
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi(0)-promoted activation of Csp2–H and Csp2–O bonds†
Sehye
Min
a,
Jonghoon
Choi
b,
Changho
Yoo
c,
Peter M.
Graham
 *d and
Yunho
Lee
*b
*d and
Yunho
Lee
*b
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
bDepartment of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: yunhochem@snu.ac.kr; Tel: +82 2 880 6653
cGreen Carbon Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon 34114, Republic of Korea
dDepartment of Chemistry, Saint Joseph's University, 5600 City Avenue, Philadelphia, PA 19131, USA
First published on 17th June 2021
Abstract
A dinickel(0)–N2 complex, stabilized with a rigid acridane-based PNP pincer ligand, was studied for its ability to activate C(sp2)–H and C(sp2)–O bonds. Stabilized by a Ni–μ–N2–Na+ interaction, it activates C–H bonds of unfunctionalized arenes, affording nickel–aryl and nickel–hydride products. Concomitantly, two sodium cations get reduced to Na(0), which was identified and quantified by several methods. Our experimental results, including product analysis and kinetic measurements, strongly suggest that this C(sp2)–H activation does not follow the typical oxidative addition mechanism occurring at a low-valent single metal centre. Instead, via a bimolecular pathway, two powerfully reducing nickel ions cooperatively activate an arene C–H bond and concomitantly reduce two Lewis acidic alkali metals under ambient conditions. As a novel synthetic protocol, nickel(II)–aryl species were directly synthesized from nickel(II) precursors in benzene or toluene with excess Na under ambient conditions. Furthermore, when the dinickel(0)–N2 complex is accessed via reduction of the nickel(II)–phenyl species, the resulting phenyl anion deprotonates a C–H bond of glyme or 15-crown-5 leading to C–O bond cleavage, which produces vinyl ether. The dinickel(0)–N2 species then cleaves the C(sp2)–O bond of vinyl ether to produce a nickel(II)–vinyl complex. These results may provide a new strategy for the activation of C–H and C–O bonds mediated by a low valent nickel ion supported by a structurally rigidified ligand scaffold.
Introduction
Nickel has recently attracted much attention as the earth-abundant alternative to precious 4d and 5d transition metals for utilization in various organometallic catalysis.1 In particular, the nickel-mediated C–H and C–O bond functionalization of various organic molecules to afford new C–X bonds (X = C, N or O) offers considerable advantages.1–3 A zero-valent nickel species is often considered to be involved in activating both C–H and C–O bonds, which is coupled with 2-electron oxidation to give a Ni(II) d8 complex.4 Employing an appropriate ligand is crucial to accessing such states of a nickel species, which generally favours tetrahedral and square planar structures for the d10 and d8 electron configuration, respectively. Accordingly, the structural flexibility of the ancillary ligands to accommodate both oxidation states of nickel is an important design principle in organonickel catalyst development.Although nickel mediated unfunctionalized C(sp2)–H bond activation of arenes such as benzene is relatively rare, a success was previously reported by the Liang group.2,5a Addition of Lewis acidic boron and aluminum to a nickel(II) hydride or alkyl species was required to facilitate benzene C–H bond activation. The van der Vlugt group reported another C–H bond activation of benzene, which involves the formation of a transient Ni(IV) nitrido complex generated from photoinitiated conversion of a nickel(II)–azide complex.5b Although oxidative addition of a C–H bond occurring at a zero-valent nickel site has been hypothetically suggested4,6 and employed in C(sp2)–H borylation of arenes,7 C–H bond activation of benzene mediated by a nickel(0) species has never been experimentally presented. Furthermore, nickel is also known to effectively cleave C–O bonds.3 In most cases, such reactions take place with C(sp2)–O bonds including aryl,3,8 vinyl,9 and acyl10 C–O bonds. An important aspect of C(sp2)–O activation is the use of Lewis acidic boron, zinc or magnesium to help overcome the high-barrier of the oxidative addition of a C–O bond.3,8–10 The Martin group has proposed that the Lewis acidity of the Grignard reagent is critical in the Ni-catalyzed Kumada–Corriu reaction of cyclic vinyl ethers.9b While the Agapie group has shown that Ni(0) reacts with an aryl ether to give a Ni(II) aryl alkoxide species, as the C–O oxidative addition product,4d,e similar studies for Ni(0) mediated C–O bond activation of vinyl ethers are lacking.
Recently, our group reported a monovalent nickel complex (acriPNP)Ni·C10H8 (1·C10H8) supported by a structurally rigidified acriPNP ligand (acriPNP− = 4,5-bis(diisopropylphosphino)-2,7,9,9-tetramethyl-9H-acridin-10-ide).11 According to the cyclic voltammogram, this metalloradical species can be further reduced to Ni(0) at −2.80 V vs. Fc/Fc+ in tetrahydrofuran (THF), which enabled the current study of C–H and C–O activation. Herein, we present the synthesis of a zero-valent nickel dinitrogen complex, {(acriPNP)Ni–(N2)–Na(THF)2}2 (2) and its reactivity with arenes and organic ethers.
Results and discussion
Synthesis and characterization of a nickel(0) dinitrogen species
A nickel(I) complex 1·C10H8 was prepared according to the literature procedure,11 and its chemical reduction in THF was conducted by using 1 equivalent of sodium naphthalide under N2 atmosphere, Scheme 1. A new diamagnetic species displaying a 31P NMR peak at 48.1 ppm was isolated as a red solid in 78% yield. The single crystal X-ray structural data reveals the resulting product is a dinickel–N2 adduct, {(acriPNP)Ni–(N2)–Na(THF)2}2 (2, Fig. 1). A Nα atom (N2 in Fig. 1) of a dinitrogen molecule coordinates to a nickel ion with an end-on binding mode, while Nβ (N3) interacts with a sodium cation, as depicted in Fig. 1. There are two sodium ions in 2, and each connects the bridging N2 ligand and the amide nitrogen (N1) of an acriPNP ligand, constructing a dimeric assembly in the solid state. Along with an elongated N–N distance of 1.138(4) Å, the short Ni–Nα distance of 1.752(3) Å, the shortest in known four coordinate Ni–N2 complexes,12,13 suggests the nickel ions in 2 are fairly reduced. The N2 coordination was also confirmed by its IR data displaying an intense band at 1944 cm−1 (see Fig. S97†). Due to the rigidity of the ligand, each nickel ion reveals a fairly distorted tetrahedral geometry based on the τ4 value of 0.71 (ref. 14a) and a distorted sawhorse geometry with the τδ value of 0.67,14b significantly different from fifteen other crystallographically characterized nickel–dinitrogen complexes.12,13 Distortion from the planarity of the acriPNP ligand can be illustrated by the displacement of the central nitrogen atom from the imaginary plane defined by the geminal atoms, see Fig. 2. In 2, N1 is 0.25 Å away from the corresponding plane, which is similar to that of a nickel(0) monocarbonyl congener, {Na}{(acriPNP)Ni(CO)}, as shown Fig. 2b.15,16 Without sodium interaction, this distance is significantly reduced to 0.14 Å, see Fig. 2c. Sodium interaction, therefore, significantly affects the planarity of 2, which is expected to influence the stability and reactivity of the nickel dimer. Furthermore, unique structural features may be coined from the interaction between N2 and Na. Interaction of alkali metal cations can assist to promote the activation of N2.17 For example, the Limberg group reported the nickel dinitrogen complex, K2[LtBuNi(μ-η1:η1-N2)NiLtBu] (LtBu = [HC(CtBuN-C6H3(iPr)2)2]−) having a potassium interaction with N2 resulting in significantly lengthening the N–N bond to 1.185(8) Å.12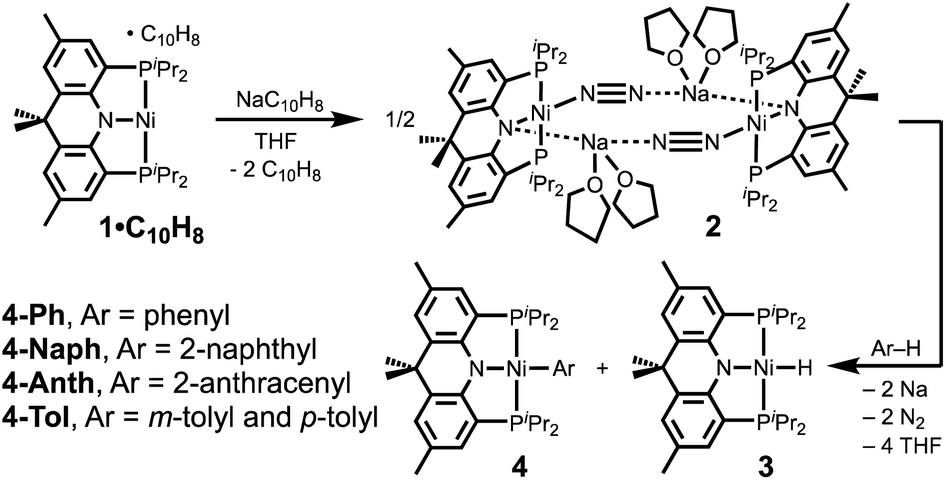 | ||
| Scheme 1 Preparation of a dimeric Ni–N2 complex 2 and its reactivity toward aromatic hydrocarbons. | ||
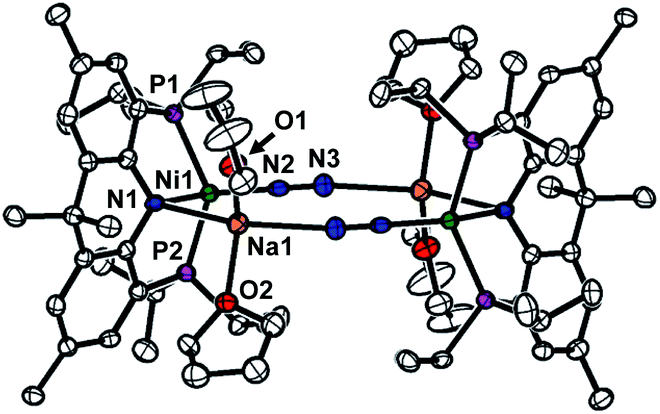 | ||
| Fig. 1 Displacement-ellipsoid (50%) representation of 2. All hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°) for 2: Ni1–N1 2.041(3), Ni1–P1 2.2022(1), Ni1–P2 2.2063(1), Ni1–N2 1.752(3), N2–N3 1.138(4), N1–Ni1–N2 126.81(1), P1–Ni1–P2 133.37(4). | ||
 | ||
| Fig. 2 Core structures of (a) 2, (b) {Na}{(acriPNP)Ni(CO)} and (c) {Na(12-C-4)2}{(acriPNP)Ni(CO)}.15 The distances of a N atom from the red plane defined by two carbon and one nickel are shown. | ||
To understand the effect of the sodium interaction, 12-crown-4 was added to the solution of 2 at −35 °C, revealing its slow decomposition, suggesting the cation interaction is crucial to its stability. To comprehend the solution-state structure, the diffusion coefficients of 2 with analogous compounds were collected in THF-d8. The data indicate a monomer, suggesting a mononuclear version of complex 2 is stabilized with Na–THF interactions, see Fig. S96 and Table S10.†
Reaction of 2 with aromatic hydrocarbons
When the red solid of 2 was dissolved in benzene at room temperature, an immediate reaction occurred, Scheme 1. The resulting mixture contains two clean products with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio according to the integration of the single-pulse 31P NMR spectrum.18 One product shows a doublet at 61.3 (JPH = 4.9 Hz) ppm in the 31P NMR spectrum, which corresponds to a nickel(II) hydride complex, (acriPNP)NiH (3).11 The other species displays a singlet at 39.3 ppm, which is (acriPNP)NiPh (4-Ph), confirmed by X-ray crystallography, as shown in Fig. 3a. This compound was independently prepared from the reaction of (acriPNP)NiCl with a phenyl Grignard reagent, see ESI.† This result is clearly different from the oxidative reactivity of 2 toward phenol or aniline resulting in the formation of 3 with a corresponding sodium salt, Scheme 2. Triphenylmethane having a moderately acidic proton was also tested. According to the product analysis, Ni–H and Ni–Ar were generated with a 1:1 ratio based on the integration of the corresponding 31P NMR signals, see Fig. S17 and S18.† In a THF solution, 2 is stable enough to be isolated, but the addition of benzene leads to activation of a benzene C–H bond. According to initial kinetic measurements using C6H6 and C6D6, a kinetic isotope effect (KIE) was determined as kH/kD = 3.5, see ESI.† The intermolecular KIE was also measured with a mixture of C6H6 and C6D6 (1:1). The value of 3.3 was obtained from the product distribution of 4-Ph and 4-C6D5 without any scrambling between products (see Fig. S58†). Both results suggest the C–H bond cleavage is the slowest step.19 This is a surprising result, which is different from the reactivity of the Ni(I) species. Compound 1 is stable in C6D6 at room temperature as well as elevated temperature at 60 °C for over 24 h. Furthermore, a Lewis acid such as a sodium cation does not induce the reactivity of 1 either, see ESI.†
:1 ratio according to the integration of the single-pulse 31P NMR spectrum.18 One product shows a doublet at 61.3 (JPH = 4.9 Hz) ppm in the 31P NMR spectrum, which corresponds to a nickel(II) hydride complex, (acriPNP)NiH (3).11 The other species displays a singlet at 39.3 ppm, which is (acriPNP)NiPh (4-Ph), confirmed by X-ray crystallography, as shown in Fig. 3a. This compound was independently prepared from the reaction of (acriPNP)NiCl with a phenyl Grignard reagent, see ESI.† This result is clearly different from the oxidative reactivity of 2 toward phenol or aniline resulting in the formation of 3 with a corresponding sodium salt, Scheme 2. Triphenylmethane having a moderately acidic proton was also tested. According to the product analysis, Ni–H and Ni–Ar were generated with a 1:1 ratio based on the integration of the corresponding 31P NMR signals, see Fig. S17 and S18.† In a THF solution, 2 is stable enough to be isolated, but the addition of benzene leads to activation of a benzene C–H bond. According to initial kinetic measurements using C6H6 and C6D6, a kinetic isotope effect (KIE) was determined as kH/kD = 3.5, see ESI.† The intermolecular KIE was also measured with a mixture of C6H6 and C6D6 (1:1). The value of 3.3 was obtained from the product distribution of 4-Ph and 4-C6D5 without any scrambling between products (see Fig. S58†). Both results suggest the C–H bond cleavage is the slowest step.19 This is a surprising result, which is different from the reactivity of the Ni(I) species. Compound 1 is stable in C6D6 at room temperature as well as elevated temperature at 60 °C for over 24 h. Furthermore, a Lewis acid such as a sodium cation does not induce the reactivity of 1 either, see ESI.†
 | ||
| Fig. 3 Displacement-ellipsoid (50%) representations of (a) 4-Ph, (b) 4-Naph, (c) 4-Anth, and (d) 4-mTol. Co-crystallized molecule and hydrogen atoms are omitted for clarity. | ||
 | ||
| Scheme 2 Reactivity of 2 toward benzene, phenol and aniline. | ||
Other polyaromatic hydrocarbons were also tested. The reaction of 2 with naphthalene in pentane led to the complete conversion to two products, (acriPNP)Ni(2-naphthyl) (4-Naph) and 3 in a 57:43 ratio18 after 40 h of stirring at room temperature, see ESI.† X-ray crystallographic data reveal that the selective C–H bond activation occurs at the 2-position of naphthalene, see Fig. 3b.20 Similarly, when anthracene was added, an analogous reaction took place to give (acriPNP)Ni(2-anthracenyl) (4-Anth). Both reactions conducted in pentane are significantly slower than those with benzene and toluene. This is probably due to the low concentration of substrates and the low solubility of a black precipitate generated from the reaction, vide infra, see ESI.† Interestingly, its X-ray structure reveals the same regioselectivity as shown in Fig. 3c. To test the possibility of C–H bond activation occurring at the 1-position of naphthalene, 1-naphthyl lithium was added to (acriPNP)NiCl. The corresponding reaction failed to give the desired product, instead resulting in decomposition, see ESI.† To assist in understanding these experimental results, geometry optimizations for the isomers of (acriPNP)Ni(naphthyl) and (acriPNP)Ni(anthracenyl) were conducted by using DFT calculations (see Fig. S107 and S108†). According to the top views shown in Fig. 4, the N–Ni–C angle of (acriPNP)Ni(1-naphthyl) is more bent than that of 4-Naph. In addition, the aryl ring with 1-naphthyl coordination is slightly tilted from the Ni–C axis, while all atoms are linearly aligned along the main axis of 4-Naph. The overall structure of (acriPNP)Ni(1-naphthyl) is noticeably twisted as indicated by the C–P–P–C dihedral angle of 118.9° because of close contact between C8 with isopropyl groups. In contrast, (acriPNP)NiCl shows the C–P–P–C dihedral angle of 129.5° with a perfect square planar nickel(II) structure. Comparing these structures, the 1-naphthyl coordination induces a significant steric influence to change the geometry of the nickel complex. The energy of (acriPNP)Ni(1-naphthyl) becomes slightly higher than that of 4-Naph by ∼2 kcal mol−1. The 1- and 2-anthracenyl analogues are almost identical to those with naphthyl complexes (see Fig. S108†). According to the DFT calculated structure of (acriPNP)Ni(9-anthracenyl), both C1 and C8 interactions with isopropyl groups significantly twist its structure, as depicted in Fig. 4d. Such steric factors may give selective product formation via the C–H bond activation at the 2-position of naphthalene and anthracene.
 | ||
| Fig. 4 Top views of optimized structures for (a) (acriPNP)NiCl,11 (b) 4-Naph, (c) (acriPNP)Ni(1-naphthyl), (d) (acriPNP)Ni(9-anthracenyl), (e) 4-pTol, (f) 4-mTol, (g) 4-oTol and (h) (acriPNP)Ni(benzyl) obtained from DFT calculations; distances between atoms in Å. | ||
To better understand the regioselectivity, the reaction of 2 with toluene having both sp2vs. sp3 C–H bonds was studied. When 2 was dissolved in toluene, an immediate conversion occurred displaying clean product formation. Two distinct (acriPNP)Ni(tolyl) complexes were detected in a 2.2:1 ratio,18 revealing two singlets at 39.0 and 39.1 ppm, along with a doublet at 61.3 ppm of 3 in the 31P NMR spectrum. To establish the identity of the resulting products, ortho-, meta- and para-tolyl and benzyl nickel(II) complexes were independently synthesized from the reactions of (acriPNP)NiCl with the corresponding reagent; tolyl lithium and a benzyl Grignard reagents, see ESI.† These complexes were fully characterized by using various spectroscopic methods and X-ray crystallography.
The product analysis established by 1H and 31P NMR spectroscopy reveals that the major and minor products are (acriPNP)Ni(m-tolyl) (4-mTol) and (acriPNP)Ni(p-tolyl) (4-pTol), respectively. Neither an ortho isomer nor a nickel benzyl species was detected, thus the selectivity is not directly related to the pKa value; that of a Cbenzyl–H bond is clearly lower than those of aryl C–H bonds, and similar results were previously observed with other metals.5a,21,22 A radical mechanism is also not likely, because a radical process would favour the weakest benzylic C–H bonds (BDE = 89.8 kcal mol−1) over aryl C–H bonds (BDE = 112.9 kcal mol−1).23 In fact, it is generally recognized that C–H bond activation of aromatic hydrocarbons is kinetically and thermodynamically favoured over aliphatic C–H bonds.24 DFT calculations support our experimental results with respect to product distribution: the energy of 4-mTol is similar, but marginally lower than that of 4-pTol by 0.2 kcal mol−1. The energies of (acriPNP)Ni(o-tolyl) (4-oTol) and (acriPNP)Ni(benzyl) are noticeably higher than that of 4-mTol by 3.8 kcal mol−1 and 7.8 kcal mol−1, respectively. While both 4-mTol and 4-pTol reveal their similarity in the solid-state structure, 4-oTol displays significant geometric distortion presumably due to a steric influence of a methyl group, Fig. 4. Due to the interaction of a phenyl ring with isopropyl groups, the benzyl coordination of (acriPNP)Ni(benzyl) dramatically distorts the planarity of the acriPNP ligand, as shown in Fig. 4h.
Detection of sodium
Interestingly, along with aryl C–H bond activation, the reaction of 2 with 1 equivalent of benzene displays two-electron oxidation for each nickel centre to generate 3 and 4-Ph. Based on the stoichiometry of this reaction, two equivalents of sodium are expected to be generated. Indeed, after dissolving 2 in neat benzene, a black precipitate noticeably formed within 15 min. If this precipitate was separated from the solution of 3 and 4-Ph after 15 min, no further reaction was detected for over 24 h, see ESI.†25 According to combustion analysis, no organic component was detected in the precipitate, see ESI.† To detect and quantify the precipitated sodium, we have employed naphthalene.26 The black powder was carefully washed with pentane, then the resulting solid was added to a naphthalene solution in THF, which reveals an immediate colour change to dark green. Upon addition of 18-crown-6 to this green solution, the UV-vis spectrum plainly displays a distinctive broad absorption at 830 nm, indicating the formation of an arene radical anion (see Fig. S94†). As controls, NaH and PhLi were subjected to a similar analysis, and no radical generation was observed. Furthermore, the corresponding naphthalide was quantified by using 1,1-diphenylethylene to give 70% yield, see ESI.† In addition, the quantitative analysis was conducted by using a benzophenone spectrophotometric method, which gave a 68% yield of sodium (see Fig. S92 and S93†). Finally, the formation of sodium(0) was further established by measuring the melting point of the resulting solid and detecting it with liquid ammonia, which gave the expected distinctive blue colour (see Fig. S95†).Based on our experimental results, we found that the initial reaction between 2 and benzene produces two equivalents of sodium, Scheme 3a. This is a striking result, because compound 2 is generated from sodium naphthalide having a potential at −3.10 V and it can reduce a sodium cation with −3.04 V vs. Fc/Fc+ in THF. If true, the Lewis acid interaction may play an important role during the C–H bond activation step, not only for cooperatively activating a C–H bond but also for accepting an electron. In order to test the Lewis acid effect, potassium and lithium analogues were also prepared, see ESI.† While these complexes are stable under ambient conditions, they react with benzene at a slower rate to give 3 and 4-Ph at elevated temperature, suggesting that the Lewis acid interaction within 2 is crucial in the bond activation, see ESI† for more information.
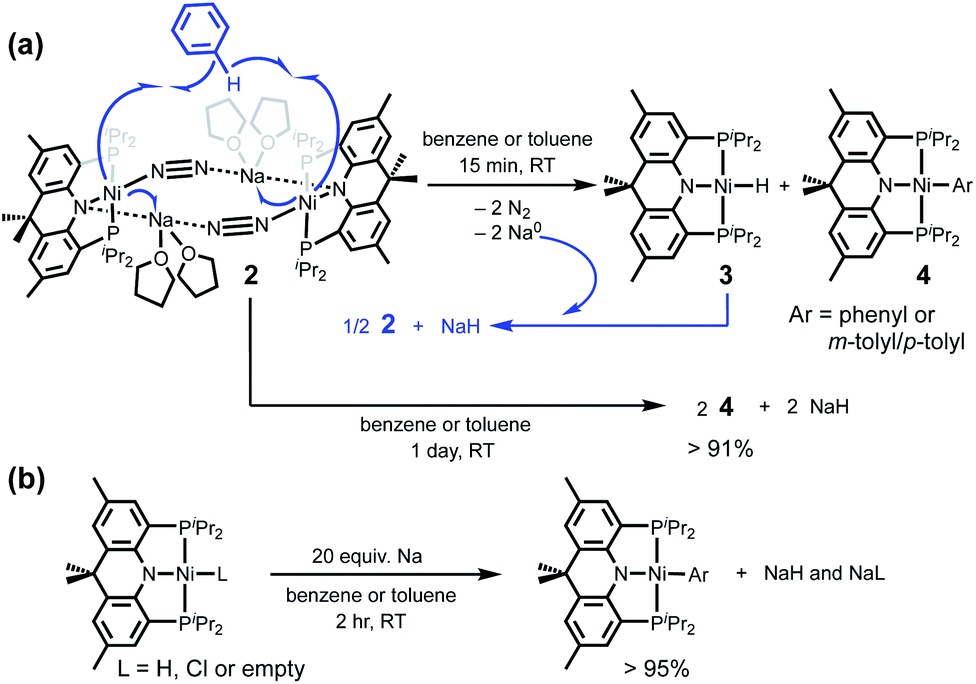 | ||
| Scheme 3 (a) Initial reaction of 2 with a benzene molecule occurring within 15 min, followed by the slow conversion to 4. (b) Quantitative preparation of nickel(II) aryl species from nickel(II or I) precursors. | ||
When the corresponding reaction was monitored for longer reaction time (>24 h), the product ratio changes with time (15 min vs. 24 h) to give a higher concentration of 4-Ph, Scheme 3a. After the reaction was completed in 24 h, a full conversion to 4-Ph was achieved with >91% yield, along with a grey precipitate.18 The corresponding solid was quantified with ethyl diethylphosphonoacetate revealing that NaH was generated in 81% yield (see Fig. S48†). Furthermore, the identity of the solid was also verified by solid-state 23Na NMR spectroscopy, see ESI.† This result suggests that the initial product 3 reacts with in situ formed sodium to re-generate a Ni(0) complex, while 4-Ph remains unchanged. According to cyclic voltammetric analysis, 3 has one irreversible wave appearing lower than −3.0 V vs. Fc/Fc+ (see Fig. S104†), which is lower than the reduction potential of −2.8 V for 1. Thus, when 3 is exposed to sodium, a nickel(0) species can be generated.
According to the net reaction, a nickel(0) dimer reacts with 1 equivalent of benzene to give Ni(II)–H (3) and Ni(II)–Ph (4-Ph) as depicted in Scheme 3a. This reaction does not proceed via the deprotonation of benzene, but occurs through the interaction of two nickel(0) ions with one benzene molecule leading to the formation of 4-Ph and 3 along with Na(0). Since 3 can be reduced by sodium, 0.5 equivalent of 2 is regenerated along with NaH. This process continues until all Na(0) is consumed to give 2 equivalents of 4-Ph and NaH, Scheme 3a. After knowing the reactivity of 2 and 3, we thought the direct synthesis of a nickel aryl complex via a nickel(0) mediated direct C–H bond activation would be possible. Indeed, when excess sodium (20 equivalents) was added to the benzene or toluene solution of (acriPNP)Ni–L (L = Cl, H or empty), the reaction was completed within 2 h at room temperature to produce corresponding (acriPNP)Ni–Ar (Ar = phenyl or tolyl) with >95% yield, Scheme 3b.
Lastly, the Ni(II)–aryl synthesis is possible under following conditions; first, the nickel(0) centre can activate a C–H bond of arene. Second, both Ni(II) and Ni(I) can be reduced to a Ni(0) species by sodium. Finally, the resulting nickel–aryl compounds are stable under reducing conditions in the absence of ether solvents. In the case of naphthalene and anthracene, excess sodium reacts with the corresponding nickel(II) aryl complexes due to their reduction potential.27
Reaction of 4-Ph with glyme and 15-crown-5
While (acriPNP)NiPh (4-Ph) is surprisingly stable towards excess sodium in benzene, it reacts with ethylene-linked ethers such as glyme and 15-crown-5, Scheme 4. For example, upon addition of 2 equivalents of sodium naphthalide to a THF solution of 4-Ph in the presence of glyme, a new yellow product featuring a 31P NMR resonance at 40.8 ppm is the major nickel-containing product, which can be isolated in >50% yield after silica chromatography, the other product is 3, see ESI.† The 1H NMR spectrum displays multiplets at 7.13, 5.89 and 5.49 ppm, that correspond to a CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 moiety. According to the COSY and HSQC 2D NMR spectral analysis (see Fig. S65 and S66†), the product is a nickel(II) vinyl complex, (acriPNP)Ni(CHCH2) (5). Single crystals were obtained by slow vapor diffusion of acetonitrile into a toluene solution of 5 at −35 °C. Its solid-state structure features two molecules of 5 in the asymmetric unit, one with crystallographic positional disorder and one without, as depicted in Fig. S88.† The N–Ni–C and P–Ni–P angles are 178.5(1)° and 172.43(3)°, respectively with the Ni–C distance of 1.890(3) Å, clearly indicating the expected Ni(II) square planar geometry, as depicted in Fig. 5a. In the disordered molecule, the vinyl group adopts two orientations that are 90° from the (acriPNP)Ni plane, as depicted in Fig. S88.† The disorder can be straightforwardly modelled but does lead to an erroneously short C–C bond (1.313(5) and 1.268(13) Å), as expected.28 Fortunately, the other molecule does not show this positional disorder, and unambiguously indicates a carbon–carbon double bond with 1.337(4) Å, a normal CC double bond. According to the DFT analysis, the corresponding ethylene moiety is orthogonal to the xy plane of nickel, due to the π interaction between a Ni dxy orbital and an ethylene π-orbital found in the HOMO-2 and HOMO-10, while the HOMO-8 reveals a σ-bonding character, as shown in Fig. 5. Nickel vinyl complexes can be prepared in more straightforward ways, e.g. by deprotonation of a cationic carbene,29 by the insertion of an alkyne into a nickel hydride,30 or via the oxidative addition of vinyl halides.31
CH2 moiety. According to the COSY and HSQC 2D NMR spectral analysis (see Fig. S65 and S66†), the product is a nickel(II) vinyl complex, (acriPNP)Ni(CHCH2) (5). Single crystals were obtained by slow vapor diffusion of acetonitrile into a toluene solution of 5 at −35 °C. Its solid-state structure features two molecules of 5 in the asymmetric unit, one with crystallographic positional disorder and one without, as depicted in Fig. S88.† The N–Ni–C and P–Ni–P angles are 178.5(1)° and 172.43(3)°, respectively with the Ni–C distance of 1.890(3) Å, clearly indicating the expected Ni(II) square planar geometry, as depicted in Fig. 5a. In the disordered molecule, the vinyl group adopts two orientations that are 90° from the (acriPNP)Ni plane, as depicted in Fig. S88.† The disorder can be straightforwardly modelled but does lead to an erroneously short C–C bond (1.313(5) and 1.268(13) Å), as expected.28 Fortunately, the other molecule does not show this positional disorder, and unambiguously indicates a carbon–carbon double bond with 1.337(4) Å, a normal CC double bond. According to the DFT analysis, the corresponding ethylene moiety is orthogonal to the xy plane of nickel, due to the π interaction between a Ni dxy orbital and an ethylene π-orbital found in the HOMO-2 and HOMO-10, while the HOMO-8 reveals a σ-bonding character, as shown in Fig. 5. Nickel vinyl complexes can be prepared in more straightforward ways, e.g. by deprotonation of a cationic carbene,29 by the insertion of an alkyne into a nickel hydride,30 or via the oxidative addition of vinyl halides.31
 | ||
| Scheme 4 Synthesis of a nickel(II) vinyl complex from glyme or 15-crown-5. | ||
 | ||
| Fig. 5 (a) Displacement-ellipsoid (50%) representation of 5. All hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°) for 5: Ni1–N1 1.932(2), Ni1–P1 2.1611(8), Ni1–P2 2.1677(8), Ni1–C30 1.890(3), C30–C31 1.337(4), N1–Ni1–C30 178.5(1), P1–Ni1–P2 172.43(3), Ni1–C30–C31 129.9(3). DFT-calculated (b) HOMO-2, (c) HOMO-8, and (d) HOMO-10 of 5. | ||
In order to probe the origin of the vinyl moiety in 5, the reduction was performed using the deuterated phenyl complex (acriPNP)Ni(Ph-d5) and glyme or 15-crown-5, which gave only a protio–vinyl complex, see ESI.† In addition, no vinyl complex was detected, when 4-Ph was reduced in the absence of glyme or 15-crown-5. Thus, the formation of 5 involves the abstraction of the ethylene bridge from glyme or crown ether, which requires the cleavage of two alkyl ether C–O bonds. Monitoring the reduction of 4-Ph in glyme by 31P, 1H NMR, and IR spectroscopy reveals that {(acriPNP)Ni–(N2)–Na(THF)2}2 (2) is initially formed, and this species persists for hours at −35 °C, even in the presence of glyme. This implies that phenyl sodium is produced and responsible for the deprotonation of ether, Scheme 5a. We believe that upon addition of one electron to the dx2–y2 orbital of a Ni(II)–Ph species, the phenyl moiety becomes more nucleophilic and/or departs from the nickel site, due to the weaker bonding character between Ni and a Ph ligand. According to the literature, such alkali alkyls and aryls are known to react with ethers to produce alkenes32 and in particular fragment glyme33 and 15-crown-5,34Scheme 5b. To confirm this, we added phenyl lithium to glyme or 15-crown-5 and found that it gives rise to alkyl vinyl ether via deprotonation followed by C–O bond cleavage, see ESI.† Importantly, when such in situ-generated vinyl ethers are exposed to 2, the vinyl species 5 is isolated in 46–60% yield along with sodium methoxide or tetraethylene glycol disodium salt as by-products, Scheme 5c. In the absence of PhLi, however, the reaction of 2 with glyme or crown only gives a trace of a vinyl species. Considering the second C–O bond cleavage, we have tested the reaction of 2 with ethyl vinyl ether, which generated 5 in 50% yield, see ESI.† This reaction does not occur with the Ni(I) species (acriPNP)Ni·C10H8 (1·C10H8), suggesting Ni(0) is crucial in the C–O cleavage of vinyl ether. To our knowledge, the isolation of a monomeric vinyl complex from the reaction of Ni(0) and a vinyl ether is unprecedented.9 In general, when acyclic vinyl ethers react with transition metal complexes, metal alkoxide species are the major product, concomitant with loss of alkene.35 Recently, the Fujita group reported that [(η5-C5Me5Ir)2(μ-bis(dimethylphosphino)methane) (μ-H)][OTf] reacts with ethyl vinyl ether to give a dimeric Ir complex featuring a bridging vinyl ligand.36 Conveniently, the vinyl complex 5 can be prepared in >93% yield via the reduction of (acriPNP)NiCl with a magnesium powder in the presence of ethyl vinyl ether, see ESI.† This result may verify that a low-valent nickel species favours nucleophilic reaction with vinyl ether to cleave a C–O bond.
 | ||
| Scheme 5 Fragmentation of glyme or crown ether by 4-Phvia2. | ||
Finally, in the presence of ether, the reduction of 4-Ph with two equivalents of sodium naphthalide generates 2 and phenyl sodium. The strongly basic phenyl anion begins the fragmentation of the ether via initial deprotonation at a carbon alpha to oxygen, which fragments to form a vinyl ether, thus providing a C(sp2)–O bond that can be broken by 2. The reaction of vinyl ether with 2 gives a nickel(II) vinyl complex. Clearly, the (acriPNP)Ni scaffold provides an electron-rich reactive nickel site not only capable of cleaving C(sp2)–H bonds but also C(sp2)–O bonds.
Conclusions
We present the discovery of an isolable sodium bridged dinuclear Ni–N2 complex capable of C(sp2)–H and C(sp2)–O bond activation. By employing a (acriPNP)Ni scaffold, a zero-valent nickel–N2 species has been prepared and stabilized by the interaction with sodium cations. Exposure of the dinuclear Ni(0) species toward arenes results in C–H bond activation at room temperature to give Ni(II)–aryl and Ni(II)–H products. Surprisingly, the electrons required to break the C–H bond are supplied by nickel as demonstrated by the by-product sodium(0), which has been carefully identified and quantified by multiple experimental methods. The reaction can be driven to produce exclusively Ni(II)–phenyl and –tolyl species using excess sodium in arene solvent. In THF, the Ni(II)–phenyl species can be reduced back to Ni(0) concomitant with the formation of a phenyl anion. This combination allows the abstraction of the ethylene linker from glyme or crown ether compounds to produce a Ni(II) vinyl complex via a C–O bond activation of in situ-generated vinyl ether. The zero-valent (acriPNP)Ni scaffold, therefore, creates a remarkably electron rich Ni core that can affect unfunctionalized arene C–H and ether C–O bond cleavage.Author contributions
S. M., J. C., C. Y., and P. M. G. performed the experiments and analysed the data. S. M., P. M. G., and Y. L. prepared the manuscript. All authors contributed to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea grant funded by the Korea government (MSIT; Grant NRF-2018R1A5A1025208), C1 Gas Refinery Program (NRF-2021M3D3A1A01022098), and LG Chemical.Notes and references
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS; (b) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed; (c) A. D. Marchese, T. Adrianov and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 2–15 CrossRef.
- (a) S. A. Johnson, Dalton Trans., 2015, 44, 10905–10913 RSC; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (c) N. A. Harry, S. Saranya, S. M. Ujwaldev and G. Anilkumar, Catal. Sci. Technol., 2019, 9, 1726–1743 RSC and references therein..
- (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC; (c) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS; (d) S.-Q. Zhang and X. Hong, Acc. Chem. Res., 2021, 9, 2158–2171 CrossRef PubMed.
- (a) S. A. Johnson, C. W. Huff, F. Mustafa and M. Saliba, J. Am. Chem. Soc., 2008, 130, 17278–17280 CrossRef CAS PubMed; (b) X.-J. Liu, Y.-Y. Tian, H.-Q. Cui and H.-J. Fan, Chem. Commun., 2018, 54, 7912–7915 RSC; (c) J. B. Diccianni and T. Diao, Trends Chem., 2019, 1, 830–844 CrossRef CAS; (d) P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480–5483 CrossRef CAS PubMed; (e) G. A. Edouard, P. Kelley, D. E. Herbert and T. Agapie, Organometallics, 2015, 34, 5254–5277 CrossRef CAS.
- (a) L.-C. Liang, P.-S. Chien and Y.-L. Huang, J. Am. Chem. Soc., 2006, 128, 15562–15563 CrossRef CAS PubMed; (b) V. Vreeken, M. A. Siegler, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 7055–7059 CrossRef CAS PubMed.
- M. Reinhold, J. E. McGrady and R. N. A. Perutz, J. Am. Chem. Soc., 2004, 126, 5268–5276 CrossRef CAS PubMed.
- (a) T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC; (b) A. Das, P. K. Hota and S. K. Mandal, Organometallics, 2019, 38, 3286–3293 CrossRef CAS.
- (a) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed; (b) H. Saito, S. Otsuka, K. Nogi and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 15315–15318 CrossRef CAS PubMed; (c) M. Tobisu, T. Takahira, T. Morioka and N. Chatani, J. Am. Chem. Soc., 2016, 138, 6711–6714 CrossRef CAS PubMed; (d) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6757 CrossRef CAS PubMed.
- (a) E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246–2247 CrossRef CAS; (b) J. Cornella and R. Martin, Org. Lett., 2013, 15, 6298–6301 CrossRef CAS PubMed; (c) M. M. Shoshani, V. Semeniuchenko and S. A. Johnson, Chem.–Eur. J., 2018, 24, 14282–14289 CrossRef CAS PubMed.
- L. Hie, N. F. Fine Nathel, X. Hong, Y.-F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810–2814 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502–9506 CrossRef CAS PubMed.
- S. Pfirrmann, C. Limberg, C. Herwig, R. Stöβer and B. Ziemer, Angew. Chem., Int. Ed., 2009, 48, 3357–3361 CrossRef CAS PubMed.
- (a) Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11561 RSC and references therein;; (b) R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12489 CrossRef PubMed; (c) Y.-E. Kim, S. Oh, S. Kim, O. Kim, S. W. Han and Y. Lee, J. Am. Chem. Soc., 2015, 137, 4280–4283 CrossRef CAS PubMed; (d) B. E. Cowie and D. J. H. Emslie, Organometallics, 2015, 34, 4093–4101 CrossRef CAS; (e) D. J. Charboneau, D. Balcells, N. Hazari, H. M. C. Lant, J. M. Mayer, P. R. Melvin, N. Q. Mercado, W. D. Morris, M. Repisky and H.-W. Suh, Organometallics, 2016, 35, 3154–3162 CrossRef CAS.
- (a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC; (b) M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 CrossRef CAS PubMed.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2017, 140, 2179–2185 CrossRef PubMed.
- With a nickel(II) ion, the central N atom is perfectly located within the plane (Fig. S89d†). To see any change in the ligand, the structural data of 2 was compared with those of 1 and 4-Ph, see Fig. S90.† Based on XRD data, there is no significant alteration in the acriPNP ligand of 2.
- G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- The quantitative analysis data for the resulting products were obtained by using single-pulse 31P NMR spectroscopy in the presence of triphenylphosphine oxide as an internal standard, see ESI.†.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS.
- The bond dissociation energies for both 1- and 2-position of naphthalene are almost identical, suggesting steric factor may induce the selectivity; D. R. Reed and S. R. Kass, J. Mass Spectrom., 2000, 35, 534–539 CrossRef CAS.
- (a) L. Johansson, O. B. Ryan, C. Rømming and M. Tilset, J. Am. Chem. Soc., 2001, 123, 6579–6590 CrossRef CAS PubMed; (b) F. Zhang, C. W. Kirby, D. W. Hairsine, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2005, 127, 14196–14197 CrossRef CAS PubMed; (c) O. Rivada-Wheelaghan, M. A. Ortuño, J. Díez, A. Lledós and S. Conejero, Angew. Chem., Int. Ed., 2012, 51, 3936–3939 CrossRef CAS PubMed; (d) M. A. Esteruelas, M. Oliván and A. Vélez, Organometallics, 2015, 34, 1911–1924 CrossRef CAS.
- For better understanding of the reaction mechanism, electron paramagnetic resonance experiments were conducted. No meaningful signal representing a paramagnetic species was detected, see ESI.† This is probably due to the dimeric nature of compound 2. This result may indicate that the mechanism is related to the previously reported reaction; W. Huang, F. Dulong, S. I. Khan, T. Cantat and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 17410–17413 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437–2450 RSC.
- To check if any reaction occurs, authentic compounds, 3 and 4-Ph, were mixed in the presence of 1 equivalent of naphthalene in C6D6. We have employed both 1H and 31P NMR spectroscopy to detect any changes in the concentration of 3 and 4-Ph relative to triphenylphosphine oxide as an internal standard. The single-pulse 31P NMR spectra were collected revealing almost identical integrations as ∼1.2 for the peak of 3 at 61 ppm and ∼1.1 for the peak of 4-Ph at 39 ppm relative to the peak of triphenylphosphine oxide at 25 ppm for 24 h at room temperature. Their 1H NMR data also do not exhibit any noticeable change. The same experiment was conducted at 60 °C revealing no reaction at the elevated temperature.
- M. Castillo, A. J. Metta-Magaña and S. Fortier, New J. Chem., 2016, 40, 1923–1926 RSC.
- The reduction potential of benzene in dimethyl ether is −3.93 V (vs. Fc/Fc+ couple); J. Mortensen and J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 84–85 CrossRef CAS . The reduction potential of naphthalene in dimethyl ether is −3.14 V and for anthracene −2.47 V (vs. Fc/Fc+ couple); N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef PubMed.
- M. B. Hall, S. Niu and J. H. Reibenspies, Polyhedron, 1999, 18, 1717–1724 CrossRef CAS.
- (a) K. Oguro, M. Wada and R. Okawara, J. Organomet. Chem., 1978, 159, 417–429 CrossRef CAS; (b) D. Xu, K. Miki, M. Tanaka, N. Kasai, N. Yasuoka and M. Wada, J. Organomet. Chem., 1989, 371, 267–277 CrossRef CAS.
- L. She, X. Li, H. Sun, J. Ding, M. Frey and H. Klein, Organometallics, 2007, 26, 566–570 CrossRef CAS.
- E. E. Marlier, S. J. Tereniak, K. Ding, J. E. Mulliken and C. C. Lu, Inorg. Chem., 2011, 50, 9290–9299 CrossRef CAS PubMed.
- D. Seyferth, Organometallics, 2006, 25, 2–24 CrossRef CAS.
- J. J. Fitt and H. W. Gschwend, J. Org. Chem., 1984, 49, 209–210 CrossRef CAS.
- (a) Z. Grobelny, A. Stolarzewicz and M. Czaja, J. Org. Chem., 1999, 64, 8990–8994 CrossRef CAS; (b) Z. Grobelny, A. Stolarzewicz and A. Maercker, J. Organomet. Chem., 2000, 604, 283–286 CrossRef CAS.
- (a) S. Komiya, R. S. Srivastava, A. Yamamoto and T. Yamamoto, Organometallics, 1985, 4, 1504–1508 CrossRef CAS; (b) B.-J. Deelman, M. Booij, A. Meetsma, J. H. Teuben, H. Kooijman and A. L. Spek, Organometallics, 1995, 14, 2306–2317 CrossRef CAS; (c) R. J. Trovitch, E. Lobkovsky, M. W. Bouwkamp and P. J. Chirik, Organometallics, 2008, 27, 6264–6278 CrossRef CAS; (d) S. Kiyota, T. Kobori, H. Soeta, Y.-i. Ichikawa, N. Komine, S. Komiya and M. Hirano, Polyhedron, 2016, 120, 3–10 CrossRef CAS.
- Y. Takahashi, T. Shimbayashi and K.-i. Fujita, Inorganics, 2019, 7, 121–131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for 2, 4-Ph, 4-Naph, 4-oTol, 4-mTol, 4-pTol and 5. CCDC 1968048–1968055 and 2078589. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02210e |
| This journal is © The Royal Society of Chemistry 2021 |