
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free arylation of sulfonamides via visible light-mediated deamination†
Yong
Luo
 *a,
Hao
Ding
b,
Jing-Song
Zhen
a,
Xian
Du
a,
Xiao-Hong
Xu
a,
Han
Yuan
a,
Yi-Hui
Li
a,
Wan-Ying
Qi
a,
Bing-Zhe
Liu
a,
Shi-Man
Lu
a,
Can
Xue
*c and
Qiuping
Ding
*b
*a,
Hao
Ding
b,
Jing-Song
Zhen
a,
Xian
Du
a,
Xiao-Hong
Xu
a,
Han
Yuan
a,
Yi-Hui
Li
a,
Wan-Ying
Qi
a,
Bing-Zhe
Liu
a,
Shi-Man
Lu
a,
Can
Xue
*c and
Qiuping
Ding
*b
aSchool of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Guangzhou, 510275, China. E-mail: luoyong5@mail.sysu.edu.cn
bKey Laboratory of Functional Small Organic Molecules, Ministry of Education, Jiangxi Normal University, Nanchang, 330022, China. E-mail: dingqiuping@jxnu.edu.cn
cSchool of Chemical Engineering and Technology, Sun Yat-sen University, Zhuhai, 519082, China. E-mail: xuecan@mail.sysu.edu.cn
First published on 5th June 2021
Abstract
A novel arylation of sulfonamides with boronic acids to afford numerous diaryl sulfones via a visible light-mediated N–S bond cleavage other than the typical transition-metal-catalyzed C(O)–N bond activation is described. This methodology, which represents the first catalyst-free protocol for the sulfonylation of boronic acids, is characterized by its simple reaction conditions, good functional group tolerance and high efficiency. Several successful examples for the late-stage functionalization of diverse sulfonamides indicate the high potential utility of this method in pharmaceutical science and organic synthesis.
Introduction
Sulfonamide drugs discovered in the 1930s first systemically used as antibacterials have continuously received renewed interest for the treatment of numerous diseases.1 Since then, a large number of synthetic routes towards diverse sulfonamides have been developed, which make the sulfonamide moiety prevalent in bioactive molecules and commercial chemicals.2 Therefore, the transformation of sulfonamide skeleton to other groups, such as sulfones, is a convenient method to construct a pharmacophore-containing molecule library for drug discovery. Moreover, the activation of sulfonamides could be used for the deprotection of sulfonyl group-protected amines,3 late-stage functionalization of sulfonamide drugs,4 and sulfonylation with sulfonamides as stable and good functional group-tolerant reagents. However, sulfonamides are usually considered as the final products of the construction of N–S bonds rather than reactive substrates because efficient strategies for the functionalization of N–S bonds are limited.Previous exploration on the N–S bond cleavage of sulfonamides mainly focused on desulfonylation to generate corresponding amines.3 The functionalization of the sulfonyl group resulted from the deamination of sulfonamides still meets great challenges. Recently, Fier and Maloney4 reported an efficient pathway to convert primary and secondary sulfonamides to sulfinate anions in the presence of an NHC catalyst or phosphine reagent (Scheme 1a). Further with the addition of electrophiles, a variety of sulfones and modified sulfonamides could be obtained. By utilizing Pyry-BF4, Cornella5 found that primary sulfonamides could react with nucleophiles to generate sulfonyl chlorides, fluorides, and sulfonic acids. Despite these significant progresses, the functionalization of tertiary sulfonamides, which are usually considered as terminal functional groups to synthesize sulfones and related compounds, has not been achieved yet.
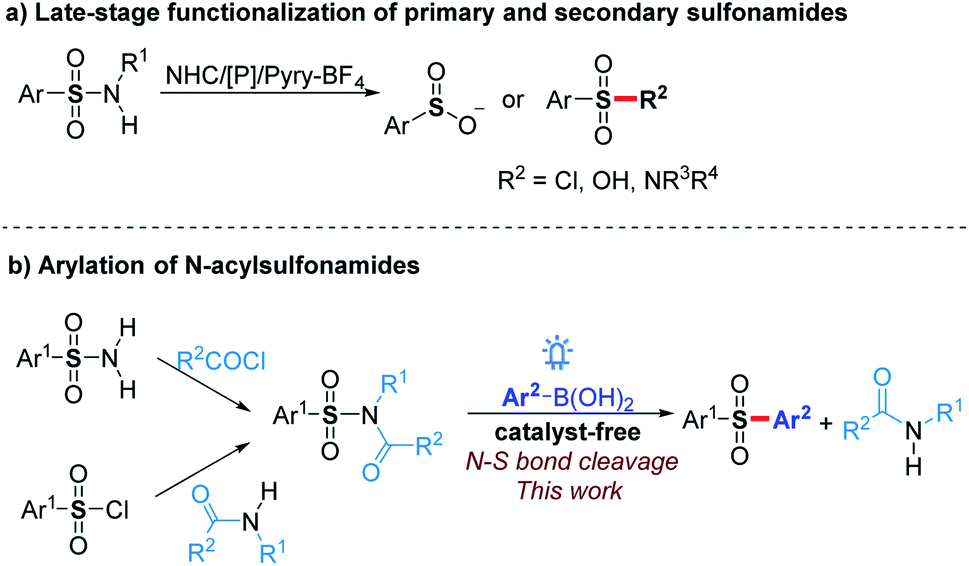 | ||
| Scheme 1 Strategies for the functionalization of sulfonamides and the synthesis of sulfones. | ||
On the other hand, despite sulfonyl chlorides,6 sodium sulfinates,7 and sulfur dioxide8 being widely used for sulfonylation towards aryl sulfones in recent years, novel sulfonylation reagents still need to be explored to extend the research in this area. The stability and good tolerance for other functional groups make tertiary sulfonamides to be good sulfonylation reagents. Herein, we report that N-acylsulfonamides as novel sulfonyl radical precursors react with aryl boronic acids to generate various diaryl sulfones via visible-light-mediated N–S bond activation without any catalyst (Scheme 1b), while these substrates typically proceed a C(O)–N bond cleavage in the presence of transition metals.9 The mechanism investigation showed that a radical cross-coupling is probably involved in our protocol, which provides an alternative strategy for the late-stage functionalization of sulfonamides. Moreover, N-acylsulfonamides synthesized from sulfonyl chlorides and amides could act as the surrogates of the sulfonyl chlorides under incompatible reaction conditions to proceed with late-stage sulfonylation.
Results and discussion
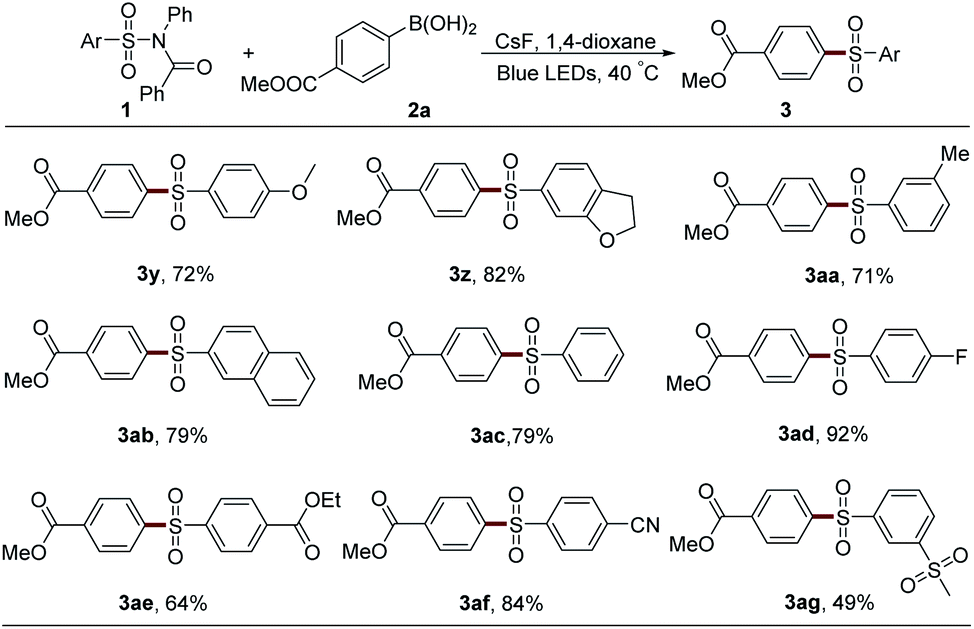
In our investigation, sulfonamide 1a went through arylation with boronic acid 2a in the presence of K3PO4 and CH3CN under blue LED irradiation, providing diaryl sulfone 3a with 57% yield (Table 1, entry 1). No ketone product was observed. Further screening showed that DMF, PhCF3, or DCE was inefficient (entries 2–4). However, ether solvents exhibited superiority in this reaction (entries 5–7), and 70% yield was obtained with 1,4-dioxane. A lower yield was detected when Cs2CO3 or K2CO3 was employed (entries 8 and 9). CsF (99.99% metal basis) gave the best result in the screening of the bases, affording the desired product 3a in 75% yield (entry 10). The decomposition of sulfonamide 1a was observed in these reactions and hampered the elevation of the product yield. Therefore, the ratio of 1a and 2a was changed to 2 to 1, resulting in a significant increase of the yield of 3a (95%, entry 11). The corresponding amide 4 was also collected and identified with a yield of 114% based on 2a owing to the visible light-mediated desulfonylation of the starting material 1a. Visible light and base were proved to be necessary since no product was detected when this reaction was performed in dark or without base (entries 12 and 13).|
|
|||
|---|---|---|---|
| Entry | Solvent | Base | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), base (0.5 mmol), solvent (2 mL), 50 W blue LED, 12 h, 40 °C, isolated yield. b 1a (0.4 mmol), 2a (0.2 mmol). c Yield of 4 in the parenthesis. d In dark. | |||
| 1 | CH3CN | K3PO4 | 57 |
| 2 | DMF | K3PO4 | Trace |
| 3 | PhCF3 | K3PO4 | n.r. |
| 4 | DCE | K3PO4 | n.r. |
| 5 | THF | K3PO4 | 66 |
| 6 | DME | K3PO4 | 61 |
| 7 | 1,4-Dioxane | K3PO4 | 70 |
| 8 | 1,4-Dioxane | Cs2CO3 | 53 |
| 9 | 1,4-Dioxane | K2CO3 | 57 |
| 10 | 1,4-Dioxane | CsF | 75 |
| 11b | 1,4-Dioxane | CsF | 95 (114c) |
| 12b,d | 1,4-Dioxane | CsF | n.r. |
| 13b | 1,4-Dioxane | — | n.r. |
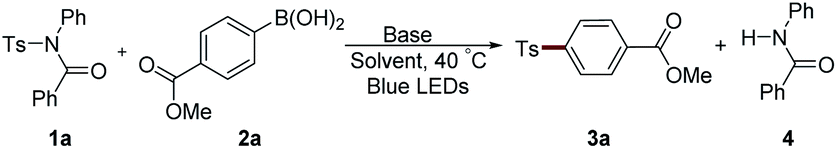
The scope of boronic acids was then explored (Table 2). Substituents with a large π system showed high efficiency. For example, 2- or 1-naphthylboronic acid provided 97% and 87% yields of the desired product, respectively (3b and 3c). Methyl group substitution had nearly no affect on this reaction (3e). Methoxy and phenyl group-attached substrates gave lower yields probably owing to the instability of these sulfonamides under blue LED irradiation (3d and 3f). It should be noted that in some cases, product 3 was inseparable from the generated amide 4. Therefore, 1b, which showed no obvious reactivity difference compared with 1a, was employed for the convenience of the isolation of product 3 (3b, 3c, 3e and 3f). Heterocycles, such as quinoline and benzofuran, are compatible in this system (3g–3k). Aldehyde, ketone, and amide group-attached aryl boronic acids also could afford moderate yields of the desired products (3l–3o), although an elevated concentration by the addition of only 1 mL solvent was necessary in several examples. Diverse electron-withdrawing (phenyl, cyano, and silane) and electron-donating group (methoxy, methyl, and amine) substituted diaryl sulfones could be synthesized using corresponding boronic acids (3p–3v). The substrate bearing an ortho methyl group could afford 3x in 68% yield. However, aliphatic boronic acids, aryl trifluoroborates, or aryl boronic acid pinacol esters could not result in any product under these reaction conditions or with a photocatalyst.
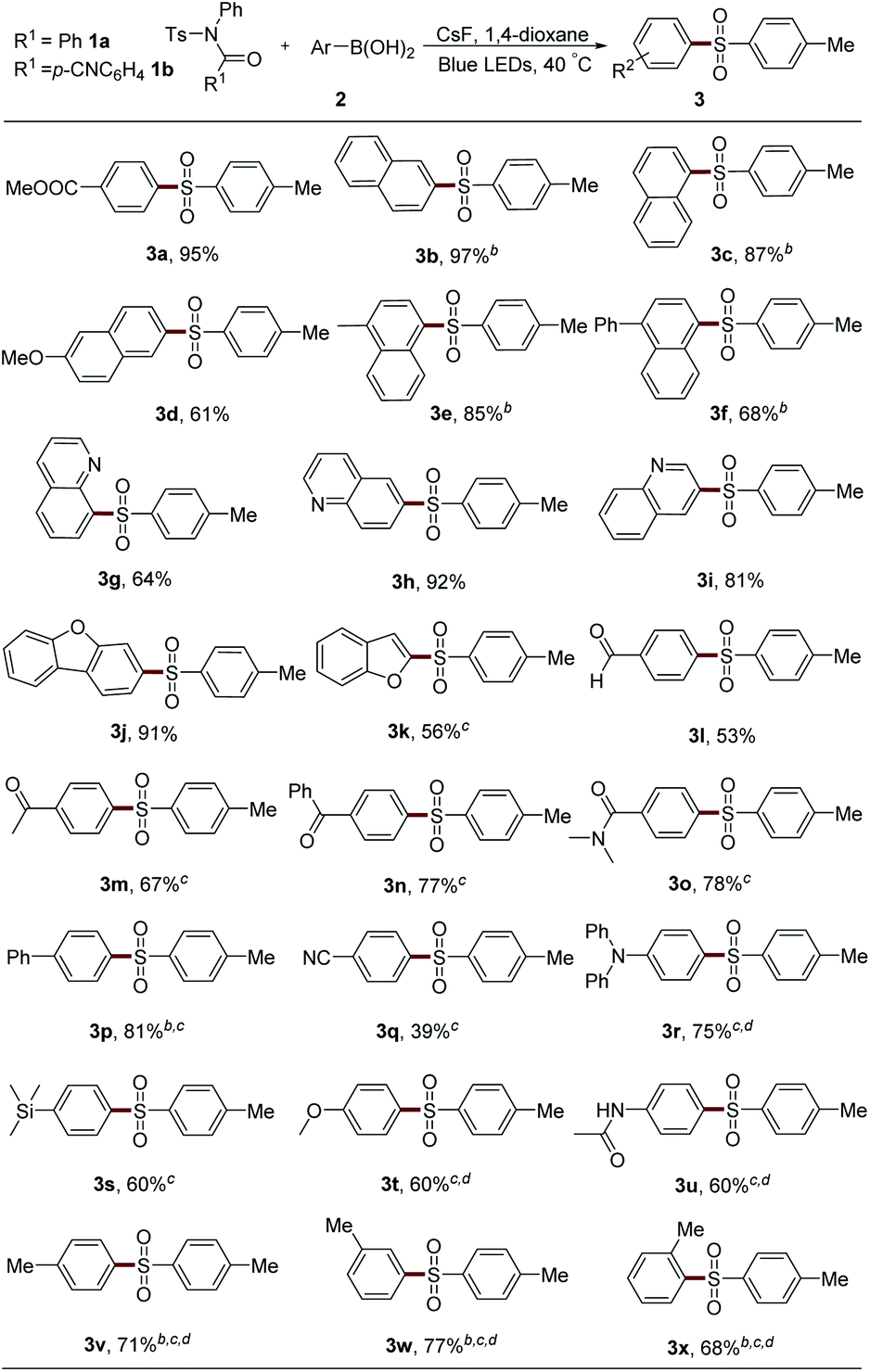
After that, the scope of sulfonyl groups was investigated (Table 3). With an electron-donating group on either para- or meta-position, sulfonamide 1 could react smoothly with boronic acid 2a to generate the desired product in good yield (3y–3aa). The arylation of naphthyl and phenyl sulfonamides also proceeded efficiently to afford sulfones 3ab and 3ac. Electron-withdrawing groups, such as fluorine, ester, cyano, and sulfone, could all be tolerated in this system very well (3ad–3ag). However, the alkyl sulfonyl group-embedded sulfonamide showed no reactivity.
| a Reaction conditions: 1 (0.4 mmol), 2a (0.2 mmol), CsF (0.5 mmol), 1,4-dioxane (2 mL), 50 W blue LED, 12 h, 40 °C. |
|---|
|
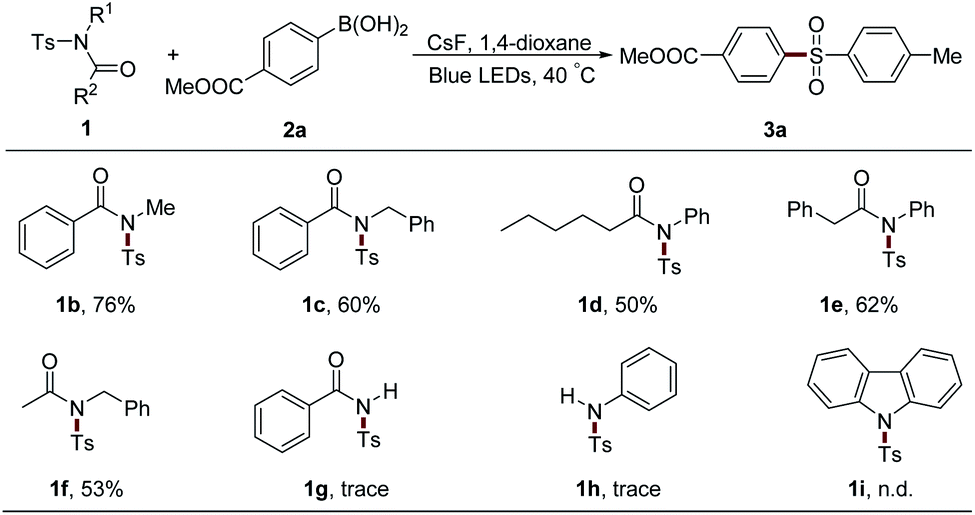
The protecting groups on the nitrogen atom were also examined (Table 4). Alkyl group-attached sulfonamides 1b and 1c exhibited an inferior efficiency than 1a, leading to 76% and 60% yields of 3a, respectively. Aliphatic acyl group-protected sulfonamides 1d and 1e could provide the desired product 3a with moderate yields. Sulfonamide 1f bearing acetyl and benzyl units, which were usually used as the protecting groups of nitrogen atom, could also result in 3a with 53% yield, which largely expands the practical utilization of our protocol in organic synthesis. However, secondary sulfonamides 1g and 1h only afforded trace amounts of 3a. Moreover, the acyl group in 1 was proved to be necessary since the two aryl group-attached substrate 1i gave no desired product under the standard conditions.
| a Reaction conditions: 1 (0.4 mmol), 2a (0.2 mmol), CsF (0.5 mmol), 1,4-dioxane (2 mL), 50 W blue LED, 12 h, 40 °C. n.d. = not detected. |
|---|
|
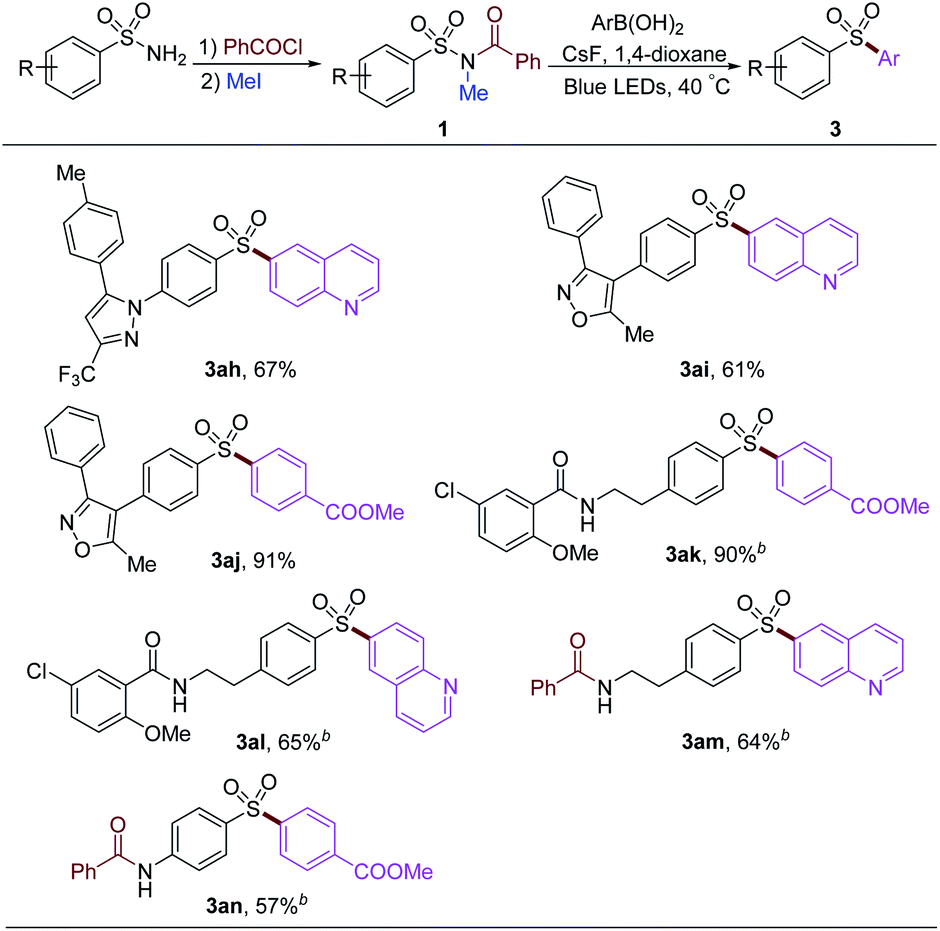
To examine our strategy in the late-stage functionalization of sulfonamides, several commercial drugs were used to proceed acylation and methylation to provide the starting material 1. After that, diverse aryl groups and heterocycles could be introduced under the standard arylation reaction conditions (Table 5). The 6-quinoline group could be installed into the drugs derived from Celecoxib and Valdecoxib with 67% and 61% yields, respectively (3ah and 3ai). 4-Methoxycarbonylphenylboronic acid could give 91% yield of the desired product (3aj). The amide group remained intact in this procedure, affording corresponding products 3ak and 3al stemmed from glibenclamide precursor in 90% and 65% yields, respectively. Free alkyl and aryl amine groups were protected by acylation under the prefunctionalization conditions, affording the products 3am and 3an in moderate yields.
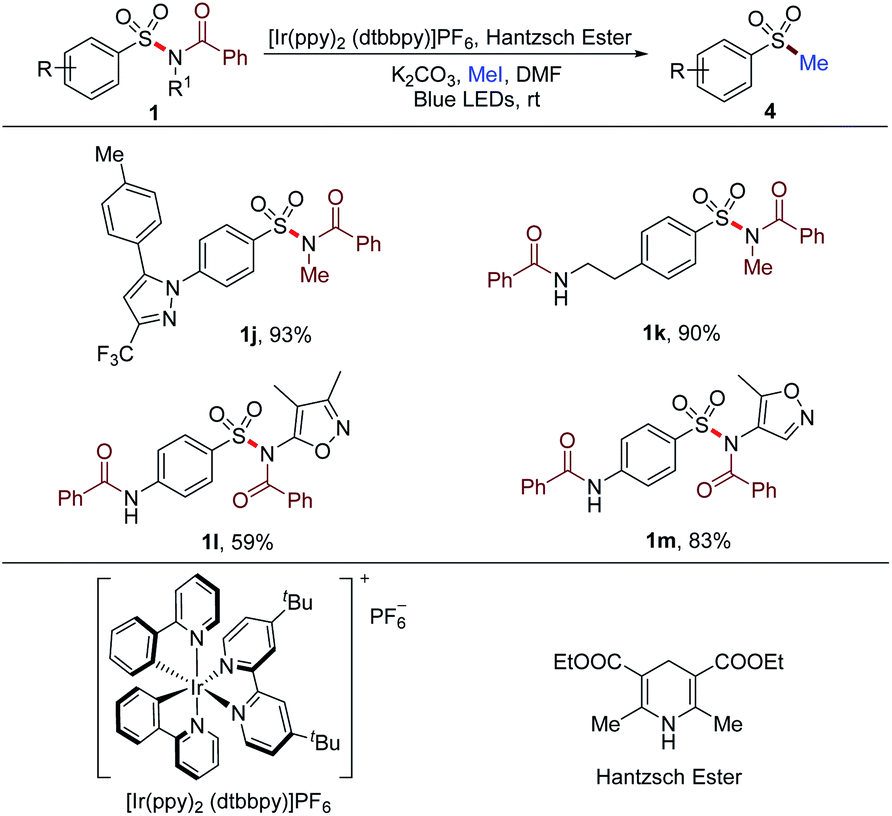
Besides the arylation of the sulfonyl group in sulfonamides, alkylation could also be achieved by altering the reaction conditions. The N–S bond of sulfonamides was cleaved by photocatalysis to deliver the sulfonyl radical. Then, the in situ methylation of the sulfinate resulted from the reduction of the sulfonyl radical proceeded to afford methyl sulfones (Table 6). Substrates 1j and 1k synthesized from primary sulfonamides furnished the methylated products with 93% and 90% yields, respectively. Secondary sulfonamide-based substrates 1l and 1m could also be prefunctionalized and methylated with this method. These successful transformations implied the potential for the rapid construction of a molecule library from not only tertiary but also primary and secondary sulfonamides for the screening of new lead compounds.
| a Reaction conditions: 1 (0.2 mmol), Hantzsch ester (0.4 mmol), [Ir(ppy)2 (dtbbpy)]PF6 (5 mol%), K2CO3 (1.0 mmol), MeI (1.0 mmol), DMF (2 mL), 50 W blue LED, 15 h, 40 °C. |
|---|
|
The mechanism of this reaction was investigated by measuring the UV-Vis absorption spectra of substrates and reaction solutions (Scheme 2). Individual solutions of sulfonamide 1a and boronic acid 2a in 1,4-dioxide showed a maximum absorption at approx. 250 nm, and a very low absorption at 460 nm (blue LED). A mixture of 1a and 2a displayed a slight red-shift of the absorption peak. When CsF was added, a further red-shift and higher absorption under a blue LED were observed. These details suggested that complex A, which was formed with 1a, 2a, and base could be directly photoexcited by blue LED without the presence of a photocatalyst (Scheme 2b). The Cs+-coordinated six-membered ring in complex A might also explain the indispensability of the acyl group (Table 4) in the substrate. This phenomenon of the combination of a substrate and a reagent affording a photo-excitable complex in situ has been reported in several studies.10 With blue LED irradiation, complex A gave a rise to the amide radical B and the sulfonyl radical C.3 After that, the attack of a nitrogen radical to the boron complex D resulted in an aryl radical E (ref. 11) with the concomitant generation of amide 4. The capture of the sulfonyl radical by E provided the product sulfone 3. This reaction was inhibited by the addition of the radical scavenger TEMPO (Scheme 2b), also indicating the involvement of a radical process.
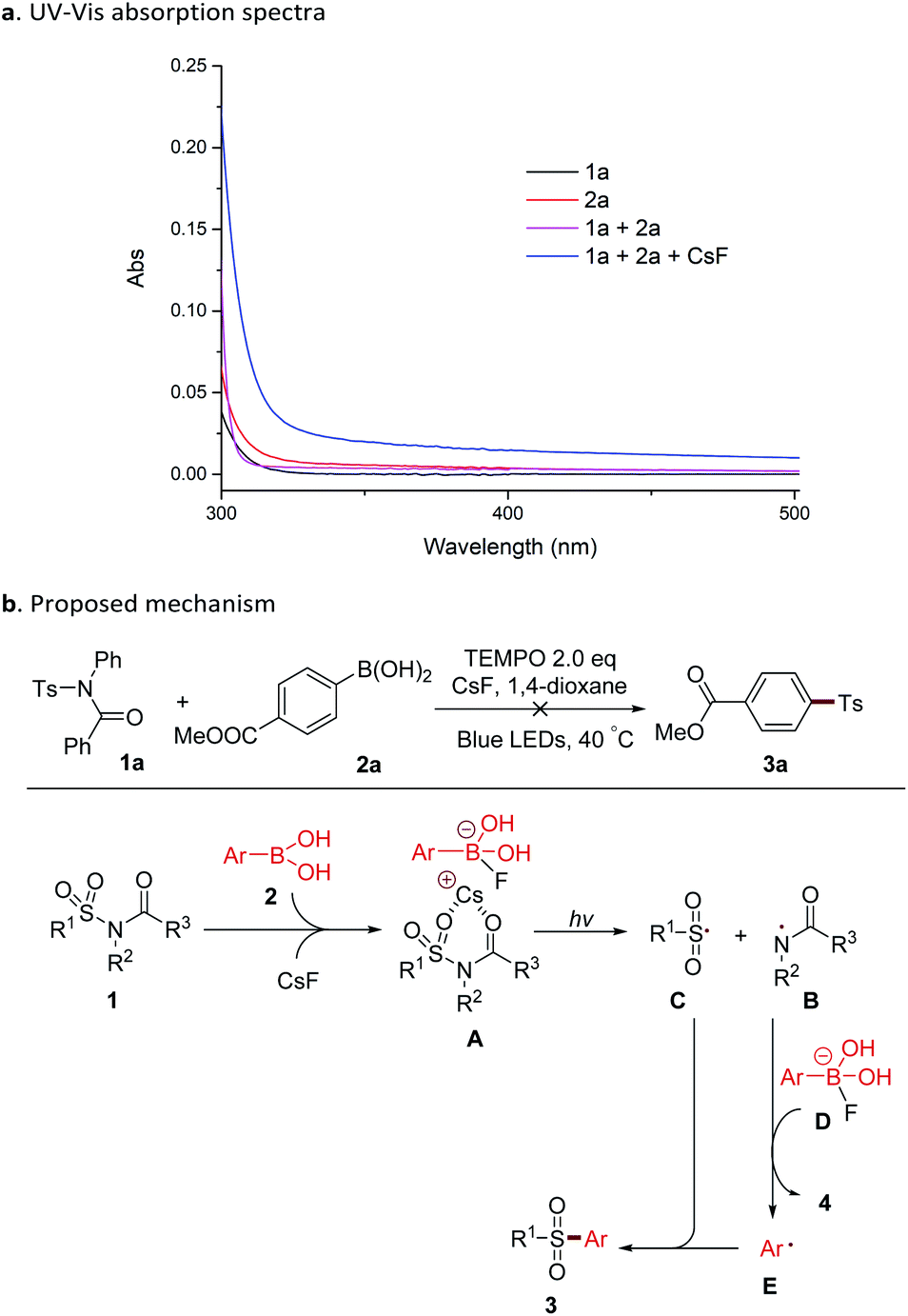 | ||
| Scheme 2 Mechanism investigation. (a) UV-Vis absorption spectra. (b) Proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient and practical strategy for the late-stage arylation of sulfonamides via a visible light-mediated N–S bond cleavage other than typical transition-metal-catalyzed C(O)–N bond activation. It also represented the first catalyst-free sulfonylation of boronic acids to synthesize aryl sulfones. The employment of diverse boronic acids and sulfonamides exhibited good functional group tolerance and high efficiency. The mechanism investigation revealed that the photo-excitable complex formed from sulfonamide, boronic acid, and base was crucial to afford sulfonyl radicals and furnish the desired products. This achievement inspired us to explore other strategies for the late-stage functionalization of sulfonamides.Data availability
The electronic supplementary information include experimental detail, NMR data and HRMS data.Author contributions
Y. Luo conceived and designed the experiments. H. Ding conducted most of the experiments. J.-S. Zhen and X. Du performed the arylation of sulfonamide drugs. X.-H. Xu, H. Yuan, Y.-H. Li, W.-Y. Qi, B.-Z. Liu and S.-M. Lu synthesized the starting materials. C. Xue contributed to the mechanism research. Q. Ding analylzed the data and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21901260, 21908256, 21662017).Notes and references
- J. E. Lesch, The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine, Oxford University Press, 2006 Search PubMed.
- K. A. Scott and J. T. Njardarson, Analysis of US FDA-Approved Drugs Containing Sulfur Atoms, Top. Curr. Chem., 2018, 376, 5 CrossRef.
- (a) R. R. Milburn and V. Snieckus, Angew. Chem., Int. Ed., 2004, 43, 892–894 CrossRef CAS PubMed; (b) S. Yoshida, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2012, 134, 19358–19361 CrossRef CAS PubMed; (c) J. Xuan, B.-J. Li, Z.-J. Feng, G.-D. Sun, H.-H. Ma, Z.-W. Yuan, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem.–Asian J., 2013, 8, 1090–1094 CrossRef CAS PubMed; (d) E. Hasegawa, N. Izumiya, T. Miura, T. Ikoma, H. Iwamoto, S. Takizawa and S. Murata, J. Org. Chem., 2018, 83, 3921–3927 CrossRef CAS PubMed; (e) E. Hasegawa, Y. Nagakura, N. Izumiya, K. Matsumoto, T. Tanaka, T. Miura, T. Ikoma, H. Iwamoto and K. Wakamatsu, J. Org. Chem., 2018, 83, 10813–10825 CrossRef CAS PubMed.
- (a) P. S. Fier and K. M. Maloney, J. Am. Chem. Soc., 2019, 141, 1441–1445 CrossRef CAS PubMed; (b) P. S. Fier, S. Kim and K. M. Maloney, J. Am. Chem. Soc., 2019, 141, 18416–18420 CrossRef CAS PubMed.
- (a) A. Gómez-Palomino and J. Cornella, Angew. Chem., Int. Ed., 2019, 58, 18235–18239 CrossRef PubMed; (b) M. Pérez-Palau and J. Cornella, Eur. J. Org. Chem., 2020, 2497–2500 CrossRef.
- Selected examples: (a) B. P. Bandgar, S. V. Bettigeri and J. Phopase, Org. Lett., 2004, 6, 2105–2108 CrossRef CAS PubMed; (b) Y. Fu, W. Zhu, X. Zhao, H. Hügel, Z. Wu, Y. Su, Z. Du, D. Huang and Y. Hu, Org. Biomol. Chem., 2014, 12, 4295–4299 RSC.
- Selected examples: (a) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188–191 CrossRef CAS PubMed; (b) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205–4208 CrossRef CAS PubMed; (c) W. Wu, S. Yi, W. Huang, D. Luo and H. Jiang, Org. Lett., 2017, 19, 2825–2828 CrossRef CAS PubMed.
- Selected examples: (a) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679–12683 CrossRef CAS; (b) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204–10208 CrossRef CAS PubMed; (c) Y. Meng, M. Wang and X. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1346–1353 CrossRef CAS PubMed; (d) S. Ye, K. Zhou, P. Rojsitthisak and J. Wu, Org. Chem. Front., 2020, 7, 14–18 RSC; (e) S. Ye, D. Zheng, J. Wu and G. Qiu, Chem. Commun., 2019, 55, 2214–2217 RSC; (f) S. Ye, G. Qiu and J. Wu, Chem. Commun., 2019, 55, 1013–1019 RSC; (g) J. Zhang, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Chem. Commun., 2020, 56, 3225–3228 RSC; (h) S. Ye, M. Yang and J. Wu, Chem. Commun., 2020, 56, 4145–4155 RSC; (i) X. Gong, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Org. Chem. Front., 2020, 7, 938–943 RSC; (j) F.-S. He, M. Yang, S. Ye and J. Wu, Chin. Chem. Lett., 2021, 32, 461–464 CrossRef CAS.
- (a) X. Li and G. Zou, Chem. Commun., 2015, 51, 5089–5092 RSC; (b) B. J. Simmons, N. A. Weires, J. E. Dander and N. K. Garg, ACS Catal., 2016, 6, 3176–3179 CrossRef CAS PubMed; (c) S. Shi, R. Lalancette, R. Szostak and M. Szostak, Org. Lett., 2019, 21, 1253–1257 CrossRef CAS PubMed.
- (a) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS; (b) S. Xie, D. Li, H. Huang, F. Zhang and Y. Chen, J. Am. Chem. Soc., 2019, 141, 16237–16242 CrossRef CAS PubMed.
- F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. V. der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02266k |
| This journal is © The Royal Society of Chemistry 2021 |