
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dioxasilepanyl group as a versatile organometallic unit: studies on stability, reactivity, and utility†
Hayate
Saito
 ,
Jun
Shimokawa
* and
Hideki
Yorimitsu
*
,
Jun
Shimokawa
* and
Hideki
Yorimitsu
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: shimokawa@kuchem.kyoto-u.ac.jp; yori@kuchem.kyoto-u.ac.jp
First published on 8th June 2021
Abstract
Organic synthesis is performed based on precise choices of functional groups and reactions employed. In a multistep synthesis, an ideal functional group should be compatible with various reaction conditions and unaltered until it is subjected to a selective conversion. The current study was set out to search for a silicon functionality that meets these criteria. Here we have established a new silicon-based synthetic methodology centred on a bulky 7-membered dialkoxysilyl group (2,4,4,7,7-pentamethyl-1,3,2-dioxasilepan-2-yl) that uniquely has both stability and on-demand reactivity. The exceptional stability of this functional group was corroborated by both experimental and computational studies which demonstrated that key factors for its stability were a 7-membered structure and steric hindrance. In turn, the dioxasilepanyl group was found to become reactive and to be easily transformed in the presence of appropriate activators. Combined with the development of easy and robust methods to introduce the dioxasilepanyl group onto aryl rings, these findings have allowed a shorter and more efficient synthesis of a bioactive molecule, thus demonstrating the potential utility of the easily accessible dioxasilepanyl group in organic synthesis.
Introduction
In conventional organic syntheses, organometallic units are expected to be temporarily reactive for desired transformations.1 As exemplified by the Grignard reaction, these transient units are usually consumed during the transformations immediately after these organometallic units are introduced to molecules. The stability of those units matters only for storage. In the case of multistep syntheses of highly functionalized molecules, ideal organometallic units are defined somewhat differently (Fig. 1a). Such an organometallic unit, represented by M, has to be orthogonal to other functional groups (FGs) across multiple transformations including aqueous workup and regular purifications with silica gel column chromatography. Also, it has to become reactive enough when it is subjected to the final designated transformation. The search for ideal Ms could be assumed to be a quest toward resolving such ambivalence that is characteristic to organometallic units (Fig. 1b). Rare examples of “tough and switchable” Ms that meet these criteria are known to be suited for modular syntheses of complex molecules. In the case of organoboron functional groups,2 the B(pin) (pinacolatoboryl) group has almost monopolized this need in modern organic synthesis since this cyclic dialkoxyborane shows moderate stability during purifications on silica gel, while having a boron centre with a vacant p-orbital that is easily activated for various transformations. Because of this contradictory nature, neither stability nor reactivity is perfect. Specifically, it is still difficult to fully avoid undesired degradation of B(pin) units during the transformations of other functional groups, and therefore B(pin) is not desirable as an M. Tougher boron units such as MIDA boronate3 and B(dan)4 are known to be tolerant to diverse reaction conditions because of their respective stabilization of the boron centre by intramolecular coordination and electron-donating amino groups. The reactivity of such protected boron units is, in turn, sacrificed and it is usually difficult to directly transform these boron functionalities without deprotection, with the exception of a few cases.5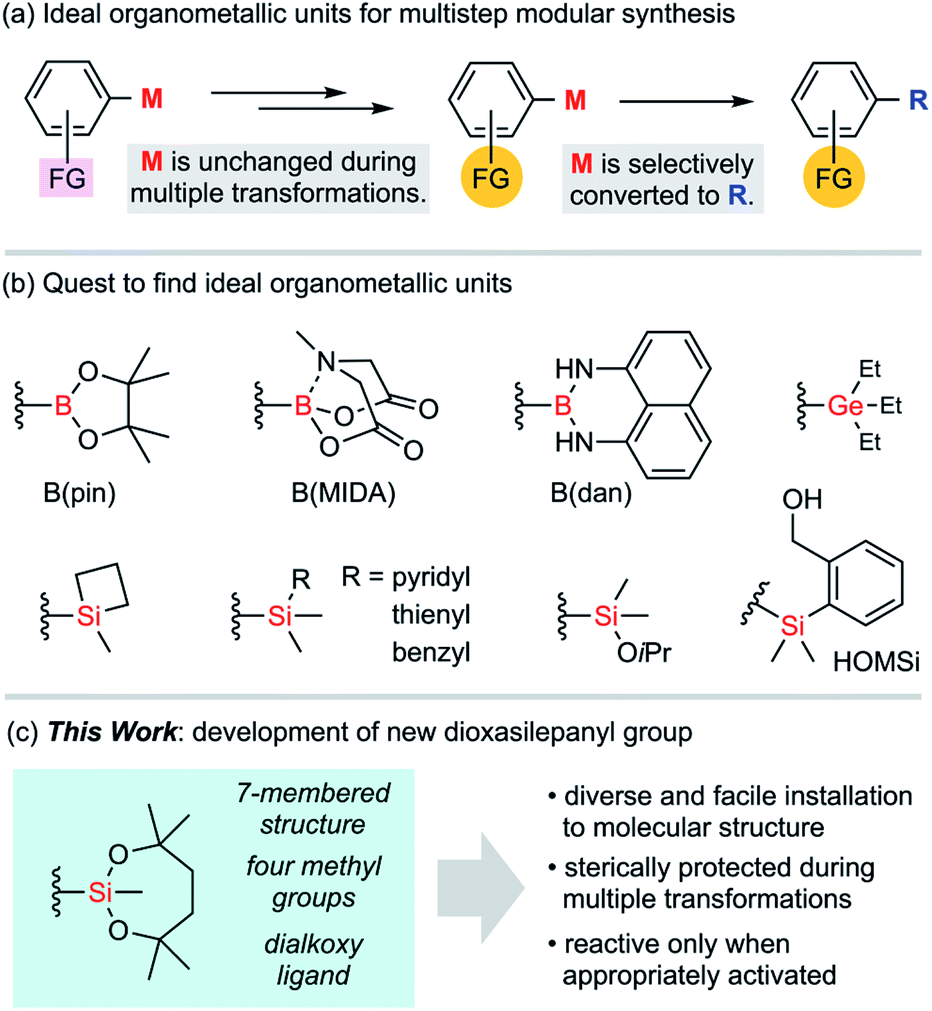 | ||
| Fig. 1 Development of ideal organometallic functional groups for multistep syntheses. | ||
Group 14 elements are also ideal organometallic unit candidates for this purpose. Although organostannyl groups were not pursued in our study due to their inherent toxicity, organogermyl and organosilyl groups are fascinating as an unconventional class of switchable organometallic units. The chemistry of organogermyl groups has recently been explored intensively to uncover their high orthogonality with other functional groups in cross-coupling reactions.6 In the case of silyl groups, the well-known potential of incorporating variable substituents onto the silicon atom is expected to tune the stability.7 Thus, it would be fair to regard silyl groups as viable key functional groups for use in multistep organic synthesis. A number of silyl groups have actually been employed as partners for cross-coupling reactions. Several triorganosilyl groups (e.g., silacyclobutyl, pyridylsilyl, thienylsilyl, and benzylsilyl groups, to name a few)8 have been developed as reactive silicon units, albeit with little use in practical synthesis. The reactive silyl group –SiMe2(OiPr) is compatible even with the generation of Grignard reagents.9 However, the known reactivity of this alkoxysilyl group is somewhat limited and transformation of the aryl–Si bond has rarely been reported.9c Among the known silyl groups, one of the most successful examples to balance reactivity and stability is the (2-hydroxymethylphenyl)dimethylsilyl group, known as HOMSi.10 This functional group can be used for transmetalation by employing intramolecular activation with a weak base instead of fluoride. Thus, the protection of the hydroxy group is indispensable for application to multistep synthesis to avoid undesired activation of the silicon centre through laborious protection/deprotection sequences.10d,e
We developed a new group M for multistep syntheses by using the architecture of an alkoxysilyl group that could easily modify the Lewis acidity of the silicon atom and the steric environment of the silyl group. Here, we would like to introduce 2,4,4,7,7-pentamethyl-1,3,2-dioxasilepan-2-yl group as a new ideal group M for multistep syntheses (Fig. 1c). This functional group is characterized by (1) a 7-membered structure that is ideally suited for a stable silicon-containing unit, (2) four methyl groups for steric protection of the silicon centre, and (3) a dialkoxysilyl group that guarantees the activation for the reaction.
Results and discussion
Experimental and theoretical studies of the dioxasilepanyl group
First, the stability of a series of alkoxysilyl groups was examined by monitoring the time courses for the hydrolysis of the corresponding phenylsilanes under basic conditions. The hydrolysis of phenylsilanes 1-I–1-X was examined in CD3CN/D2O in the presence of 10 mol% NaOD and the remaining phenylsilanes were quantified by 1H NMR analysis. The decrease of the phenylsilanes occurred only through the hydrolysis of their Si–O bonds and decomposition through the protodesilylation of C–Si bonds was not observed in all cases. Scheme 1 shows the amounts of the remaining alkoxyphenylsilanes 1-I–1-X after 4 hours of hydrolysis. Detailed time-dependent decreases of alkoxyphenylsilanes 1-I–1-X are shown in Fig. S1–S3 in the ESI.† In all cases, bulkier alkoxy groups on the silicon atom showed higher stability of silyl groups, irrespective of the number of oxygen atoms (I > II, III > IV, and IX > X). This tendency is consistent with the fact that the –Si(OiPr)3 group (I) bearing three bulky alkoxy groups survives strong hydrolytic conditions.11 We also tested a disiloxysilyl group (–SiMe(OSiMe3)2, V), which has been attracting attention as a versatile organometallic unit for facile introduction and transformation.12 Although arylsilanes bearing V are generally known to be stable,12a1-V underwent hydrolysis faster than PhSi(OEt)3 (1-II) under the current conditions. We next examined cyclic alkoxysilyl groups bearing bulky substituents around the silicon atom in an attempt to stabilize the alkoxy groups. We tested 6-membered (2,4,4,6,6-pentamethyl-1,3,2-dioxasilynan-2-yl, VI) and 7-membered (2,4,4,7,7-pentamethyl-1,3,2-dioxasilepan-2-yl, VII) cyclic dialkoxysilyl groups whose structures can be easily constructed from the corresponding tertiary diols. Compounds bearing the dioxasilynanyl moiety were reported to exhibit certain stability toward hydrolysis,13 which suggested us that 1-VI would be a promising candidate for having high stability. In contrast to our prospects, the dioxasilynanyl group in 1-VI was easily hydrolyzed to recover only 17% of 1-VI after 4 hours. In contrast, the dioxasilepanyl group 1-VII showed excellent stability under the same conditions and 93% of 1-VII survived. A simplified dioxasilepanyl group 1-VIII without four methyl groups was found to be far less stable than 1-VII with a lifetime of less than 30 minutes under the same conditions. This means that both the 7-membered cyclic structure and the steric hindrance around the silicon atom are indispensable for the high stability of 1-VII.14 | ||
| Scheme 1 Stabilities of alkoxyphenylsilanes and a siloxyphenylsilane under basic conditions. | ||
To figure out the origin of the difference in the rates of hydrolysis among acyclic silyl III, dioxasilynanyl VI, and dioxasilepanyl VII groups, we conducted computational estimation of the stabilities of bulky dialkoxyphenylsilanes (i.e. PhSiMe(OtBu)2 (1-III), 1-VI, and 1-VII) by calculating the relative free energies of the formation of the corresponding dialkoxyfluorosilicates. All reasonable conformers of these phenylsilanes and their corresponding fluorosilicates were automatically explored using the GRRM17 program15 by the SC-AFIR/PM7 method16 and reoptimized at the level of ωB97X-D/jun-cc-pVTZ17,18 in THF (SMD).19 The conformers of pentacoordinated fluorosilicates could be classified into one of the substitution patterns on trigonal bipyramidal geometrical configurations around the silicon atom. For all of the three silyl groups examined (III, VI, and VII), each of the most favorable conformers exhibited the same pattern in which two apical positions were occupied by a fluoro group and an alkoxy group. Their calculated relative free energies at 298.15 K are shown in Fig. 2. The detailed structures and relative free energies of the most stable conformers of each substitution patterns are described in the ESI.†1-VII-F was calculated to be higher in free energy by +8.8 kcal mol−1 than 1-VII and F−, which is similar to the ΔG value of 1-III-F (+10.1 kcal mol−1). In contrast, 1-VI-F required a smaller energy (+3.2 kcal mol−1) to form the corresponding silicate. Combined with the fact that the optimized structures of silicates 1-VI-F and 1-VII-F showed no significant difference in their bond angles and lengths around their silicon and oxygen atoms, the difference between the stabilities of 1-VI and 1-VII would mainly be due to the intrinsically higher ring strain of the 6-membered dioxasilynane ring in VI20 in contrast to the unstrained formation of the 7-membered dioxasilepanyl moiety in VII.
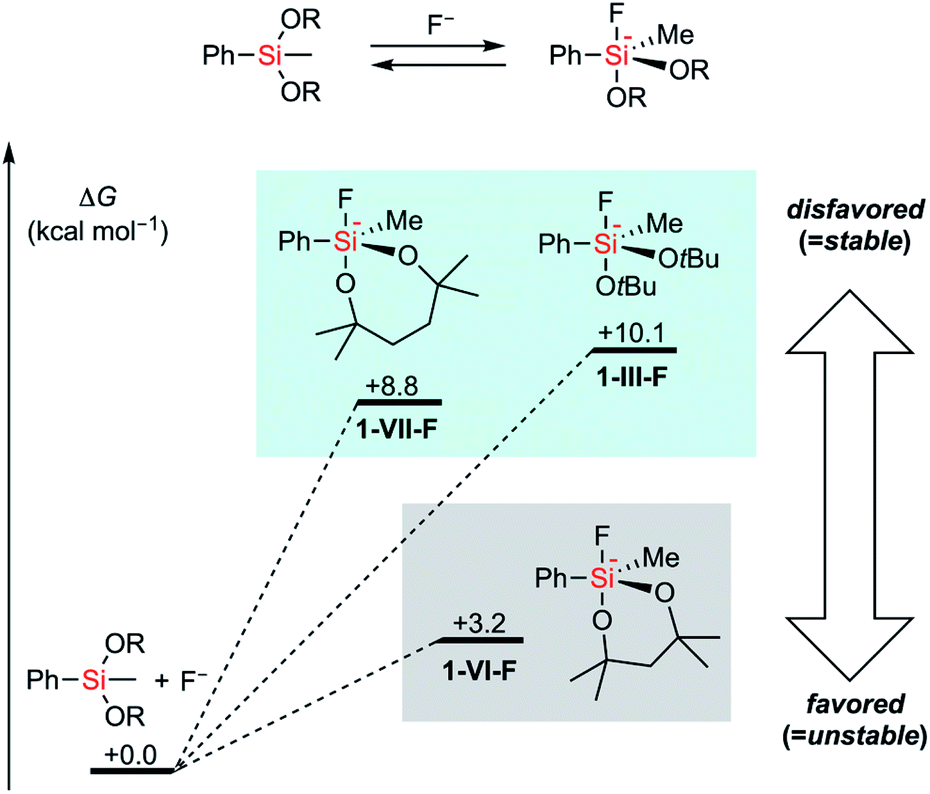 | ||
| Fig. 2 Calculated relative free energies of dialkoxyfluorosilicates. | ||
Interestingly, arylsilanes bearing the dioxasilepanyl group VII tend to be obtained as solids unlike those with common organosilanes. The crystal structure of 5a shown in Fig. 3 reveals that the dioxasilepane ring in the solid state adopts a twisted chair-like conformation that is reported to be the most stable for cycloheptane.21 A similar structure is also found in another reported X-ray structure of a dioxasilepane ring.22 Meanwhile, the 1H NMR spectrum of 1-VII shows that the four ethylene protons of the dioxasilepane core appear as one broad signal. This is in contrast to the methylene protons in the dioxasilynane moiety of 1-VI that show two independent signals in the 1H NMR spectrum. This difference confirms that the 7-membered skeleton of VII in a twisted-chair form is flexible enough to smoothly flip back and forth even at room temperature while the 6-membered skeleton of VI is sluggish to flip.
 | ||
| Fig. 3 X-ray structure of 5a (chlorine atoms and hydrogens except for those on the C2H4 unit are omitted for clarity). | ||
In addition to the thermodynamic stabilities of dialkoxysilanes, steric hindrance around their silicon atoms in a series of silyl groups (I, III, IV, VI, VII, and VIII) was evaluated by calculating a percent buried volume (% Vbur) which is useful to discuss the spatial filling of the whole sphere around the coordination centre (Fig. 4).23 Each silicon atom was set as the centre of a sphere whose diameter is 7.0 Å. To exactly determine the hindrance of the alkoxide unit around the central silicon atom, the phenyl ring and the silicon atom were omitted and the hydrogen atoms on the silyl groups were considered for the calculation of % Vbur. In these steric maps, the larger the appearance of the red region, the deeper the alkoxide unit penetrates into the side of the hemisphere containing the phenyl ring (z < 0).
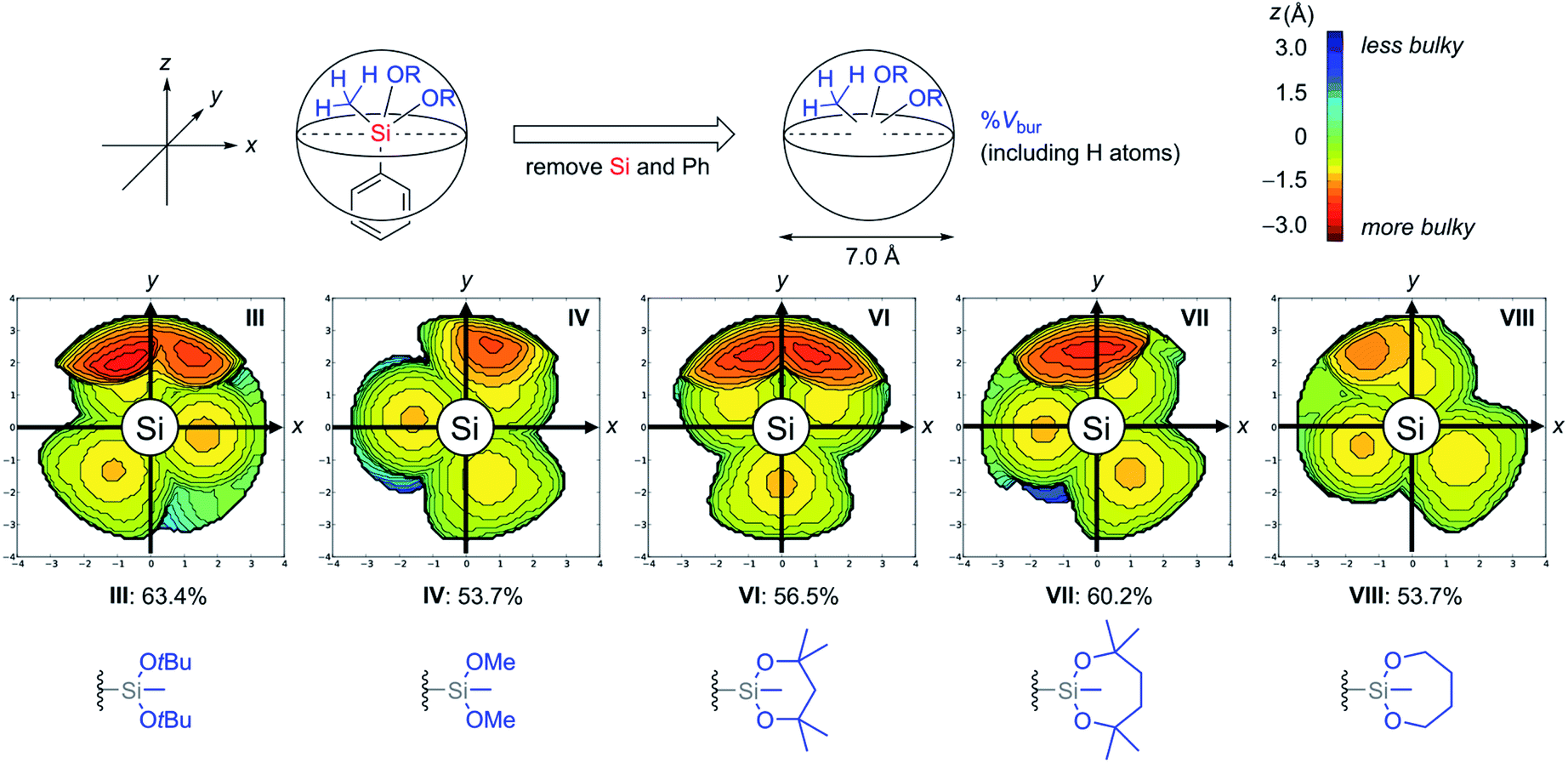 | ||
| Fig. 4 % Vbur and steric maps of silyl groups. | ||
We observed a tendency of cyclic structures to have a reduced % Vbur compared with acyclic structures due to their tight conformations. For example, cyclic 1-VIII showed the same value (53.7%) as acyclic 1-IV despite the additional C2H4 components of 1-VIII. This tendency is consistent with the decrease of the red region in the steric map of VIII compared with that of IV. The tendency of the cyclic system to decrease % Vbur is also found in cyclic 1-VI (56.5%) and 1-VII (60.2%) compared with 1-III (63.4%). The difference of the values of % Vbur between VII (60.2%) and VIII (53.7%) indicates that the four methyl groups on the dioxasilepane ring provided efficient steric protection around the silicon atom.24 To obtain more insights into the stability of VII, 29Si NMR analysis of 1-III, 1-IV, 1-VI, and 1-VII was also conducted. The result revealed no significant correlation between the structure and their chemical shifts in the current state (see the ESI†). Based on the discussion above, we concluded that the stability of the dioxasilepanyl group VII can be explained by two factors: (1) thermodynamic stability of the dioxasilepanyl group that avoids the formation of silicate, (2) steric protection around the silicon atom provided by the bulky alkoxy moieties.
Synthetic applications of the dioxasilepanyl group: preparation and orthogonality
We hypothesized that the stability of the silyl group VII would have advantages, especially in multistep synthesis. Although the structure and the synthesis of phenylsilane 1-VII have been reported in the literature,25 its reactivity and detailed synthetic utility have not been investigated. We aimed to establish preparative methods for silanes bearing the dioxasilepanyl group VII in order to evaluate their reactivity as well as compatibility with various transformations.The dioxasilepanyl group VII could be easily and reliably constructed from the corresponding chlorosilanes and an inexpensive diol in the presence of Et3N (Scheme 2). Similar to the synthesis of phenylsilane 1-VII, methoxysilane 2-VII and hydrosilane 3-VII were also synthesized. Silanes 1-, 2-, and 3-VII were selectively obtained without forming detectable amounts of polymeric byproducts, likely because the reaction tends to form a kinetically favored 7-membered dioxasilepane ring. Thus, from the three chlorides of trichloromethylsilane, only two reacted to form a dioxasilepanyl moiety, which cleanly left one chloro group for the subsequent reaction with methanol to afford 2-VII.
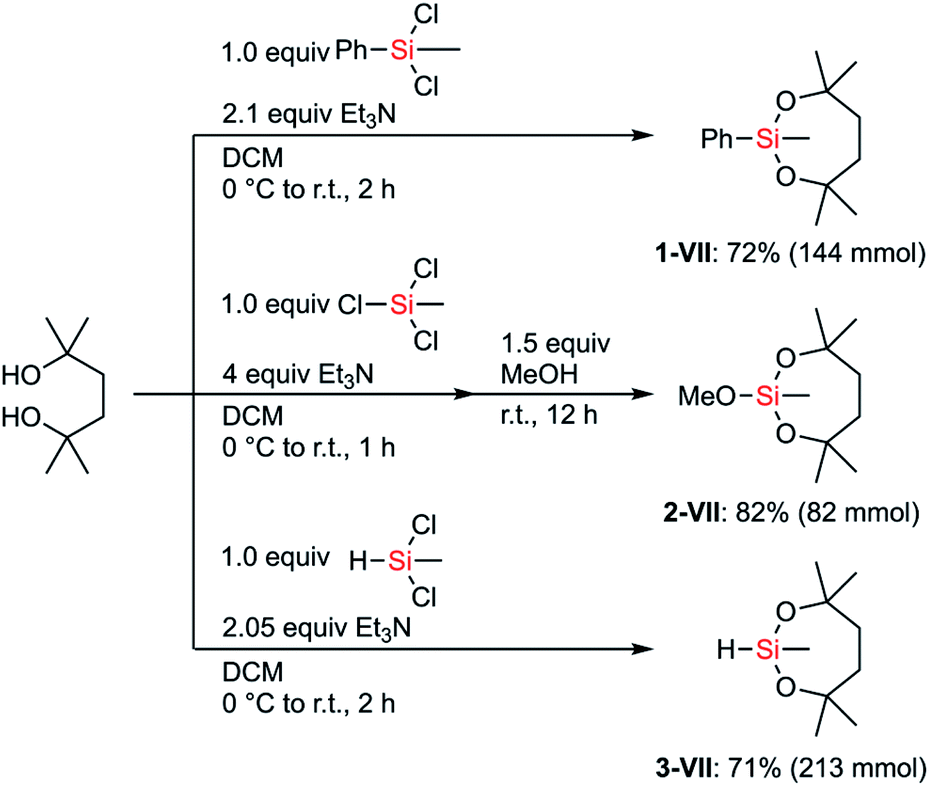 | ||
| Scheme 2 General methods for the construction of the dioxasilepanyl group VII and precursors for introducing VII. | ||
To develop a general preparation methodology for the compound with the dioxasilepanyl group, we examined the reaction of aryllithiums with methoxysilane 2-VII that is much easier to handle than the chlorosilanes. When 2-VII reacted with 4-tBu-C6H4Li, the substitution of the methoxy group of 2-VII proceeded to give 4a as the sole product while retaining the dioxasilepanyl structure; neither multiple substitution products nor ring-opening products were observed even though an excessive amount of aryllithium was added to the reaction mixture (Scheme 3). Using this method, the electron-rich arylsilane 4a (96%) and electron-deficient p-fluorophenylsilane 4b (76%) were synthesized. In the case of sterically demanding 2-biphenylyllithium, it was necessary to increase the reactivity of the aryllithium in the presence of TMEDA. The corresponding silane 4c was obtained in moderate yield (58%). A heteroaryllithium prepared by the deprotonation of benzofuran was also applicable to this method, and 2-benzofurylsilane 4d was synthesized in 83% yield.
 | ||
| Scheme 3 Reaction of methoxysilane 2-VII with aryllithiums; a1.5 equiv. TMEDA was added before lithiation. | ||
The dioxasilepanyl group VII is reactive to aryllithiums only when the leaving group (OMe) is attached and no reaction was observed after the first substitution. Interestingly, the cyclic structure was kept intact despite the remaining 0.5 equivalent of an aryllithium, which implies that the ring opening would require a higher activation energy than the substitution of the methoxy moiety.
Next, we focused on Ir-catalyzed C–H silylation reactions26 of arenes to provide an opportunity to synthesize a broad range of arylsilanes, especially the ones bearing delicate functional groups. In Hartwig's report,27,28 only disiloxyhydrosilane (HSiMe(OSiMe3)2) 3-V showed sufficient reactivity for the silylation reaction under the optimized conditions ([Ir(OMe)(cod)]2, 2,4,7-trimethylphenanthroline, cyclohexene, THF, and 80 °C). Other hydrosilanes such as HSi(OEt)3, HSiMe(OEt)2, HSiMe2OEt, HSiEt3, and HSi(SiMe3)3 did not work at all in Hartwig's reaction. Hydrosilane 3-VII is potentially viable as a silylation reagent to introduce the silyl group VII to arenes via C–H activation. We wondered whether our dioxasilepanyl group VII would exhibit different reactivity from other conventional alkoxysilyl groups for the C–H silylation reaction.
When Hartwig's original reaction conditions were applied to 3-VII, the corresponding silylation products were obtained in low yield. We eventually found that a higher reaction temperature (80 °C to 100 °C) and the use of 3,3-dimethyl-2-butene28a as an alternative hydrogen acceptor improved the efficiency of the silylation (Scheme 4). An optimized silylation reaction of arenes with hydrosilane 3-VII demonstrated good compatibility with functionalities. Halogen and ester groups survived, which ensured the possibility for further transformations after the silylation (5a–5d). Electron-deficient substrates, such as pentafluorobenzene and 2,3-di(methoxycarbonyl)naphthalene, were also applicable (5e and 5f). Substrates bearing heteroaromatic cores such as benzoxazole, indole, thiophene, pyridine, and quinoline also underwent the silylation reaction without apparent drawbacks (5g–5k). Notably, in most cases, a single mono-silylated product was exclusively obtained, except for 5g (7-silyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6-silyl = 92:8). A gram-scale synthesis of 5c was successfully conducted even with a reduced loading of the catalyst.
:6-silyl = 92:8). A gram-scale synthesis of 5c was successfully conducted even with a reduced loading of the catalyst.
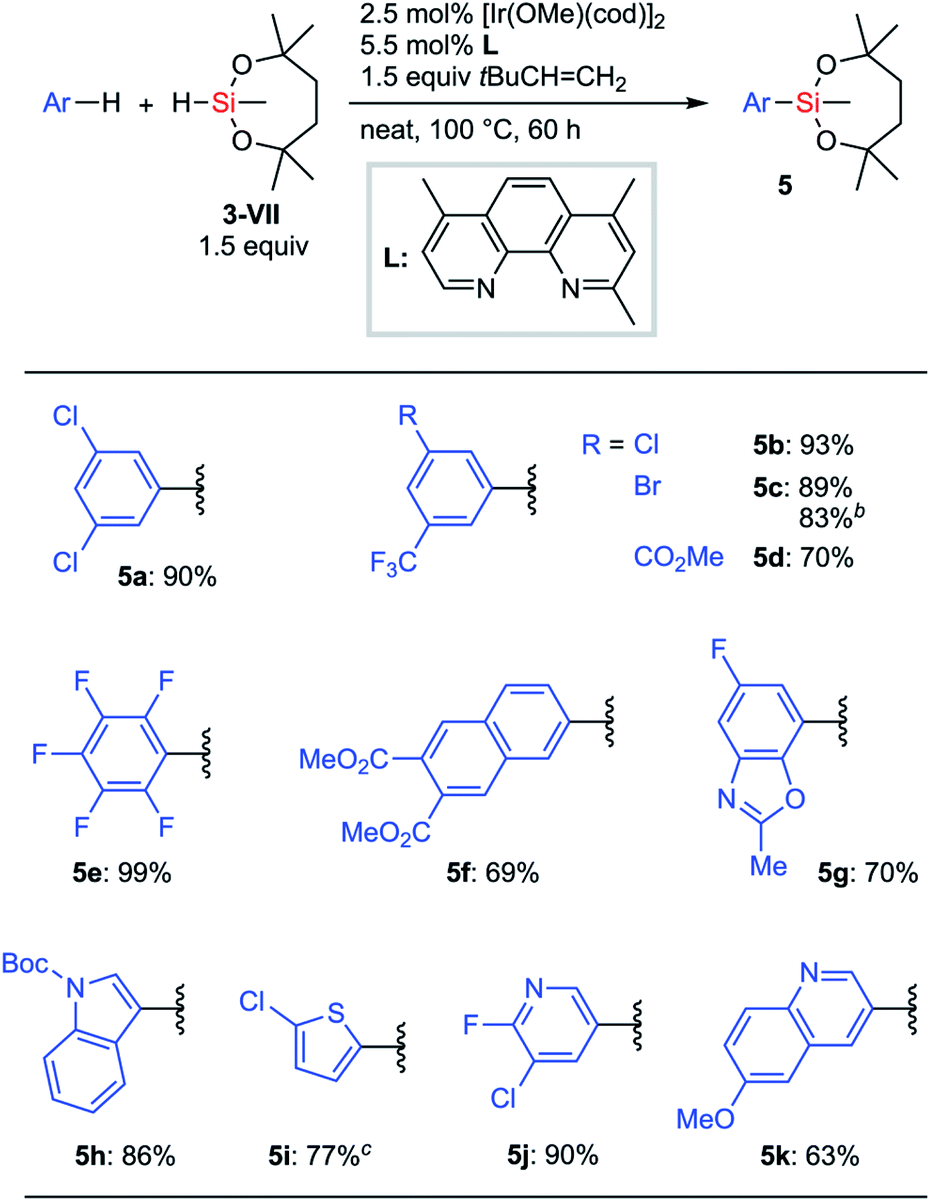 | ||
| Scheme 4 C–H silylation of arenes with 3-VIIa. a1 mmol substrate. b10 mmol substrate, [Ir(OMe)(cod)]2 (1 mol%), 2,4,7-trimethylphenanthroline (L) (2.2 mol%). c1 equiv. 3-VII. | ||
To gain insight into the reasons for the exceptional reactivity of 7-membered hydrosilane 3-VII, other hydrosilanes shown in Scheme 5 were also tested as silylation reagents under the new conditions for the C–H silylation of m-dichlorobenzene. Due to the exceptionally high reactivity of disilyloxyhydrosilane 3-V for C–H silylation, the comparison of the reactivity of hydrosilanes has been limited. Under the current conditions, hydrosilanes bearing two tert-butoxy groups (3-III) and one adamantyloxy group (3-XI) gave the corresponding silylation products in low to moderate yield. In contrast, silanes bearing smaller alkoxy groups, tri- and diethoxyhydrosilane (3-II and 3-IV) did not react at all without any formations of the corresponding disilanes.29 These results indicate that the bulkier alkoxy moieties on the silicon atom increase the reactivity of hydrosilanes in this C–H silylation reaction.
 | ||
| Scheme 5 C–H silylation of m-dichlorobenzene with a series of hydrosilanes under the optimized conditions. Ad: 1-adamantyl. | ||
The compatibility of the silyl group VII with various synthetic transformations was examined. The silyl group VII in 5d survived the reduction of the ester moiety with an excess amount of LiAlH4 (Scheme 6a). This is in sharp contrast to the report that diethoxysilanes are reduced to dihydrosilanes under similar conditions.30 The resulting benzyl alcohol 6 could be oxidized under TEMPO/PhI(OAc)2 conditions to give aromatic aldehyde 7 without affecting the silyl group.31 Aldehyde 7 successfully underwent reductive amination with pyrrolidine to give benzylamine 8 in the presence of NaBH3CN and AcOH under mildly acidic conditions.32 It is noteworthy that 1-VII could tolerate acidic conditions of 10 mol% trifluoroacetic acid (TFA) even at 60 °C in CDCl3. In the case of methanesulfonic acid (MsOH), slight degradation of the alkoxy moiety was observed even at room temperature, probably via generation of the tertiary carbocation by the elimination of silanol species. Nonetheless, 89% of 1-VII was recovered after 1 hour. To compare with other base-resistant silyl groups, non-cyclic silanes 1-III and 1-IX were similarly examined under acidic conditions (10 mol% acid and r.t. in CDCl3). 1-III and 1-IX were recovered only in moderate yield (89% and 48%) with TFA even at r.t., and in reduced yield (61% and 33%) with MsOH. These results revealed the importance of the 7-membered dialkoxysilyl moiety for the stability under acidic conditions (see the ESI†).
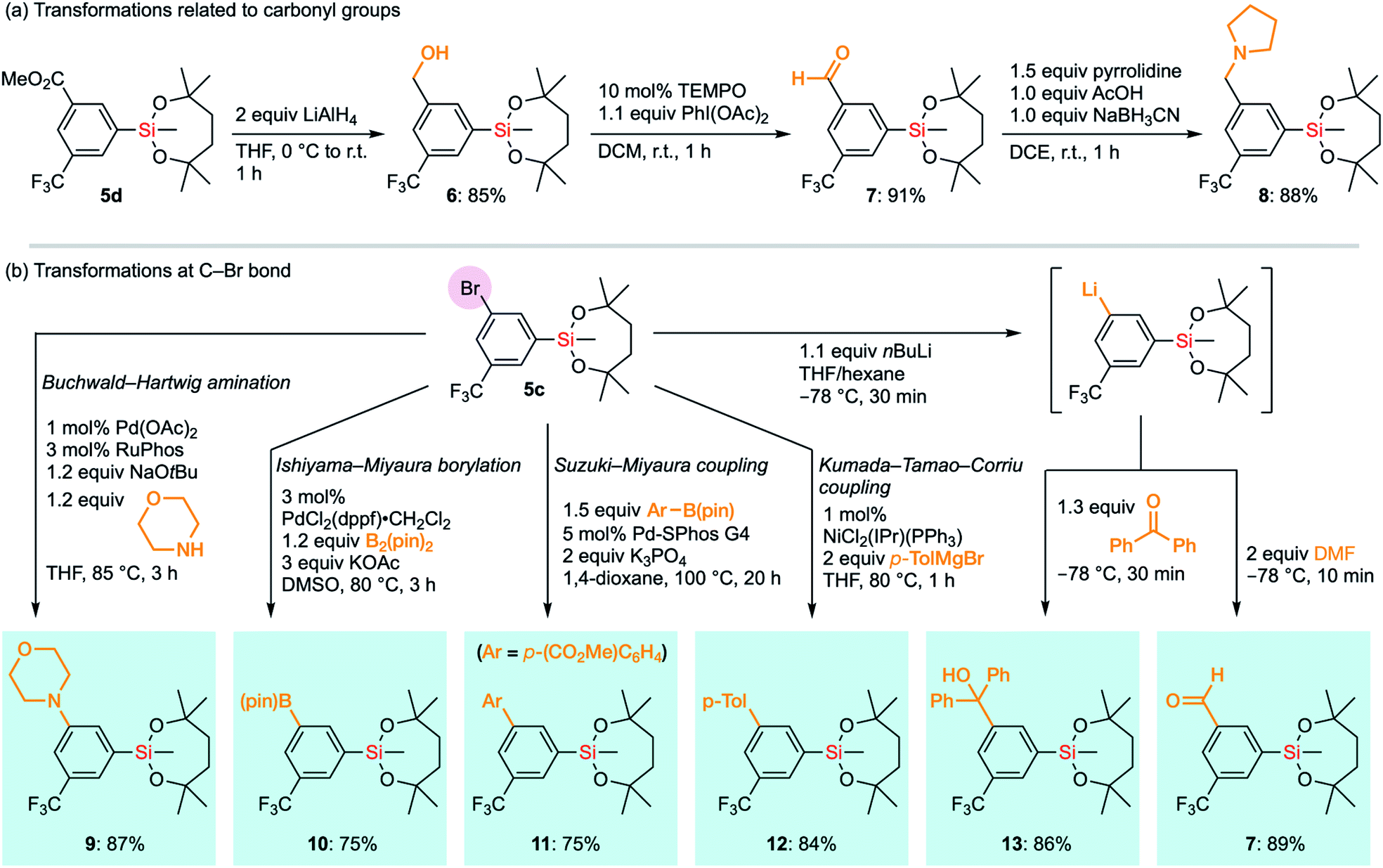 | ||
| Scheme 6 Transformations of functional groups in 5d and 5c without affecting the silyl group VII. TEMPO: 2,2,6,6-tetramethylpiperidine 1-oxyl. | ||
Next, we envisioned a series of transformations of carbon–halogen bonds in arylsilane 5c without interfering with the silyl group VII (Scheme 6b). Pd-Catalyzed Buchwald–Hartwig amination33 successfully took place to give 9 without any decomposition of VII even in the presence of a strong base and a highly nucleophilic amine. Under the same conditions, the triethoxysilyl group (II) in p-bromophenyltriethoxysilane did not survive the coupling reaction, which underscores the higher stability of VII. VII also survived the transformation of boron functional groups. Bromide 5c underwent Pd-catalyzed borylation with B2(pin)2 to afford 10,34 which bears two organometallic units on one aromatic ring. The Suzuki–Miyaura cross-coupling reaction with an arylboronic acid ester to give biaryl 11 was also compatible with the silyl group VII.35 We also exposed arylsilane 5c to stronger carbon nucleophiles, with which ordinary alkoxysilanes could not survive. Kumada–Tamao–Corriu cross-coupling of 5c with p-tolylmagnesium bromide gave the coupling product 12 without any decomposition of the silyl group despite being under heating conditions.36 Surprisingly, even aryllithium species bearing the silyl group VII could be generated from 5c through a halogen–lithium exchange reaction. Aryllithium species thus formed could be used for subsequent nucleophilic additions to benzophenone and DMF to afford 13 and 7, respectively. Accordingly, the dioxasilepanyl group VII has been confirmed to be very stable and reliable during various transformations.
Synthetic applications of the dioxasilepanyl group: transformations into other functional groups
With the information on the stability of VII in hand, we next attempted to establish methods for the transformations of C–Si bonds of arylsilanes bearing VII to prove their utility in organic synthesis. Due to the great stability of the silyl group VII, conventional conditions37 for Tamao–Fleming oxidation employing basic aq. H2O2 were not effective. Fortunately, a combination of tert-butyl hydroperoxide and KOtBu38 successfully promoted the hydroxylation of arylsilane 4a to afford the corresponding phenol 14 (Scheme 7). The treatment of 4a with bromine led to ipso-bromination to give aryl bromide 15 in high yield.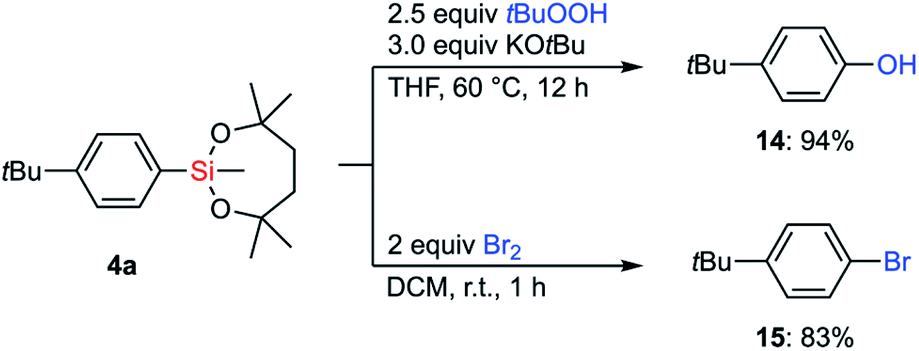 | ||
| Scheme 7 Hydroxylation and bromination of arylsilane 4a. | ||
The reactivity of the dioxasilepanyl group VII was also examined in the Hiyama–Denmark cross-coupling reaction.39 In an initial attempt of the cross-coupling reaction of phenylsilane 1-VII with p-iodoanisole, nBu4NF was employed as an activator of the silyl group VII to give the coupled product 16a in 62% yield with the concomitant formation of bianisyl (28%) (Table 1, entry 1). Biaryl byproducts are often problematic in palladium-catalyzed cross-couplings of arylsilanes.40 To reduce the amount of the undesired dimer, bianisyl, we optimized the reaction conditions. While CsF is often employed as an activator of organosilicon compounds, it did not promote the reaction at all in this case (entry 2). When AgF was employed as a base, the generation of bianisyl was slightly suppressed and 16a was observed in 67% yield (entry 3). We explored other solvents, 1,4-dioxane and toluene (entry 4 and 5), and finally found toluene to be the best solvent for avoiding the formation of bianisyl. Thus, the cross-coupling product 16a was obtained in 91% yield. The yield decreased drastically (10%) in the absence of CuCl(IPr) (entry 6). Both Cu(I) and Ag(I) salts are known to mediate the transmetalation of arylsilanes.41 The current result indicates that copper species assist the transmetalation from arylsilane. We also confirmed that trimethylphenylsilane (1-SiMe3) was kept intact under the optimized conditions for the current Hiyama–Denmark cross-coupling, which highlighted the higher on-demand reactivity of 1-VII.

Orthogonality in cross-coupling reactions by selective activations between VII and B(pin) moieties was achieved by simply changing fluoride sources (Scheme 8). For a mixture of 1-VII and an arylboronic acid pinacol ester, Hiyama–Denmark cross-coupling with an iodoarene selectively proceeded under our optimized conditions with AgF as a base. In contrast, CsF promoted only Suzuki–Miyaura cross-coupling. In both cases, the other organometallic reagent was recovered without consumption. Both electron-rich (R = OMe) and electron-deficient (R = CO2Et) iodoarenes were confirmed to be applicable to this orthogonal cross-coupling.
 | ||
| Scheme 8 Orthogonal cross-coupling between dioxasilepanyl VII and B(pin) switched by fluoride sources. aThe recovery of organosilane or organoboron was expressed based on the molar ratio to p-iodoanisole (the maximum recovery is 150%). | ||
Both electron-rich and electron-deficient aryl iodides were applicable to cross-coupling reactions with phenylsilane 1-VII (Scheme 9, 16a and 16b). The aldehyde moiety was compatible with the reaction conditions (16c). 1-Naphthyl and heteroaryl iodides could also be used as coupling partners (16d–16f). C–H silylation products could be employed as coupling reagents. Thus, 5b was employed in cross-coupling with p-iodoanisole to give 16g keeping the chloro and CF3 groups intact. This difference in reactivity among halogen atoms should be useful in further transition metal-catalyzed transformations. Heteroaromatic silepane 4d was also employed for the coupling reaction to afford 16h in 87% yield.
 | ||
| Scheme 9 Hiyama–Denmark cross-coupling of various iodoarenes with phenylsilanes. | ||
The more organometallic units are resistant to unexpected transformations, the more flexible strategies we could plan for the synthesis. To demonstrate the advantages of the silyl group VII in organic synthesis, we examined an efficient preparation of a bioactive molecule via a route that is shorter than the known synthetic route. Diarylmethanol 19 is known as a BET bromodomain inhibitor and several synthetic approaches to 19 have already been reported.42 We decided to develop an original synthetic approach to 19 by taking full advantage of the chemistry of the dioxasilepanyl group VII (Scheme 10). The synthesis of 19 using VII started by employing iterative halogen–lithium exchange reactions, during which the silyl group VII could survive. The monolithiation of 3,5-dibromoanisole by nBuLi in THF/hexane followed by treatment with methoxysilane 2-VII gave silylated bromoarene 17 in moderate yield. The second halogen–lithium exchange from 17 cleanly gave the corresponding aryllithium species, which was subjected to the subsequent addition to benzaldehyde to provide benzylic alcohol 18. Finally, 18 underwent Hiyama–Denmark cross-coupling without the protection of the hydroxy group to furnish the target molecule 19 in 69% yield in only three overall steps, which is shorter than the known four-step synthesis42b from the commercially available starting material. Of note, the diaryl methanol moiety survived the cross-coupling reaction without forming any ketone product. Analogs of 19 could be easily prepared by simply changing aldehydes or aryl halides, which makes the current convergent synthetic scheme of 19 advantageous toward further derivatization of this bioactive molecule.
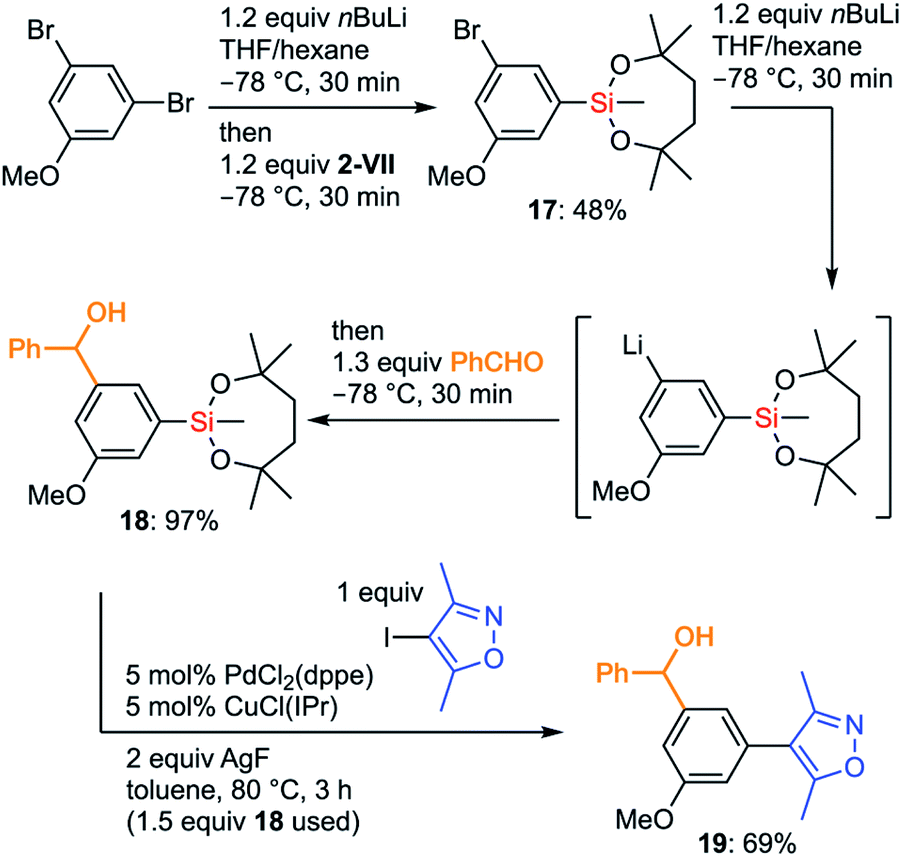 | ||
| Scheme 10 3-Step synthesis of the BET bromodomain inhibitor 19. | ||
Conclusions
Ideal organometallic units for modular multistep syntheses are required to be generally stable, and when desired, they should be easily activated for reactions. Boronic acid derivatives have dominated such chemistry and silyl functionalities have been less utilized despite their potential. In the current study, we searched for silyl groups that are suitable for multistep syntheses and have revealed that the 7-membered dialkoxysilyl group VII shows outstanding stability among a series of alkoxysilyl groups. The dioxasilepanyl group VII was also found to be a good organometallic unit for various transformations, which makes this silyl group an ideal functional group for multistep synthesis. The stability of VII was estimated both experimentally and theoretically to gain the insight that VII is not only kinetically protected by its steric hindrance but also thermodynamically stabilized by the 7-membered structure. Silyl groups VII on aryl rings are stable and can survive strong nucleophiles such as LiAlH4 and organolithiums that are not usually compatible with conventional alkoxysilyl groups. VII could be easily installed onto functionalized arenes by using new silyl sources methoxysilane 2-VII and hydrosilane 3-VII, which could be easily prepared on a large scale and handled without particular care. Under specifically designed conditions, arenes bearing VII were found to be applicable to various transformations that cleave an aryl–Si bond. The advantages of such properties of VII were demonstrated in the synthesis of a bioactive molecule in short steps. Thus, the silyl group VII has unveiled a new flexible synthetic strategy that allows the introduction and transformation of the silyl group in any stage of the synthesis.Author contributions
J. S. and H. S. conceived the project. J. S. and H. Y. directed the research. H. S. performed the experiments and computational studies. H. S. and J. S. composed the manuscript and the ESI section.† All authors contributed to the editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP18J22838, JP19H00895, and JS21H01934 and partly by JST CREST Grant Number JPMJCR19R4, Japan. H. S. acknowledges a JSPS Predoctoral Fellowship. H. S. deeply thanks Prof. Dr Satoshi Maeda and Dr Yosuke Sumiya for instructing him on how to use the GRRM17 program. J. S. acknowledges the support by The Sumitomo Foundation.Notes and references
- (a) Organometallics in Synthesis, ed. M. Schlosser, Wiley, Hoboken, NJ, 2013 Search PubMed; (b) Handbook of Functionalized Organometallics, ed. P. Knochel, Wiley, Weinheim, 2005 Search PubMed.
- (a) A. Suzuki and H. C. Brown, Organic Syntheses Via Boranes, Aldrich Chemical Company, Milwaukee, WI, 2003, vol. 3 Search PubMed; (b) N. Miyaura, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004 Search PubMed; (c) Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed; (d) Applied Cross-Coupling Reactions, ed. Y. Nishihara, Springer, Heidelberg, Germany, 2013 Search PubMed; (e) H.-Y. Sun and D. G. Hall, in Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, Springer, Heidelberg, Germany, 2015, p. 221 Search PubMed.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed; (b) E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17 CAS; (c) J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297 CrossRef CAS PubMed; (d) A. M. Kelly, P.-J. Chen, J. Klubnick, D. J. Blair and M. D. Burke, Org. Lett., 2020, 22, 9408 CrossRef CAS PubMed.
- (a) H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758 CrossRef CAS PubMed; (b) H. Noguchi, T. Shioda, C.-M. Chou and M. Suginome, Org. Lett., 2008, 10, 377 CrossRef CAS PubMed; (c) N. Iwadate and M. Suginome, Chem. Lett., 2010, 39, 558 CrossRef CAS; (d) T. Yamamoto, A. Ishibashi, M. Koyanagi, H. Ihara, N. Eichenauer and M. Suginome, Bull. Chem. Soc. Jpn., 2017, 90, 604 CrossRef CAS.
- (a) H. Yoshida, M. Seki, S. Kamio, H. Tanaka, Y. Izumi, J. Li, I. Osaka, M. Abe, H. Andoh, T. Yajima, T. Tani and T. Tsuchimoto, ACS Catal., 2020, 10, 346 CrossRef CAS; (b) Y. Mutoh, K. Yamamoto and S. Saito, ACS Catal., 2020, 10, 352 CrossRef CAS.
- (a) N. Konami, K. Matsuoka, T. Yoshino and S. Matsunaga, Synthesis, 2018, 50, 2067 CrossRef; (b) C. Fricke, A. Dahiya, W. B. Reid and F. Schoenebeck, ACS Catal., 2019, 9, 9231 CrossRef CAS PubMed; (c) C. Fricke, K. Deckers and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 132, 18876 CrossRef; (d) G. J. Sherborne, A. G. Gevondian, I. Funes-Ardoiz, A. Dahiya, C. Fricke and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 15543 CrossRef CAS PubMed; (e) C. Fricke and F. Schoenebeck, Acc. Chem. Res., 2020, 53, 2715 CrossRef CAS PubMed.
- (a) Organosilicon Chemistry, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019 Search PubMed; (b) T. Hiyama, in Organometallics in Synthesis Third Manual, ed. M. Schlosser, Wiley, Hoboken, 2013, p. 373 Search PubMed; (c) Science of Synthesis, ed. I. Fleming, Thieme, Stuttgart, 2002, vol. 4 Search PubMed; (d) M. A. Brook, in Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed; (e) Organosilicon Chemistry Set: From Molecules to Materials, ed. N. Auner and J. Weis, Wiley-VCH, Weinheim, 1994–2005 Search PubMed.
- (a) S. E. Denmark and J. Y. Choi, J. Am. Chem. Soc., 1999, 121, 5821 CrossRef CAS; (b) K. Itami, T. Nokami and J. Yoshida, J. Am. Chem. Soc., 2001, 123, 5600 CrossRef CAS PubMed; (c) K. Hosoi, K. Nozaki and T. Hiyama, Chem. Lett., 2002, 31, 138 CrossRef; (d) B. M. Trost, M. R. Machacek and Z. T. Ball, Org. Lett., 2003, 5, 1895 CrossRef CAS PubMed.
- (a) F. Lerouge, G. Cerveau and R. J. P. Corriu, New J. Chem., 2006, 30, 272 RSC; (b) T. Iwai, R. Tanaka, T. Harada and M. Sawamura, Chem.–Eur. J., 2014, 20, 1057 CrossRef CAS PubMed; (c) L. Li and N. Navasero, Org. Lett., 2004, 6, 3091 CrossRef CAS PubMed.
- (a) Y. Nakao, H. Imanaka, A. K. Sahoo, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 6952 CrossRef CAS PubMed; (b) Y. Nakao, A. K. Sahoo, A. Yada, J. Chen and T. Hiyama, Sci. Technol. Adv. Mater., 2006, 7, 536 CrossRef CAS; (c) Y. Nakao, M. Takeda, T. Matsumoto and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4447 CrossRef CAS PubMed; (d) Y. Nakao, J. Chen, M. Tanaka and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 11694 CrossRef CAS PubMed; (e) Y. Minami, K. Shimizu, C. Tsuruoka, T. Komiyama and T. Hiyama, Chem. Lett., 2014, 43, 201 CrossRef CAS; (f) Y. Nakao and T. Hiyama, J. Synth. Org. Chem., Jpn., 2011, 69, 1221 CrossRef CAS.
- (a) T. Shimada, K. Aoki, Y. Shinoda, T. Nakamura, N. Tokunaga, S. Inagaki and T. Hayashi, J. Am. Chem. Soc., 2003, 125, 4688 CrossRef CAS PubMed; (b) Y. Maegawa, M. Waki, A. Umemoto, T. Shimada and S. Inagaki, Tetrahedron, 2013, 69, 5312 CrossRef CAS; (c) M. P. Kapoor, S. Inagaki, S. Ikeda, K. Kakiuchi, M. Suda and T. Shimada, J. Am. Chem. Soc., 2005, 127, 8174 CrossRef CAS PubMed.
- (a) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed; (b) J. Morstein, H. Hou, C. Cheng and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 8054 CrossRef CAS PubMed; (c) J. Morstein, E. D. Kalkman, C. Bold, C. Cheng and J. F. Hartwig, Org. Lett., 2016, 18, 5244 CrossRef CAS PubMed.
- C. N. Foley and J. L. Leighton, Org. Lett., 2015, 17, 5858 CrossRef CAS PubMed.
- We also attempted the synthesis of phenylsilanes 1 bearing a 5-membered pinacolatosilyl group or 8-membered dioxasilocanyl group. Disappointingly, the pinacolatosilyl group that formed in the synthesis instantly decomposed upon contact with water. In contrast, the dioxasilocanyl moiety on 1 was not observed under the current synthetic conditions..
- S. Maeda, Y. Harabuchi, Y. Sumiya, M. Takagi, K. Suzuki, M. Hatanaka, Y. Osada, T. Taketsugu, K. Morokuma and K. Ohno, GRRM17, http://iqce.jp/GRRM/index_e.shtml, 1 Aug, 2018 Search PubMed.
- (a) S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC; (b) S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- These differences in ΔG values are not directly reflected in the empirically observed reaction rates of the decomposition of alkoxysilyl groups because this reaction consists of the initial nucleophilic attack of the silicon centre followed by the elimination of alkoxide. Despite the difficulties in the detailed computation of all transition states of the two consequential reactions, the formation of silicate is thermodynamically uphill in all three cases. Thus, based on Hammond's postulate, the order of activation barriers of nucleophilic attack was presumed to be proportional to the order of energies to form silicates..
- K. B. Wiberg, J. Org. Chem., 2003, 68, 9322 CrossRef CAS PubMed.
- A. Roushanbakhti, Y. Liu, P. C. M. Winship, M. J. Tucker, W. M. Akhtar, D. S. Walter, G. Wrigley and T. J. Donohoe, Angew. Chem., Int. Ed., 2017, 56, 14883 CrossRef CAS PubMed.
- (a) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679 CrossRef CAS; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872 CrossRef CAS PubMed; (c) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759 CrossRef CAS.
- A selective ligand transfer reaction that capitalizes on the stability and the bulkiness of the dioxasilepanyl skeleton has been reported: T. Ikeuchi, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2021, 143, 4879 CrossRef CAS PubMed.
- A. A. Toutov, K. N. Betz, M. C. Haibach, A. M. Romine and R. H. Grubbs, Org. Lett., 2016, 18, 5776 CrossRef CAS PubMed.
- (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS PubMed; (b) Y. Yang and C. Wang, Sci. China: Chem., 2015, 58, 1266 CrossRef CAS.
- (a) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592 CrossRef CAS PubMed; (b) C. Karmel, Z. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 7063 CrossRef CAS PubMed; (c) C. Karmel and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 10494 CrossRef CAS PubMed.
- For related work on non-directed C–H silylations with hydrosilanes, (a) Y. Minami, T. Komiyama and T. Hiyama, Chem. Lett., 2015, 44, 1065 CrossRef CAS; (b) M. Murai, K. Takami and K. Takai, Chem.–Eur. J., 2015, 21, 4566 CrossRef CAS PubMed.
- Monohydrosilanes are known to hardly undergo dehydrogenative homo-coupling of hydrosilanes. Actually, di- and triethoxyhydrosilanes we tested did not give the corresponding disilanes under our C–H silylation conditions. The exact reasons of the different reactivities toward C–H silylation among hydrosilanes are not clear in the current study. For details of dehydrogenative formation of disilanes from hydrosilanes, (a) K. A. Brown-Wensley, Organometallics, 1987, 6, 1590 CrossRef CAS; (b) L. Rosenberg, C. W. Davis and J. Yao, J. Am. Chem. Soc., 2001, 123, 5120 CrossRef CAS PubMed.
- P. Gigler, W. A. Herrmann and F. E. Kühn, Synthesis, 2010, 1431 CAS.
- A. De Mico, R. Margarita, L. Parlanti, A. Vescovi and G. Piancatelli, J. Org. Chem., 1997, 62, 6974 CrossRef CAS.
- (a) R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897 CrossRef CAS; (b) A. F. Abdel-Magid, C. A. Maryanoff and K. G. Carson, Tetrahedron Lett., 1990, 31, 5595 CrossRef CAS.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57 RSC.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508 CrossRef CAS.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- K. Matsubara, K. Ueno and Y. Shibata, Organometallics, 2006, 25, 3422 CrossRef CAS.
- (a) K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS; (b) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29 RSC.
- (a) J. H. Smitrovich and K. A. Woerpel, J. Org. Chem., 1996, 61, 6044 CrossRef CAS; (b) A. Kuznetsov and V. Gevorgyan, Org. Lett., 2012, 14, 914 CrossRef CAS PubMed.
- (a) Y. Hatanaka and T. Hiyama, Synlett, 1991, 845 CrossRef CAS; (b) T. Hiyama, J. Organomet. Chem., 2002, 653, 58 CrossRef CAS; (c) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (d) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS PubMed.
- M. E. Mowery and P. DeShong, J. Org. Chem., 1999, 64, 1684 CrossRef CAS PubMed.
- (a) H.-Q. Luo and W. Dong, Synth. Commun., 2013, 43, 2733 CrossRef CAS; (b) R. Müller, M. Dressler and C. Dathe, J. Prakt. Chem., 1970, 312, 150 CrossRef; (c) K. Tamao, H. Matsumoto, T. Kakui and M. Kumada, Tetrahedron Lett., 1979, 20, 1137 CrossRef.
- (a) D. S. Hewings, O. Fedorov, P. Filippakopoulos, S. Martin, S. Picaud, A. Tumber, C. Wells, M. M. Olcina, K. Freeman, A. Gill, A. J. Ritchie, D. W. Sheppard, A. J. Russell, E. M. Hammond, S. Knapp, P. E. Brennan and S. J. Conway, J. Med. Chem., 2013, 56, 3217 CrossRef CAS PubMed; (b) C. P. Seath, J. W. B. Fyfe, J. J. Molloy and A. J. B. Watson, Angew. Chem., Int. Ed., 2015, 54, 9976 CrossRef CAS PubMed; (c) C. W. Muir, J. C. Vantourout, A. Isidro-Llobet, S. J. F. Macdonald and A. J. B. Watson, Org. Lett., 2015, 17, 6030 CrossRef CAS PubMed; (d) L. E. Jennings, M. Schiedel, D. S. Hewings, S. Picaud, C. M. C. Laurin, P. A. Bruno, J. P. Bluck, A. R. Scorah, L. See, J. K. Reynolds, M. Moroglu, I. N. Mistry, A. Hicks, P. Guzanov, J. Clayton, C. N. G. Evans, G. Stazi, P. C. Biggin, A. K. Mapp, E. M. Hammond, P. G. Humphreys, P. Filippakopoulos and S. J. Conway, Bioorg. Med. Chem., 2018, 26, 2937 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterisation data, details of the X-ray analyses of compounds 5a, details of the computational study and NMR spectra. CCDC 1966652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02083h |
| This journal is © The Royal Society of Chemistry 2021 |