
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfated poly-amido-saccharides (sulPASs) are anticoagulants in vitro and in vivo†
Maria
Varghese
 a,
Rae S.
Rokosh
b,
Carolyn A.
Haller
b,
Stacy L.
Chin
a,
Jiaxuan
Chen
b,
Erbin
Dai
b,
Ruiqing
Xiao
a,
Elliot L.
Chaikof
*b and
Mark W.
Grinstaff
*a
a,
Rae S.
Rokosh
b,
Carolyn A.
Haller
b,
Stacy L.
Chin
a,
Jiaxuan
Chen
b,
Erbin
Dai
b,
Ruiqing
Xiao
a,
Elliot L.
Chaikof
*b and
Mark W.
Grinstaff
*a
aDepartments of Chemistry, Biomedical Engineering, and Medicine, Boston University, Boston, MA 02215, USA. E-mail: mgrin@bu.edu; mariavar@bu.edu; stacy.chin@hydroglydecoatings.com; xiaorq@mit.edu
bDepartment of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Wyss Institute of Biologically Inspired Engineering of Harvard University, Boston, MA, USA. E-mail: echaikof@bidmc.harvard.edu; rsrokosh@bidmc.harvard.edu; challer@bidmc.harvard.edu; jchen13@bidmc.harvard.edu; edai@bidmc.harvard.edu; mailto:echaikof@bidmc.harvard.edu
First published on 26th August 2021
Abstract
Anticoagulant therapeutics are a mainstay of modern surgery and of clotting disorder management such as venous thrombosis, yet performance and supply limitations exist for the most widely used agent – heparin. Herein we report the first synthesis, characterization, and performance of sulfated poly-amido-saccharides (sulPASs) as heparin mimetics. sulPASs inhibit the intrinsic pathway of coagulation, specifically FXa and FXIa, as revealed by ex vivo human plasma clotting assays and serine protease inhibition assays. sulPASs activity positively correlates with molecular weight and degree of sulfation. Importantly, sulPASs are not degraded by heparanases and are non-hemolytic. In addition, their activity is reversed by protamine sulfate, unlike small molecule anticoagulants. In an in vivo murine model, sulPASs extend clotting time in a dose dependent manner with bleeding risk comparable to heparin. These findings support continued development of synthetic anticoagulants to address the clinical risks and shortages associated with heparin.
Introduction
Anticoagulants control, prevent, and treat clotting disorders, and are routinely used in surgery for the treatment of thromboembolic disorders.1,2 Unfractionated heparin (UFH), a sulfated glycosaminoglycan extracted from porcine mucosa, is the most widely used anticoagulant.3,4 Today, there is an imminent risk of a global shortage of heparin due to the current epidemic of African Swine Fever.5,6 This virus has wiped out over one-quarter of the world's pig population.7 In 2008, a similar porcine ailment in China resulted in global shortages and in emergence of adulterated UFH and low molecular weight heparin (LMWH), leading to patient deaths.8,9Heparin is primarily used for thromboprophylaxis to prevent venous or arterial thrombus (i.e., clot) formation due to cancer, thrombophilia, or immobility.10 Prophylactic use of LMWH, prepared by chemical or enzymatic degradation of unfractionated heparin11 prevents the occurrence of deep vein thrombosis and pulmonary embolism,12 one of the most prevalent, but preventable, causes of death in hospitalized patients. Unlike UFH, which requires diligent monitoring due to its highly variable activity13 and patient-dependent dose-responses,11,14,15 LMWH displays less variability in its bioactivity, as well as higher bioavailability.16 In spite of its advantages and widespread use, LMWH, similar to UFH leads to heparin induced thrombocytopenia (HIT)17–19 in 1–5% of patients,20 and its anticoagulant effect is only partially neutralized using protamine sulfate.21 All told, the development of synthetic heparin alternatives is of keen interest and of clinical importance.
Fondaparinux is a synthetic penta-saccharide consisting of the minimal antithrombin binding region of heparin.22 While fondaparinux is not associated with HIT,23 unlike UFH or LMWH, it cannot be effectively reversed, which, along with its high plasma half-life of ≈19 hours is associated with an increased bleeding risk.24 The synthesis of fondaparinux is a multi-step (20+) procedure requiring protected monosaccharides and precise control of stereo orientation during the glycosylation reactions.22,25 A variety of sulfated polymers26 have been explored as heparin substitutes, including linear polymers of sulfated dextran27 and fucosylated chondroitin sulfate.28 Promising in vivo activity has been observed but the ability to rapidly and reliably reverse the anticoagulant activity of these polymers has not been reported. Completely synthetic mimetics are less well studied and include polymers comprised of a non-carbohydrate backbone with pendant sulfated sugars29–32 and branched sulfated polyglycerols.33 These sulfated polymers possess limited in vitro anticoagulant activity as compared to UFH and few are reversed by protamine sulfate. Moreover, in vivo studies are very rarely reported.
Given the biological activity of heparin arises from electrostatic and hydrogen bond interactions between the sulfate groups on a rigid carbohydrate backbone and antithrombin III,34,35 we envision that a synthetic polymer that structurally and functionally mimics heparin will provide insights into the design and applications of new polymeric anticoagulants, with the ultimate goal of a reversible, heparanase resistant agent with higher potency and greater selectivity of action to maximize therapeutic benefits while mitigating associated risks. Herein, we report the first synthesis and characterization of a heparin-mimetic, sulfated glucose-derived poly-amido-saccharide (sulPAS; Fig. 1). PASs are enantiopure polymers composed of sugar units connected by an α-1,2-amide peptide36 linkage as opposed to an ether linkage37 and possess a linear helical conformation.38–42 Further we evaluate the anticoagulant activity using in vitro serine protease inhibition assays, ex vivo human plasma clotting assays, and an in vivo murine bleeding model. sulPAS anticoagulant activity depends upon the degree of sulfation and chain length, and is reversed by treatment with protamine sulfate.
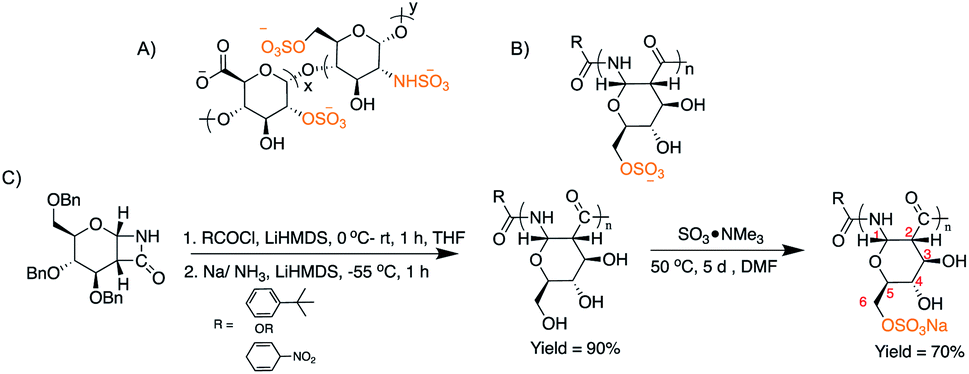 | ||
| Fig. 1 (A) Structure of heparin; (B) structure of sulPAS; (C) synthetic scheme of sulPAS. | ||
Results and discussion
Synthesis and chemical characterization of sulPASs
We synthesized a library of PAS derivatives by anionic ring opening polymerization (AROP) of a glucose derived β-lactam monomer38 with t-butyl benzoyl chloride as the initiator. Reduction of the benzyl-protected polymers followed by dialysis purification and lyophilization afforded the desired product (Fig. 1 and Table 1). Similarly, we prepared a galactose PAS (galPAS, DP = 12). Estimation of the molecular weight by 1H NMR spectroscopy for gluPAS-12 and galPAS-12 agreed with GPC data (Fig. S1 and S4†).| Sample | PAS | sulPAS | ||||
|---|---|---|---|---|---|---|
| M n (kDa) | Đ | M n (kDa) | Đ | DSb | IU mg−1c | |
| a GPC analyses were conducted in aqueous buffer versus dextran standards. b Elemental analyses were conducted to estimate DS. c IU mg−1 was calculated from APTT assay. | ||||||
| Glu-12 | 2.0 | 1.2 | 2.4 | 1.2 | 1.0 (LS) | 3.15 ± 0.5 |
| Glu-12 | 2.0 | 1.2 | 4.6 | 1.2 | 1.9 (HS) | 5.03 ± 1.1 |
| Glu-25 | 3.0 | 1.3 | 9.2 | 1.4 | 1.3 (HS) | 6.79 ± 3.0 |
| Glu-50 | 4.1 | 1.4 | 14.2 | 1.4 | 1.4 (HS) | 6.96 ± 1.8 |
| Gal-12 | 2.6 | 1.1 | 4.24 | 1.2 | 1.6 (HS) | 2.91 ± 0.7 |
We explored several sulfation strategies to synthesize the sulPAS post polymerization, including the use of different sulfation reagents, such as SO3·NMe3, SO3·DMF, SO3·EtOH, SO3·pyridine, pyridine/HClSO3; solvents such as DMF, pyridine or DMSO; reaction times ranging from 5 hours to 7 days; and temperatures from 25–70 °C. Based on the results of this screen, we dissolved PASs in anhydrous DMF and added 3 eq. or 5 eq. of SO3·NMe3 to the reaction mixture followed by stirring at 50 °C for 5 days (Fig. 1C),43 to access low sulfated PAS (sulPAS-LS) or high sulfated PAS (sulPAS-HS), respectively. We quenched the reaction with NaOH, filtered it through a 0.4 μm syringe filter, and removed the residual trimethyl amine by high-vacuum prior to neutralization with HCl, dialysis against distilled water for 24 hours and subsequent lyophilization. Following this procedure, we also prepared a galactose sulPAS (DP = 12) starting from galactose PAS (Table 1). sulPASs are water-soluble white powders with degrees of sulfation (DS; average number of sulfate groups per monomer; calculated from elemental analysis; Table 1) from 1 to 1.9. Symmetric (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2 and S–O–C stretching bands at ∼1200 cm−1 and in the range 1000–769 cm−1, respectively, are present in the IR spectrum of sulPAS indicative of O-sulfation (Fig. S15 and S16†). From the cross-peaks in the HSQCAD spectrum, the shift of methylene C6 position in glu-sulPAS (which are distinct in HSQCAD from methine C3, C4 and C5) as compared to gluPAS is consistent with C6 sulfation (Fig. S10–S14†). However, ascertaining the next position of sulfation in polymers with DS > 1 was not possible because of the difficulty of assigning multiple new cross-peaks in the spectra. The sulPAS displays a prominent peak shift to earlier retention times (Fig. S17†) in aqueous GPC characterization due to an increase in the molecular weight (MW) and anionic charges on the sulPAS structure in contrast to the neutral gluPAS samples.44 Circular dichroism (CD) spectra of glu-12-LS and glu-12-HS show a maxima at 186 nm and minima at 220 nm (Fig. S18†), consistent with the sulPASs adopting an α-helical secondary structure in aqueous environments similar to PASs.
O)2 and S–O–C stretching bands at ∼1200 cm−1 and in the range 1000–769 cm−1, respectively, are present in the IR spectrum of sulPAS indicative of O-sulfation (Fig. S15 and S16†). From the cross-peaks in the HSQCAD spectrum, the shift of methylene C6 position in glu-sulPAS (which are distinct in HSQCAD from methine C3, C4 and C5) as compared to gluPAS is consistent with C6 sulfation (Fig. S10–S14†). However, ascertaining the next position of sulfation in polymers with DS > 1 was not possible because of the difficulty of assigning multiple new cross-peaks in the spectra. The sulPAS displays a prominent peak shift to earlier retention times (Fig. S17†) in aqueous GPC characterization due to an increase in the molecular weight (MW) and anionic charges on the sulPAS structure in contrast to the neutral gluPAS samples.44 Circular dichroism (CD) spectra of glu-12-LS and glu-12-HS show a maxima at 186 nm and minima at 220 nm (Fig. S18†), consistent with the sulPASs adopting an α-helical secondary structure in aqueous environments similar to PASs.
In vitro anticoagulant activity of sulPASs
We first determined the activated partial thromboplastin time (APTT) and prothrombin time (PT) for sulPAS in human plasma as a measure of compound inhibition of the intrinsic and extrinsic pathways of the coagulation system, respectively (see Fig. S19† for the coagulation cascade).45 Varying concentrations of sulPAS, heparin, or fondaparinux were added to normal human plasma (NHP), and we measured the clotting times46 (Fig. 2A). Heparin (1 IU mL−1) increases both APTT and PT as compared to the HEPES buffered saline (HBS) control; effecting both intrinsic and extrinsic pathways (Fig. 2). In contrast, the sulPAS only increases APTT, but not PT (Fig. 2), indicating selective inhibition of the intrinsic pathway of coagulation. The graph is also plotted with polymer concentration expressed in IU mL−1 to aid in the analysis of activity (Fig. S20†). The pentasaccharide fondaparinux (1 μg mL−1) also selectively increases APTT and not PT. Importantly, the non-sulfated PAS controls does not affect APTT or PT (Fig. S21†). APTT increases with greater sulPAS chain length and degree of sulfation. Additionally, the longer clotting time for the glucose-based PAS, compared to the galactose-based PAS, suggests that the anticoagulant activity of sulPAS depends on the spatial orientation of the sulfate group. Finally, we estimated the anticoagulant activity of the polymers in IU mg−1 using a standard curve of APTT versus heparin concentration (Fig. S22†). | ||
| Fig. 2 (A) Schematics of ex vivo APTT and PT assay protocol in normal human plasma; (B) APTT increases with increase in concentration, chain length and degree of sulfation; (C) no statistically significant change in PT compared to HBS was observed with increase in concentration, chain length and concentration. Data represent mean ± SD, n = 3, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001 (post hoc Student's t-test). Concentration of heparin control used is 1 IU mL−1. Concentration of fondaparinux is 1 μg mL−1. There was no significant difference observed from HBS for all data points in (C) except the ones that are represented. | ||
To further investigate the mechanism of action, we assessed the antithrombin-III (AT-III) dependent inhibition of amidolytic activity of factor Xa (FXa) and thrombin in the presence of sulPAS (see ESI† for assay procedure). FXa and thrombin are downstream in the coagulation cascade and heparin (180 IU mg−1, MW = 15 kDa) inhibits these enzymes with an IC50 of 0.02 μM (Fig. S23†) and 0.04 μM (Fig. S24†), respectively. The sulPAS with greatest anticoagulant activity according to APTT, glu-50-HS (6.96 ± 1.8 IU mg−1) exhibits an IC50 of 64.7 μM for FXa (Fig. 3A), whereas thrombin inhibition by sulPAS shows an ascending limb, pseudo plateau and a descending limb, indicative of a template mechanism of inhibition47 with optimal concentration for maximal inhibition ∼30.3 μM (Fig. S24†). We also studied the inhibition of amidolytic activity of FXa and thrombin by glu-50-HS in the absence of AT-III (Fig. S25 and S26,† respectively). glu-50-HS does not inhibit FXa in the absence of AT-III, even at high concentrations (up to 3.57 mM, Fig. S25†), thus confirming the requirement of AT-III for FXa inhibition by sulPASs. On the other hand, thrombin inhibition exhibits an ascending limb, pseudo plateau and a descending limb, in the absence of AT-III (Fig. S26†), as is observed in the presence of AT-III. The percent maximum inhibition of thrombin by glu-50-HS is higher in the presence of AT-III (∼57% vs. ∼40% in the absence of AT-III). This result suggest glu-50-HS follows a template mechanism for inhibition of thrombin in the presence and absence of AT-III, with the presence of AT-III slightly improving the percent maximum inhibition.
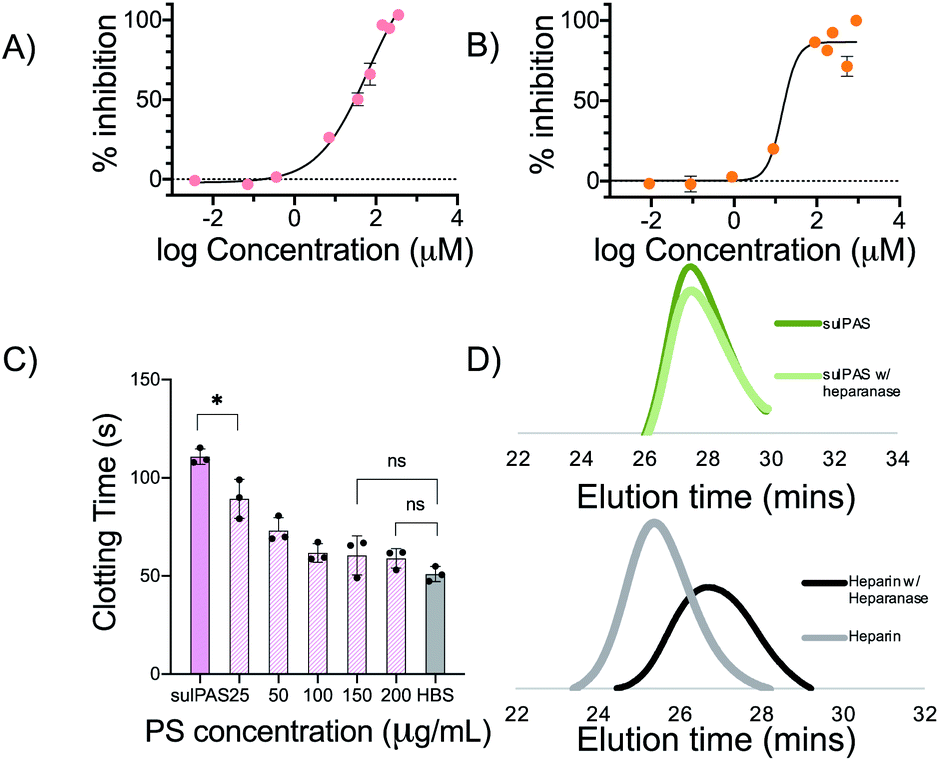 | ||
| Fig. 3 (A) glu-50-HS inhibits FXa with an IC50 of 64.7 μM. (B) glu-50-HS inhibits FXIa with an IC50 of 14.58 μM. (C) Statistically significant difference in APTT of 100 μg mL−1 glu-50-HS is observed with the addition of 25 μg mL−1 protamine sulfate (PS). No significant difference in APTT with respect to HBS was observed with 100 μg mL−1 PS. (D) Representative elution profile of unfractionated heparin and glu-25-HS in the presence and absence of heparanase. a, b and c data represent mean ± SD, n = 3, *P ≤ 0.05 (unpaired Student's t-test). | ||
These relatively high IC50 values of FXa and thrombin are inconsistent with the prolonged APTT, suggesting that other proteases in the intrinsic pathway are involved in the anticoagulant effect of sulPAS. Therefore, we examined the inhibitory effect of sulPAS on FXIa and FXIIa – two serine proteases in the intrinsic pathway, unaffected by heparin, identified as potential novel anticoagulant targets.48–50 glu-50-HS inhibits FXIa with an IC50 of 14.58 μM (Fig. 3B). This result is similar to the FXIa inhibition effect of dextran sulfate reported by Hack et al.51 The sulPAS does not inhibit FXIIa at lower concentrations and moderately activates the enzyme at high concentrations (Fig. S28†). We propose that the anticoagulant activity of sulPAS, observed in APTT assay, arises primarily from inhibition of FXIa, along with a modest inhibitory effect on FXa and thrombin.
Elevated plasma levels of heparanases, observed in patients with diabetes and renal insufficiency, may contribute to variable bioactivity.52 Hence, we evaluated the susceptibility of sulPAS to heparanase degradation. Heparanase degrades unfractionated heparin but not sulPAS as determined by size-exclusion chromatography SEC (Fig. 3D). Given the molecular weight of the sulPAS, we propose that the sulPAS polymer will be cleared by the kidneys, as the glomerular filtration cut-off is 50–60 kDa.53 However, this will need to be experimentally determined via a radiolabeled pharmacokinetics and biodistribution study.
One of the concerns pertaining to the use of LMWH and fondaparinux is the lack of reversal agents. Thus, we investigated the ability of protamine sulfate (PS), a cationic polypeptide that clinically reverses unfractionated heparin, to inhibit sulPAS' anticoagulant activity. We added varying concentrations (25–200 μg mL−1) of PS to a solution of NHP and 100 μg mL−1 of glu-50-HS and measured APTT. The prolonged clotting time of NHP with sulPAS shortens with the addition of 25 μg mL−1 PS and normalizes (i.e., not statistically significant from HBS control) with the addition of 150 μg mL−1 of PS (Fig. 3C). A similar assay using 1 IU mL−1 UFH shows a statistically significant decrease in APTT with 25 μg mL−1 of PS (Fig. S29†).54
We also evaluated the cytotoxicity of sulPAS in comparison with UFH, gluPAS, and dextran using a standard MTS colorimetric assay. NIH 3T3 fibroblast mammalian cells were incubated in the presence of these reagents at varying concentrations ranging from 1.0 to 0.05 mg mL−1 for 24 hours. All samples display minimal cytotoxicity with results comparable to media control, particularly at low concentrations. However, sulPASs are slightly more cytotoxic at high concentration of 1.0 mg mL−1 (Fig. S30†).
We subsequently evaluated the hemolytic activity of sulPAS by incubating the polymers with human red blood cells (RBCs) at varying concentrations (0.125–1 mg mL−1) of glu-50-HS and unfractionated heparin at 37 °C for 24 hours. Heparin and glu-50-HS show only a minimal hemolytic effect (Fig. S31†).
In vivo anticoagulant activity of sulPASs
Given these in vitro results, we evaluated the performance of glu-50-HS in vivo. Intravenous administration of glu-50-HS at doses ranging from 50 μg to 200 μg in a mouse affords a dose dependent increase in APTT (Fig. 4A), consistent with the ex vivo dose-dependent anticoagulant effect of glu-50-HS. We next determined the effect of glu-50-HS on hemostasis in a mouse tail vein transection bleeding time assay. glu-50-HS (200 μg) and low dose heparin (1 IU) did not significantly prolong bleeding time in comparison to saline vehicle (Fig. 4B). These results suggest that sulPAS can be dosed to achieve a clinically relevant increase in APTT with comparable bleeding risk to heparin.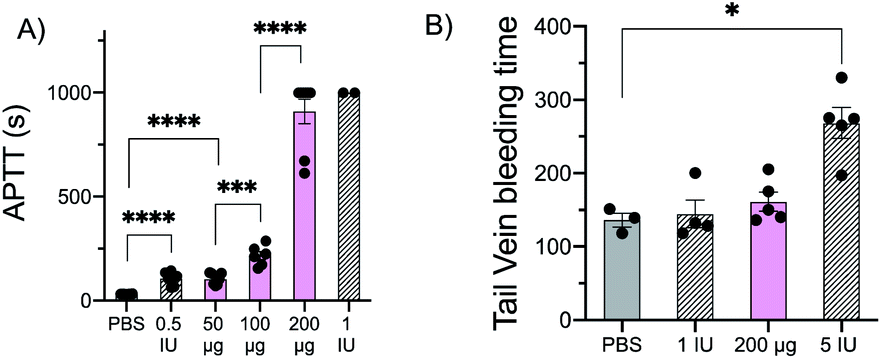 | ||
| Fig. 4 (A) PBS, heparin (0.5 IU, 1.0 IU), or glu-50-HS (50–200 μg) were administered (IV) to C57BL/6 mice 3 minutes prior to blood draw, and plasma clotting time was measured on a microcoagulometer. glu-50-HS demonstrated a dose dependent increase in APTT. (B) Tail vein transection was performed 3 min post IV administration; significant prolongation of bleeding time was only observed after administration of positive control (5 IU heparin). Data represent mean ± SEM, n = 3–6 mice per agent, *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001 (ANOVA and post hoc Student's t-test). | ||
Conclusions
In summary we describe the synthesis of heparin mimicking sulPASs. sulPAS anticoagulant activity positively correlates with molecular weight and degree of sulfation. Mechanistically, sulPASs inhibit the amidolytic activity of FXa and FXIa, with minimal effect on thrombin and FXIIa. sulPAS are not susceptible to heparanase degradation and their anticoagulant activity is reversed by protamine sulfate. When administered in vivo, sulPASs demonstrate a dose dependent increase in clotting time. The current limitations of animal derived heparin warrant the development of novel heparin analogs to include bioengineered analogs,55,56 small molecule agents,22 and polymer anticoagulant therapeutics.Experimental
Synthetic procedure
gluPAS (n = 12, 25 and 50) and galPAS (n = 12) were prepared as previously described.39,57Biological assays
A standard curve was plotted by estimating the clotting time of NHP in the presence of varying concentration of heparin (0.2–2.5 IU mL−1) and fitted to exponential growth model. The anticoagulant activity of each sulPAS concentrations in IU was calculated using the equation of the curve. By dividing the activity thus obtained with the concentration used, the activity in IU mg−1 was calculated (Table S2†).
where Ai is the slope of absorbance at 405 nm vs. time for varying concentration of analyte, A0 is the slope in the absence of any inhibitor (positive control), Ab is the slope when FXIa was completely inhibited by adding 10 μL of 20% acetic acid before addition of the substrate (negative control).

Data availability
The raw data required to reproduce these findings are available to upon request from the authors. The processed data required to reproduce these findings are available to download from the ESI.†Author contributions
M. V. wrote the manuscript, performed all synthetic procedures, performed chemical characterization, was involved in data collection for Fig. 2, 3, S1–S29, S31 and Tables 1, S1–S4† and prepared all figures. S. L. C. with input from R. X. performed data collection for Fig. S30.† C. H., R. R., J. C. and E. D., performed data collection for Fig. 4 and R. R. helped edit the manuscript. E. C. and M. W. G. provided research direction, mentorship, and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Hannah Caputo and Dr Anant S. Balijepalli for discussions in synthesis and Dr C. James McKnight of the BU School of Medicine for help in obtaining HSQCAD spectra. This work was supported in part by Boston University, DoD Uniformed Services University (HU0001810012), NIH 5T32HL007734 fellowship funding to Rae Rokosh, and Beth Israel Deaconess Medical Center.Notes and references
- M. B. Gorbet and M. V. Sefton, Biomaterials, 2004, 25, 5681–5703 CrossRef CAS PubMed.
- W. Geerts and R. Selby, Chest, 2003, 124, 357S–363S CrossRef PubMed.
- H. Chu, N. R. Johnson, N. S. Mason and Y. Wang, J. Controlled Release, 2011, 150, 157–163 CrossRef CAS PubMed.
- M. F. Maitz, U. Freudenberg, M. V. Tsurkan, M. Fischer, T. Beyrich and C. Werner, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- E. Vilanova, A. M. F. Tovar and P. A. S. Mourão, J. Thromb. Haemostasis, 2019, 17, 254–256 CrossRef PubMed.
- J. Fareed, W. Jeske and E. Ramacciotti, Clin. Appl. Thromb./Hemostasis, 2019, 25, 1–3 Search PubMed.
- J. Gale, Deadly Pig Disease Sparks Fear of Heart Drug Shortage: QuickTake – Bloomberg, https://www.bloomberg.com/news/articles/2019-11-11/deadly-pig-disease-sparks-fear-of-heart-drug-shortage-quicktake, accessed 19 February, 2020 Search PubMed.
- H. Liu, Z. Zhang and R. J. Linhardt, Nat. Prod. Rep., 2009, 26, 313–321 RSC.
- K. Hedlund, D. Coyne, D. Sanford and J. Huddelson, Perfusion, 2013, 28, 61–65 CrossRef CAS PubMed.
- R. L. Bick, E. P. Frenkel, J. Walenga, J. Fareed and D. A. Hoppensteadt, Hematology/Oncology Clinics of North America, 2005, 19, 1–51 CrossRef PubMed.
- R. D. Rosenberg and L. Lam, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1218–1222 CrossRef CAS PubMed.
- C. Kearon, Chest, 2003, 124, 386S–392S CrossRef PubMed.
- H. Bussey, J. L. Francis and T. H. Consensus Group, Pharmacotherapy, 2004, 24, 103S–107S CrossRef CAS PubMed.
- C. Kroon, W. R. ten Hove, A. de Boer, J. M. Kroon, J. M. van der Pol, E. J. Harthoorn-Lasthuizen, H. C. Schoemaker, F. J. van der Meer and A. F. Cohen, Circulation, 1992, 86, 1370–1375 CrossRef CAS PubMed.
- L. Manson, J. I. Weitz, T. J. Podor, J. Hirsh and E. Young, J. Lab. Clin. Med., 1997, 130, 649–655 CrossRef CAS.
- J. Dawes, Acta Chir. Scand., 1990, 68–74 CAS.
- T. E. Warkentin, M. N. Levine, J. Hirsh, P. Horsewood, M. K. Gent and J. G. Kelton, N. Engl. J. Med., 1995, 332, 1330–1335 CrossRef CAS PubMed.
- M. Hussain, H. P. Wendel and F. K. Gehring, International Journal of Blood Disorders & Diseases, 2017, 1, 7–9 Search PubMed.
- T. E. Warkentin, J. A. Sheppard, P. Horsewood, P. J. Simpson, J. C. Moore and J. G. Kelton, Blood, 2000, 96, 1703–1708 CrossRef CAS PubMed.
- M. Alquwaizani, L. Buckley, C. Adams and J. Fanikos, Curr. Emerg. Hosp. Med. Rep., 2013, 1, 83–97 CrossRef PubMed.
- M. Wolzt, A. Weltermann, M. Nieszpaurlos, B. Schneider, A. Fassslt, K. Lechner, H. G. Eichler and P. A. Kyrle, Thromb. Haemostasis, 1995, 73, 439–443 CrossRef CAS.
- K. A. Bauer, D. W. Hawkins, P. C. Peters, M. Petitou, J. M. Herbert, A. A. Van Boeckel and D. G. Meuleman, Cardiovasc. Drug Rev., 2002, 20, 37–52 CrossRef CAS PubMed.
- T. E. Warkentin, Expert Rev. Hematol., 2010, 3, 567–581 CrossRef CAS PubMed.
- A. Greinacher, T. Thiele and K. Selleng, Thromb. Haemostasis, 2015, 113, 931–942 CrossRef PubMed.
- C. H. Chang, L. S. Lico, T. Y. Huang, S. Y. Lin, C. L. Chang, S. D. Arco and S. C. Hung, Angew. Chem., Int. Ed., 2014, 53, 9876–9879 CrossRef CAS PubMed.
- S. J. Paluck, T. H. Nguyen and H. D. Maynard, Biomacromolecules, 2016, 17, 3417–3440 CrossRef CAS PubMed.
- S. Charef, E. Petit, D. Barritault, J. Courty and J.-P. Caruelle, J. Biomed. Mater. Res., Part A, 2007, 83, 1024–1031 CrossRef PubMed.
- R. J. C. Fonseca and P. A. S. Mourão, Thromb. Haemostasis, 2006, 96, 822–829 CrossRef CAS.
- M. Akashi, N. Sakamoto, K. Suzuki and A. Kishida, Bioconjugate Chem., 1996, 7, 393–395 CrossRef CAS PubMed.
- Y. I. Oh, G. J. Sheng, S. K. Chang and L. C. Hsieh-Wilson, Angew. Chem., Int. Ed., 2013, 52, 11796–11799 CrossRef CAS PubMed.
- D. Grande, S. Baskaran, C. Baskaran, Y. Gnanou and E. L. Chaikof, Macromolecules, 2000, 33, 1123–1125 CrossRef CAS.
- Y. Huang, L. Taylor, X. Chen and N. Ayres, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5230–5238 CrossRef CAS.
- H. Türk, R. Haag and S. Alban, Bioconjugate Chem., 2004, 15, 162–167 CrossRef PubMed.
- M. Lima, T. Rudd and E. Yates, Molecules, 2017, 22, 1–11 CrossRef PubMed.
- J. E. Turnbull and J. T. Gallagher, Biochem. J., 1991, 273(Pt 3), 553–559 CrossRef CAS PubMed.
- M. Metzke, N. O'Connor, S. Maiti, E. Nelson and Z. Guan, Angew. Chem., Int. Ed., 2005, 44, 6529–6533 CrossRef CAS PubMed.
- L. Li, E. A. Franckowiak, Y. Xu, E. McClain and W. Du, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3693–3699 CrossRef CAS.
- E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- E. L. Dane, S. L. Chin and M. W. Grinstaff, ACS Macro Lett., 2013, 2, 887–890 CrossRef CAS PubMed.
- S. E. Stidham, S. L. Chin, E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2014, 136, 9544–9547 CrossRef CAS PubMed.
- S. L. Chin, Q. Lu, E. L. Dane, L. Dominguez, C. J. McKnight, J. E. Straub and M. W. Grinstaff, J. Am. Chem. Soc., 2016, 138, 6532–6540 CrossRef CAS PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 391–412 CrossRef PubMed.
- Y. Suhara, M. Ichikawa, J. E. K. Hildreth and Y. Ichikawa, Tetrahedron Lett., 1996, 37, 2549–2552 CrossRef CAS.
- E. S. P. Bouvier and S. M. Koza, TrAC, Trends Anal. Chem., 2014, 63, 85–94 CrossRef CAS.
- V. Kumar, A. K. Abbas and J. C. Aster, in Robbins Basic Pathology, 2013 Search PubMed.
- D. Tilley, I. Levit and J. A. Samis, J. Visualized Exp., 2012, 1–7 Search PubMed.
- G. M. Oosta, W. T. Gardner, D. L. Beeler and R. D. Rosenberg, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 829–833 CrossRef CAS PubMed.
- W. A. Schumacher, J. M. Luettgen, M. L. Quan and D. A. Seiffert, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 388–392 CrossRef CAS PubMed.
- M. L. Quan, D. J. P. Pinto, J. M. Smallheer, W. R. Ewing, K. A. Rossi, J. M. Luettgen, D. A. Seiffert and R. R. Wexler, J. Med. Chem., 2018, 61, 7425–7447 CrossRef CAS PubMed.
- M. Larsson, V. Rayzman, M. W. Nolte, K. F. Nickel, J. Björkqvist, A. Jämsä, M. P. Hardy, M. Fries, S. Schmidbauer, P. Hedenqvist, M. Broomé, I. Pragst, G. Dickneite, M. J. Wilson, A. D. Nash, C. Panousis and T. Renné, Sci. Transl. Med., 2014, 6, 1–11 Search PubMed.
- W. A. Wuillemin, E. Eldering, F. Citarella, C. P. De Ruig, H. Ten Cate and C. Erik Hack, J. Biol. Chem., 1996, 271, 12913–12918 CrossRef CAS PubMed.
- N. J. Nasser, G. Sarig, B. Brenner, E. Nevo, O. Goldshmidt, E. Zcharia, J. P. Li and I. Vlodavsky, J. Thromb. Haemostasis, 2006, 4, 560–565 CrossRef CAS PubMed.
- G. Arturson and G. Wallenius, Scand. J. Clin. Lab. Invest., 1964, 16, 81–86 CrossRef CAS PubMed.
- T. F. Kresowik, T. W. Wakefield, R. D. Fessler 2nd and J. C. Stanley, J. Surg. Res., 1988, 45, 8–14 CrossRef CAS.
- M. Suflita, L. Fu, W. He, M. Koffas and R. J. Linhardt, Appl. Microbiol. Biotechnol., 2015, 99, 7465–7479 CrossRef CAS PubMed.
- J. A. Englaender, Y. Zhu, A. N. Shirke, L. Lin, X. Liu, F. Zhang, R. A. Gross, M. A. G. Koffas and R. J. Linhardt, Appl. Microbiol. Biotechnol., 2017, 101, 2843–2851 CrossRef CAS PubMed.
- E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- D. Hanjaya-Putra, C. Haller, X. Wang, E. Dai, B. Lim, L. Liu, P. Jaminet, J. Yao, A. Searle, T. Bonnard, C. E. Hagemeyer, K. Peter and E. L. Chaikof, JCI Insight, 2018, 3, 1–18 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02302k |
| This journal is © The Royal Society of Chemistry 2021 |