15 on Pd(II)/norbornene-promoted C2-selective C–H alkylation of indole.16 Different from previous synthetic strategies, this sequential C–H activation logic enables the asymmetric synthesis of indole DKPs from the inexpensive and commercially available L-alanine as the chiral pool, avoiding the employment of expensive chiral catalysts/auxiliaries. | ||
| Fig. 2 Retrosynthetic analysis. | ||
Total synthesis of tryprostatin A via a sequential C–H activation strategy
We have previously reported the practical synthesis of β-aryl-α-amino acids through a Pd(II)-catalyzed β-methyl C(sp3)–H monoarylation of 8-aminoquinoline derived alanine 6 with a broad range of aryl iodides,11 which has been successfully applied to the total synthesis of tetrahydroisoquinoline alkaloids.10c,d The robustness of this protocol encouraged us to examine if 3-iodoindole derivatives were compatible with this protocol. We commenced the synthesis with the examination of different N-protected 3-iodo-6-methoxyindoles with the monoarylation of alanine derivative 6 (Scheme 1).11 We were delighted to find that the use of Ac, Ts and Ns as the protecting groups gave the desired 6-OMeTrps in moderate to high yields (8d, R = Ac, 55%; 9, R = Ts, 83%; 10, R = Ns, 82%), whereas the use of free indole or N-Boc and N-PMB-protected ones resulted in no detectable products (8a–8c). Considering the scalable synthesis (Scheme 1, 10, 80%, 4.32 g) and ease of removal, N-Ns-protected iodoindole 7f was chosen as the iodination reagent. Of note, the N-Phth protecting group is crucial for the high efficiency of C–H arylation as reported in previous studies.11,12 Removal of the 8-aminoquinoline group of Trp 10 was completed by treating with BF3·Et2O in methanol (Scheme 2).12b,17 Trp derivative 11 was obtained in 89% yield along with the recovery of 8-aminoquinoline in 80% yield. Subsequent removal of the p-Ns group with mercaptoacetic acid/K2CO3 gave N-phthaloyl-6-OMeTrp 12 in 96% yield.18 | ||
| Scheme 1 Synthesis of tryptophan derivatives by Pd(II)-catalyzed C(sp3)–H monoarylation. a0.2 mmol 6. b8 mmol 6. | ||
 | ||
| Scheme 2 Synthesis of tryprostatin A. | ||
With N-phthaloyl-6-OMeTrp 12 in hand, we set out to install the C2-prenyl moiety. The selective introduction of prenylation at the C2 position of the indole ring is a daunting challenge. In the synthesis of tryprostatin B, Danishefsky group elegantly developed a C2-selective nucleophilic prenylation of in situ generated 3-chloroindolenine with prenylstannane and BCl3 in 83% yield.19 However, when we tried to apply this method to the C2-prenylation of N-phthaloyl-6-OMeTrp 12, the desired C2-prenylation product 13 was only obtained in 46% yield, largely due to the formation of chlorinated byproducts caused by the electron-donating methoxy group at the C6-position.
In 2011, Bach and co-workers elegantly reported a Pd(II)/norbornene-promoted C2-selective C–H alkylation of indoles15a,b and extended it to the regioselective alkylation at the C2 indole position of tryptophan.15c Inspired by these pioneering studies, we rationalized that a Pd-catalyzed, norbornene-mediated C2-selective prenylation might be possible by finely tuning the conditions. Firstly, N-phthaloyl-6-OMeTrp 12 was converted to N-Boc-6-OMeTrp 15 in 95% yield over two steps. Screening experiments were conducted with 15 and prenyl bromide (Table S1, see the ESI† for details). Although the previous conditions only gave trace C2-prenylation product 16, we found that the prenylation reaction was dramatically affected by bases and solvents, with a stronger base, Cs2CO3, and a mixture of acetonitrile and water as the solvent, being the optimal conditions. Finally, the prenylation product 16 was obtained in 68% yield when the reaction was conducted under the following optimized conditions: Pd(OAc)2 (10 mol%), PPh3 (20 mol%), 3 equiv. prenyl bromide, 5 equiv. norbornene, 3 equiv. Cs2CO3, CH3CN/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 50 °C for 24 hours under N2.
:1) at 50 °C for 24 hours under N2.
Cleavage of N-Boc followed by coupling with N-Boc-Pro-OH, gave dipeptide 17 in 98% yield. Subsequently, deprotection of N-Boc followed by an intramolecular cyclization accomplished the total synthesis of tryprostatin A (1). The 1H and 13C NMR spectra of our synthetic tryprostatin A (1) were identical to that reported by Osada.3b The optical rotation values also matched (our synthetic product: [α]20D = −67.5 (c 0.825, CHCl3); lit:3b [α]27D = −69.7 (c 0.70, CHCl3)). The concise total synthesis of tryprostatin A was achieved in 12 linear steps from alanine with 25% overall yield using Pd(II)-catalyzed monoarylation of methyl C(sp3)–H and Pd/norbornene-promoted C–H prenylation as key steps.
Removal of the N-phthaloyl group from 13 with ethylenediamine, followed by coupling with N-Boc-Pro-OH also furnished dipeptide 17 in 95% yield. Thus, an alternative total synthesis of tryprostatin A could also be achieved in 11 linear steps from alanine with 18% overall yield using Danishefsky's C2-selective nucleophilic prenylation as a key step.
Total syntheses of maremycins A and B via a sequential C(sp3)–H activation strategy
Encouraged by these results, the total syntheses of maremycins A and B via sequential C(sp3)–H activation was conducted. The major challenge was the efficient synthesis of the β-MeTrp unit (Fig. 2, I). We envisioned that the key unit could be obtained from 8-aminoquinoline-derived alanine 6via a sequential Pd(II)-catalyzed methyl C(sp3)–H monoarylation followed by methylene C(sp3)–H methylation. Following our discovery in the synthesis of tryprostatin A, N-Ns-3-iodoindole 18 was used as an efficient arylation reagent to react with alanine derivative 6, giving Trp 19 in 82% yield (Scheme 3, 10.55 g), which was subjected to a subsequent Pd(II)-catalyzed methylene C(sp3)–H methylation. However, unsatisfactory yield and stereoselectivity were obtained under previously established conditions for aliphatic amides (Table 1, entry 1, 29% yield, d.r. = 3:1).12a Screening of ligands was carried out since we have observed that ligands are essential for both the reactivity and diastereoselectivity. Several sulfonamides were tested and moderate yields with poor diastereoselectivities were observed (entries 2–4). The use of pivalic acid and 1-adamantanecarboxylic acid could significantly improve the yields, albeit with very low diastereoselectivities (entries 5 and 6).
 | ||
| Scheme 3 Total syntheses of maremycins A and B. | ||
|
|
||||
|---|---|---|---|---|
| Entrya | Ligand | [Ag] | d.r. | Yield (%) |
| a Reaction conditions: 19 (0.15 mmol), Pd(OAc)2 (10 mol%), ligand (30 mol%), [Ag] (1.5 equiv.), MeI (10.0 equiv.), NaOCN (2.0 equiv.), 1,4-dioxane, 80 °C, 24 h. Isolated yield. b 4.0 mmol scale. c 1.0 equiv. Ag3PO4. | ||||
| 1 | p-ClPhSO2NH2 | Ag2CO3 | 3:1 |
29 |
| 2 | PhSO2NH2 | Ag2CO3 | 3:1 |
64 |
| 3 | p-MePhSO2NH2 | Ag2CO3 | 5:2 |
56 |
| 4 | p-CF3PhSO2NH2 | Ag2CO3 | 2:1 |
44 |
| 5 | PivOH | Ag2CO3 | 1:1 |
80 |
| 6 | AdCO2H | Ag2CO3 | 9:7 |
66 |
| 7 | N-Boc-glycine | Ag2CO3 | 3:1 |
44 |
| 8 | N-Cbz-glycine | Ag2CO3 | 3:1 |
90 |
| 9 | N-Fmoc-glycine | Ag2CO3 | 12:1 |
40 |
| 10 | N-Fmoc-glycine | AgOAc | 12:1 |
52 |
| 11 | N-Fmoc-glycine | Ag3PO4 | 12:1 |
89(84)b,c |
| 12 | N-Fmoc-glycine | AgOCN | 12:1 |
77 |
| 13 | N-Fmoc-glycine | AgOTs | 12:1 |
93(52)b |
| 14 | N-Fmoc-glycine | AgTFA | 12:1 |
62 |
| 15 | N-Fmoc-glycine | AgOMs | 12:1 |
68 |
| 16 | N-Fmoc-glycine | AgBF4 | 12:1 |
0 |
| 17 | None | Ag2CO3 | — | Trace |
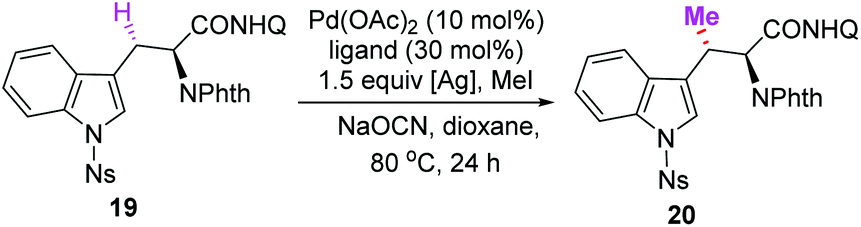
It has been reported that N-monoprotected amino acids could act as efficient ligands to promote C–H activation reactions.20 Further investigations revealed that the diastereoisomeric ratio could be significantly improved to 12:1 when using N-Fmoc-glycine as the ligand (entry 9, 40% yield). Various silver salts were then tested (entries 10–16). Although AgOTs was found to be the most efficient additive when the reaction was conducted in 0.15 mmol scale (entry 13, 93% yield), it was unsuitable for the scale-up reaction (4 mmol, 52% yield). Fortunately, Ag3PO4 was found to be the optimal oxidant for the scale-up reaction, giving the same high yields (entry 11, 0.15 mmol scale, 89% yield; 4 mmol, 84% yield, 12:1 d.r.). Trace product was observed when the reaction was conducted in the absence of the ligand, indicating the remarkable facilitation of ligands (entry 17). The absolute configuration of methylated product 20 was confirmed unambiguously by X-ray crystallographic analysis (Scheme 3, CCDC 2056344†).21
Removal of the 8-aminoquinoline directing group under BF3·Et2O/MeOH gave methyl ester 21 in 65% yield.12b,17 After removal of the N-Ns group followed by N-methylation of indole, Trp 23 was obtained in 72% yield over two steps. The direct oxidation of N-methyl-indole 23 to 3-hydroxy-2-indolone under IBX/CeCl3 turned out to be unsuccessful due to the steric bulkiness of the β-methyl group.22 Inspired by Jia's work,7d a two-step oxidation sequence was adopted. N-Methyl-indole 23 was first oxidized by DMSO in an acidic environment to furnish indolone 24a/b in 99% yield as a 1:1 mixture of diastereomers. The N-Phth group was then transformed to N-Boc to give 25a/b, which was oxidized with O2 and subsequently underwent lactonization to give the spiro-lactone 27.23
Finally, deprotection of N-Boc in 27, followed by condensation with 28 using HATU gave dipeptide product 29, which upon treating with silica gel in boiling p-xylene, accomplished the total synthesis of maremycin A (2a) in 36% yield and maremycin B (2b) in 38% yield over three steps. All the spectroscopic data of our synthetic maremycin A were identical to those previously reported {see Table S2† for NMR data comparison; our synthetic product: [α]20D = −111.3 (c 0.24, MeOH); lit:6 [α]25D= −120.95 (c 0.21, MeOH); lit:7d [α]25D = −115.5 (c 0.30, MeOH)}. The NMR spectroscopic data of maremycin B (2b) also matched those reported (see Table S3† for NMR comparison).6,7d However, the optical rotation of our synthetic 2b {[α]20D = +69.8 (c 0.78, MeOH)} was identical to that of Jia's synthetic sample {[α]25D = +78.3 (c 0.28, MeOH)}7d but differed from that of Laatsch's report {[α]25D = +2.94 (c 0.21, MeOH)}.6
Conclusions
In summary, we have reported the development of a unified strategy for the synthesis of three Trp-containing DKPs, namely tryprostatin A, and maremycins A and B, via a sequential C–H activation strategy. The key Trp skeletons were synthesized from the inexpensive, readily available alanine via a Pd(II)-catalyzed β-methyl C(sp3)–H monoarylation. A subsequent C2-selective prenylation of the resulting 6-OMe-Trp by Pd/norbornene-promoted C–H activation led to the total synthesis of tryprostatin A in 12 linear steps from alanine with 25% overall yield. Meanwhile, total syntheses of maremycins A and B were successfully accomplished using a sequential Pd-catalyzed methylene C(sp3)–H methylation as the key step in 15 linear steps from alanine. These achievements showcase the robustness of C–H activation in natural product total synthesis. Further application of the C–H activation strategy in the synthesis of other indole DKP alkaloids is ongoing in our laboratory.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterisation and copies of 1H and 13C NMR spectra. Crystallographic data for compound 20 has been deposited under CCDC 2056344.Author contributions
X.-S. Yin and W.-Y. Qi contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (21925109 and 21772170) and Outstanding Young Talents of Zhejiang Province High-level Personnel of Special Support (ZJWR0108) is gratefully acknowledged.Notes and references
- (a) C. J. Dinsmore and D. C. Beshore, Tetrahedron, 2002, 58, 3297–3312 CrossRef CAS; (b) M. B. Martins and I. Carvalho, Tetrahedron, 2007, 63, 9923–9932 CrossRef CAS; (c) A. D. Borthwick, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed; (d) Y. Wang, P. Wang, H. Ma and W. Zhu, Expert Opin. Ther. Pat., 2013, 23, 1415–1433 CrossRef CAS; (e) P. Umberto and P. Silvia, Targets Heterocycl. Syst., 2013, 17, 215–239 Search PubMed; (f) R.-M. Huang, X.-X. Yi, Y. Zhou, X. Su, Y. Peng and C.-H. Gao, Mar. Drugs, 2014, 12, 6213–6235 CrossRef PubMed; (g) A. D. Borthwick and J. Liddle, Med. Res. Rev., 2011, 31, 576–604 CrossRef CAS PubMed; (h) I. Bellezza, M. J. Peirce and A. Minelli, Trends Mol. Med., 2014, 20, 551–558 CrossRef CAS.
- Y.-M. Ma, X.-A. Liang, Y. Kong and B. Jia, J. Agric. Food Chem., 2016, 64, 6659–6671 CrossRef CAS PubMed.
- Isolation and biological activity of tryprostatin A: (a) C.-B. Cui, H. Kakeya, G. Okuda, R. Onose, M. Ubukata, I. Takahashi, K. Inoso and H. Osada, J. Antibiot., 1995, 48, 1382–1384 CrossRef CAS PubMed; (b) C.-B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 534–540 CrossRef CAS; (c) C.-B. Cui, H. Kakeya, G. Okada, R. Onose and H. Osada, J. Antibiot., 1996, 49, 527–533 CrossRef CAS PubMed; (d) H. Woehlecke, H. Osada, A. Herrmann and H. Lage, Int. J. Cancer, 2003, 107, 721–728 CrossRef CAS PubMed; (e) H. D. Jain, C.-C. Zhang, S. Zhou, H. Zhou, J. Ma, X.-X. Liu, X.-B. Liao, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, K. S. Smith, T. L. Macdonal, H. Kakeya, H. Osada and J. M. Cook, Bioorg. Med. Chem., 2008, 16, 4626–4651 CrossRef CAS PubMed.
- Synthesis of tryprostatin A: (a) T. Gan and J. M. Cook, Tetrahedron Lett., 1997, 38, 1301–1304 CrossRef CAS; (b) T. Gan, R. Liu, P. Yu, S. Zhao and J. M. Cook, J. Org. Chem., 1997, 62, 9298–9304 CrossRef CAS; (c) S. Zhao, T. Gan, P. Yu and J. M. Cook, Tetrahedron Lett., 1998, 39, 7009–7012 CrossRef CAS; (d) C. Ma, X. Liu, X. Li, J. Flippen-Anderson, S. Yu and J. M. Cook, J. Org. Chem., 2001, 66, 4525–4542 CrossRef CAS PubMed; (e) T. Yamakawa, E. Ideue, J. Shimokawa and T. Fukuyama, Angew. Chem., Int. Ed., 2010, 49, 9262–9265 CrossRef CAS; (f) T. Yamakawa, E. Ideue, Y. Iwaki, A. Sato, H. Tokuyama, J. Shimokawa and T. Fukuyama, Tetrahedron, 2011, 67, 6547–6560 CrossRef CAS.
- For examples of synthesis of tryprostatin B: (a) K. M. Depew, S. J. Danishefsky, N. Rosen and L. Sepp-Lorenzino, J. Am. Chem. Soc., 1996, 118, 12463–12464 CrossRef CAS; (b) J. M. Schkeryantz, J. C. G. Woo, P. Siliphaivanh, K. M. Depew and S. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11964–11975 CrossRef CAS; (c) B. Wang, L. Chen and K. Kim, Tetrahedron Lett., 2001, 42, 1463–1466 CrossRef CAS; (d) S. Zhao, K. S. Smith, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, T. L. Macdonald and J. M. Cook, J. Med. Chem., 2002, 45, 1559–1562 CrossRef CAS PubMed; (e) E. Caballero, C. Avendaño and J. C. Menéndez, J. Org. Chem., 2003, 68, 6944–6951 CrossRef CAS PubMed; (f) M. Huisman, M. Rahaman, S. Asad, S. Oehm, S. Novin, A. L. Rheingold and M. M. Hossain, Org. Lett., 2019, 21, 134–137 CrossRef CAS.
- W. Balk-Bindseil, E. Helmke, H. Weyland and H. Laatsch, Liebigs Ann. Chem., 1995, 7, 1291–1294 CrossRef.
- (a) T. Ueda, M. Inada, I. Okamoto, N. Morita and O. Tamura, Org. Lett., 2008, 10, 2043–2046 CrossRef CAS; (b) G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 971–974 CrossRef CAS; (c) J. Dugal-Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 4963–4967 CrossRef CAS PubMed; (d) Y. H. Liu, L. R. Zhang and Y. Jia, Tetrahedron Lett., 2012, 53, 684–687 CrossRef CAS; (e) T. Ueda, M. Inada, N. Morita and O. Tamura, Heterocycles, 2015, 90, 1179–1195 CrossRef CAS; (f) Y. Lan, Y. Zou, T. Huang, X. Wang, N. L. Brock, Z. Deng and S. Lin, Sci. China: Chem., 2016, 59, 1224–1228 CrossRef CAS; (g) Y. Zou, Q. Fang, H. Yin, Z. Liang, D. Kong, L. Bai, Z. Deng and S. Lin, Angew. Chem., Int. Ed., 2013, 52, 12951–12955 CrossRef CAS PubMed; (h) M. Herger, P. van Roye, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, J. Am. Chem. Soc., 2016, 138, 8388–8391 CrossRef CAS.
- (a) T. Huang, X. Wang, W. Guo and S. Lin, Tryptophan-Derived Microbial Alkaloids, in Comprehensive Natural Products III: Chemistry and Biology, ed. H.-W. Liu and T. P. Begley, 2020, vol. 2, pp. 393–445 Search PubMed; (b) L. M. Alkhalaf and K. S. Ryan, Chem. Biol., 2015, 22, 317–328 CrossRef CAS PubMed; (c) C. M. Santiveri and M. A. Jiménez, Biopolymers, 2010, 94, 779–790 CrossRef CAS PubMed.
- (a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed; (d) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (e) K. Chen and X. Lei, Current Opinion in Green and Sustainable Chemistry, 2018, 11, 9–14 CrossRef; (f) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS PubMed; (g) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767–15790 CrossRef CAS.
- (a) G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 CrossRef CAS PubMed; (b) J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352–3356 CrossRef CAS PubMed; (c) S.-L. Fang, M.-X. Jiang, S. Zhang, Y.-J. Wu and B.-F. Shi, Org. Lett., 2019, 21, 4609–4613 CrossRef CAS PubMed; (d) W.-Y. Qi, S.-L. Fang, X. Xu, K. Zhang and B.-F. Shi, Org. Chem. Front., 2021, 8, 1802–1807 RSC.
- K. Chen, S.-Q. Zhang, J.-W. Xu and B.-F. Shi, Chem. Commun., 2014, 50, 13924–13927 RSC.
- For selected reports on Pd(II)-catalysed AQ-directed C–H arylation of α-amino acids, see: (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS PubMed; (b) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (c) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3952 RSC. For reviews on modification of α-amino acids and peptides via C–H activation, see: (d) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed; (e) X. Lu, B. Xiao, R. Shang and L. Liu, Chin. Chem. Lett., 2016, 27, 305–311 CrossRef CAS; (f) W. Wang, M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS PubMed; (g) B.-B. Zhan, M.-X. Jiang and B.-F. Shi, Chem. Commun., 2020, 56, 13950–13958 RSC; (h) Z.-B. Bai and H. Wang, Synlett, 2020, 31, 199–204 CrossRef CAS; (i) H. Tong, B. Li, G. Li, G. He and G. Chen, CCS Chem., 2021, 3, 1797–1820 CrossRef CAS.
- (a) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS PubMed; (b) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906–3911 RSC.
- For the pioneering development of 8-aminoquinoline, see: V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- (a) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990–12993 CrossRef CAS PubMed; (b) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS PubMed; (c) H. K. Potukuchi and T. Bach, J. Org. Chem., 2013, 78, 12263–12267 CrossRef CAS PubMed.
- (a) L. Cao, Y. Hua, H.-G. Cheng and Q. Zhou, Org. Chem. Front., 2021, 8, 3883–3914 RSC; (b) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed; (c) N. Della Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef CAS PubMed; (d) J.-T. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed.
- S. Miltsov, L. Rivera, C. Encinas and J. Alonso, Tetrahedron Lett., 2003, 44, 2301–2303 CrossRef CAS.
- E. Fillion and A. M. Dumas, J. Org. Chem., 2008, 73, 2920–2923 CrossRef CAS PubMed.
- K. Depew, M. Samuel, J. Danishefsky, N. Rosen and L. S. Lorenzino, J. Am. Chem. Soc., 1996, 118, 12463–12464 CrossRef CAS.
- (a) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed; (b) K. M. Engle, Pure Appl. Chem., 2016, 88, 119–138 CAS.
- CCDC 2056344 (20) contains the ESI† crystallographic data for this paper.
- J. S. Yadav, B. V. Subba Reddy, C. Suresh Reddy and A. D. Krishna, Tetrahedron Lett., 2007, 48, 2029–2032 CrossRef CAS.
- Compounds 24–27 were all obtained in a 1:1 mixture of diastereomers and could be used directly for the total synthesis. The configuration at C3-position of indolones 24 and 25 was not determined since it would not be retained during the oxidation (25 → 27). The configuration of 27 was identified by finishing the total synthesis. See ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectral data, NMR data, and HPLC data. CCDC 2056344. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02343h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |