
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An intramolecularly self-templated synthesis of macrocycles: self-filling effects on the formation of prismarenes†
Paolo
Della Sala‡
 a,
Rocco
Del Regno‡
a,
Luca
Di Marino
a,
Carmela
Calabrese
a,
Carmine
Palo
a,
Carmen
Talotta
a,
Silvano
Geremia
*b,
Neal
Hickey
b,
Amedeo
Capobianco
a,
Placido
Neri
a and
Carmine
Gaeta
*a
a,
Rocco
Del Regno‡
a,
Luca
Di Marino
a,
Carmela
Calabrese
a,
Carmine
Palo
a,
Carmen
Talotta
a,
Silvano
Geremia
*b,
Neal
Hickey
b,
Amedeo
Capobianco
a,
Placido
Neri
a and
Carmine
Gaeta
*a
aDipartimento di Chimica e Biologia, “A. Zambelli” Università di Salerno Via Giovanni Paolo II, I-84084, Fisciano, Italy. E-mail: cgaeta@unisa.it
bCentro di Eccellenza in Biocristallografia, Dipartimento di Scienze Chimiche e Farmaceutiche Università di Trieste Via L. Giorgieri 1, I-34127, Trieste, Italy. E-mail: sgeremia@units.it
First published on 3rd June 2021
Abstract
Ethyl- and propyl-prism[6]arenes are obtained in high yields and in short reaction times, independent of the nature and size of the solvent, in the cyclization of 2,6-dialkoxynaphthalene with paraformaldehyde. PrS[6]Et or PrS[6]nPr adopt, both in solution and in the solid state, a folded cuboid-shaped conformation, in which four inward oriented alkyl chains fill the cavity of the macrocycle. On these bases, we proposed that the cyclization of PrS[6]Et or PrS[6]nPr occurs through an intramolecular thermodynamic self-templating effect. In other words, the self-filling of the internal cavity of PrS[6]Et or PrS[6]nPr stabilizes their cuboid structure, driving the equilibrium toward their formation. Molecular recognition studies, both in solution and in the solid state, show that the introduction of guests into the macrocycle cavity forces the cuboid scaffold to open, through an induced-fit mechanism. An analogous conformational change from a closed to an open state occurs during the endo-cavity complexation process of the pentamer, PrS[5]. These results represent a rare example of a thermodynamically controlled cyclization process driven through an intramolecular self-template effect, which could be exploited in the synthesis of novel macrocycles.
Introduction
Since the birth of supramolecular chemistry, the field of macrocyclic hosts and related host–guest complexes has grown considerably.1,2 The internal cavity of macrocyclic hosts is reminiscent of the binding sites of natural bioreceptors and for this reason macrocycles have been the focus of biomimetic chemistry research.3 The equilibrium of inclusion of a guest inside the host cavity is thermodynamically influenced by many factors, such as host–guest desolvation processes, ion pair rupture, conformational control of the host and guest, and host/guest weak interactions.4,5 The strength of such non-covalent interactions in guest@host complexes plays a crucial role in overcoming the other unfavorable thermodynamic factors.4–6 In general, an extended contact area4,5 between the host and guest and a high level of preorganization of the host can lead to stronger complexation. Aromatic cyclophane7 macrocycles, such as calixarenes,8 resorcinarenes,7 and pillararenes,9 show a variety of deep cavities that can wrap the included guest. Recently, naphthol-based macrocycles with a deeper π-cavity have generated a great deal of interest. Thus, oxatubarenes, naphthotubes, and naphthocages were reported by Jiang and coworkers, which show interesting conformational properties and recognition abilities.10–16The scientific interest toward the design of new macrocyclic hosts17 with deep cavities and conformational adaptive behavior16 has recently prompted us to synthesize new examples of naphthol-based macrocycles named calix[2]naphth[2]arene18 and prismarenes (Fig. 1).19,20 The first report on prismarenes was mainly focused on the synthesis and recognition properties of the pentameric PrS[5]Me macrocycle (Fig. 1) constituted by 1,5-methylene bridged 2,6-dimethoxynaphthalene units.19 With the aim to explore the supramolecular properties of the larger hexameric PrS[6] macrocycle, we decided to investigate its efficient synthesis by changing the solvent, templating agents, and alkoxy-chains. In this way, a rare and peculiar intramolecularly self-templated synthesis was discovered. 2,6-Dimethoxynaphthalene-based prism[n]arenes PrS[n]Me (Fig. 1) were obtained by a thermodynamically controlled cyclization process (Scheme 1).21a,22
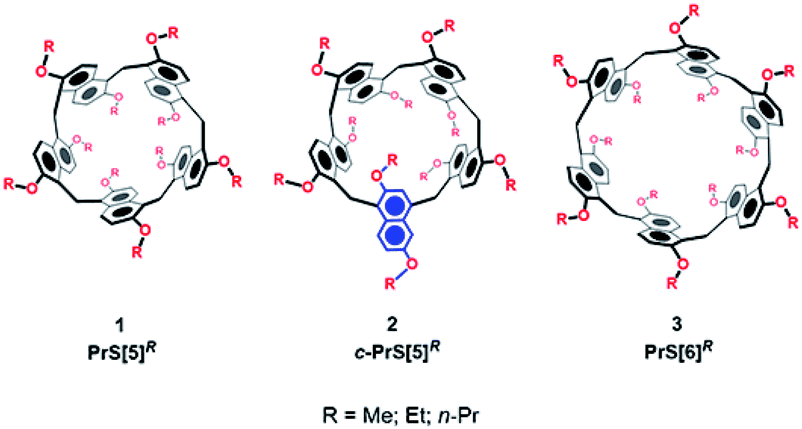 | ||
| Fig. 1 The prism[n]arene family. | ||
 | ||
| Scheme 1 Synthesis of PrS[n]R prismarenes. | ||
Starting from 4a, under the conditions reported in Scheme 1, the isomeric prismarenes PrS[5]Me and c-PrS[5]Me (the isomer with one 1,4 methylene bridge unit) were, respectively, the kinetic and the thermodynamic adduct.
Specifically, the pentamer PrS[5]Me was formed faster than its confused isomer c-PrS[5]Me, and DFT calculations confirmed that the transition state for the formation of the confused isomer was higher in energy than that for PrS[5]Me.19 In addition, DFT calculations confirmed the crucial role played by the solvent 1,2-DCE in the macrocyclization steps for the formation of the two isomeric PrS[5]Me and c-PrS[5]Me pentamers.19 Under the conditions reported in Scheme 1, and in the presence of the solvent 1,2-DCE, the most stable product c-PrS[5]Me prevailed over time (Table 1, entry 1). When the template 52+ or 6+ (Scheme 1 and Table 1, entries 2 and 3) was added to the reaction mixture in 1,2-DCE, the formation of endo-cavity inclusion complexes, 52+@PrS[5]Me or 6+@PrS[6]Me, respectively, stabilized the desired product via supramolecular interactions,19 and shifted the equilibrium toward PrS[5]Me (Table 1, entry 2) or PrS[6]Me (Table 1, entry 3), respectively (thermodynamic templating21b).19 The crucial factor which determines the greater thermodynamic stability of c-PrS[5]Me in the equilibrium mixture (Scheme 1), in the absence of guest 52+ or 6+, is its ability to self-fill the internal cavity.19 In fact, the 1,4-naphthalene ring assumes an orientation in which the 2-OMe group is inward oriented with respect to the cavity and completely fills it.19
| Entrya | Solvent | Monomer | Time | c-PrS[5]R | PrS[5]R | PrS[6]R |
|---|---|---|---|---|---|---|
| a [Monomer] = 5 mM; TFA and paraformaldehyde; 70 °C. b In the presence of 52+. c In the presence of 6+. d In the presence of 72+ (see Scheme 2) as an iodide salt; derivatives PrS[5]nPr and PrS[6]nPr were obtained in 10% and 47% yield, respectively. | ||||||
| 1 | 1,2-DCE | 4a (ref. 19) | 22 h | 40 | 0.3 | — |
| 2 | 1,2-DCE | 4a (ref. 19)b | 22 h | 16 | 47 | — |
| 3 | 1,2-DCE | 4a (ref. 19)c | 72 h | 6.0 | 0.3 | 20 |
| 4 | 1,2-DCE | 4b | 45 min | — | — | 75 |
| 5 | 1,2-DCE | 4c | 90 min | — | — | 65 |
| 6 | 1,2-DCE | 4b | 22 h | — | 10 | 35 |
| 7 | 1,2-DCE | 4b | 22 h | — | — | 60 |
| 8 | 1,2-DCE | 4c | 22 h | — | 25 | 25 |
| 9 | 1,2-DCE | 4c , | 22 h | — | — | 55 |
| 10 | 1,2-DCE | 4d | 40 min | — | — | 30 |
| 11 | 1,2-DCE | 4e | 12 h | — | — | 8 |
| 12 | Cl-CyHex | 4b | 40 min | — | — | 71 |
| 13 | Cl-CyHex | 4c | 2 h | — | — | 64 |
| 14 | Toluene | 4a | 24 h | 11 | — | — |
| 15 | Toluene | 4b | 3 h | — | — | 74 |
| 16 | Toluene | 4c | 8 h | — | — | 57 |
| 17 | CyHex | 4a | 24 h | 7 | — | — |
| 18 | CyHex | 4b | 30 min | — | — | 76 |
| 19 | CyHex | 4c | 40 min | — | — | 65 |
| 20 | Decaline | 4b | 30 min | — | — | 75 |
| 21 | Decaline | 4c | 45 min | — | — | 70 |
Considering these results, the question arises as to whether the pentamers and their confused isomers are still the kinetic and thermodynamic products, respectively, when starting from 4b–e bearing longer alkoxy groups. In other words, can the length of alkoxy substituents influence the synthesis, the conformational, and supramolecular properties of prismarenes?
Results and discussion
Synthesis of ethoxy- and propoxy-prismarenes
As previously reported, when methoxy-derivative 4a (ref. 19) was reacted in the presence of paraformaldehyde and TFA in 1,2-DCE as the solvent, c-PrS[5]Me and its PrS[5]Me isomer were formed in 40% and 0.3% yield (Table 1, entry 1), respectively, while no trace of PrS[6]Me hexamer was detected in the reaction mixture.19 We showed that under these conditions, c-PrS[5]Me was the thermodynamic product, while PrS[5]Me was the kinetic one and its formation was templated by the 1,2-DCE solvent. Thus, under these conditions, 1,2-DCE didn't act as a template for the prism[6]arene methoxy derivative.19 Differently, when 2,6-diethoxynaphthalene 4b was reacted in 1,2-DCE (Table 1, entry 4), unexpectedly, the PrS[6]Et hexamer was formed in a very high yield (75%) after 45 min. Analogously, when 2,6-dipropoxynaphthalene 4c was used as the precursor, the PrS[6]nPr hexamer was again the favored product (65% yield) after 90 min (Table 1, entry 5). These results were unexpected because, according to the data previously reported by Ogoshi22 and by us,19 the 1,2-DCE solvent does not play any role as a template for the larger pillar[6]arene and prism[6]arene methoxy-derivatives. The formation of ethoxy- and propoxy-prismarenes PrS[n]R (n = 5 and 6, R = Et and nPr) as described in Scheme 1 and Table 1 was monitored as a function of time by HPLC (ESI†). The formation of the cyclic oligomers proceeded through many linear intermediate compounds that, after being initially formed, were then consumed as the cyclization reaction progressed.In the case of 4b in 1,2-DCE (Scheme 1 and Table 1, entry 4), PrS[6]Et was formed faster than the pentamer PrS[5]Et (ESI†). In fact, the hexamer started to appear after 10 min and remained the favored product over time, whereas an equilibrium value of 75% was reached after 45 min. HPLC analysis (ESI†) revealed that the pentamer appeared after 15 min but vanished after 30 min. Analogous results were obtained by monitoring the formation of PrS[6]Et and PrS[5]Et over time using different solvents (ESI†), such as chloro-cyclohexane (Cl-CyHex, Table 1, entry 12), cyclohexane (CyHex, Table 1, entry 18), toluene (Table 1, entry 15), and decaline (Table 1, entry 20). Independent of the nature and size of the solvent, the hexamer PrS[6]Et is always kinetically and thermodynamically favored with respect to PrS[5]Et, and its formation occurs in very high yields (71–76%) in short reaction times (30–45 min). Similar results were observed for the reaction in Scheme 1 starting from propoxy precursor 4c. In this case, the hexamer PrS[6]nPr is the kinetically and thermodynamically favored product independent of the nature of the solvent (Table 1, entries 5, 13, 16, 19, and 21). These results are surprising; in fact, as reported in the literature, only halogenated solvents play a fundamental role as templates in the formation of π-electron rich macrocycles as in the synthesis of pillararenes.22,23 To confirm the thermodynamic nature of the hexamers, PrS[5]Et or PrS[5]nPr (vide infra) was heated to 70 °C in 1,2-DCE, in the presence of TFA. Under these conditions, a conversion to PrS[6]Et and PrS[6]nPr in 83% and 85% yield, respectively, was observed after 22 h, confirming their higher thermodynamic stability.
As previously reported,19 starting from the methoxy-derivative 4a in 1,2-DCE c-PrS[5]Me is preferentially formed, while no evidence of the hexamer was detected in the reaction mixture (Table 1, entry 1). Therefore, we further investigated whether PrS[6]Me would be formed using the more bulky cyclohexane or toluene as the solvent. Starting from methoxy-derivative 4a in toluene, the pentamer c-PrS[5]Me was formed in a very low yield (11%, Table 1, entry 14) in addition to a copious quantity of polymeric material. Analogous results were obtained in cyclohexane (7% of c-PrS[5]Me, Table 1, entry 17).
Under these conditions, no hexamer PrS[6]Me was detected. When the reaction in Scheme 1 was performed using a monomer with even longer alkyl chains than propyl, namely 2,6-dibutoxy- and 2,6-dipentoxynaphthalene 4d and 4e in 1,2-DCE, hexamers PrS[6]nBu and PrS[6]nPent were formed in lower yields of 30% and 8%, respectively (Table 1, entries 10 and 11). Thus, in summary, in the presence of small methoxy groups, the hexamer PrS[6]Me is not formed in any solvent (CyHex, toluene, or 1,2-DCE), whereas in the presence of ethoxy and propoxy groups the hexamer PrS[6]R is always formed in high yields (75 and 65%) independent of the nature and size of the solvent. Interestingly, the yield of prism[6]arene decreases (from 75% to 8%) as the chain length increases (entries 4, 5, 10, and 11) from ethoxy (4b, entry 4, Table 1) to pentoxy (4e, entry 11, Table 1). In addition, the pentamer PrS[5]Me is the kinetically favored product while its confused isomer prevails under thermodynamic conditions. Differently, in the presence of ethoxy and propoxy functions the hexamers PrS[6]R are both kinetically and thermodynamically favored products. These results led us to conclude that the length of the alkyl chains on the rims of prismarenes plays a “special role”, driving their cyclization process. In detail, we can invoke a kinetically and thermodynamically self-templating effect by the Et and nPr alkyl chains in the synthesis of prismarene hexamers. At this point, we studied the role of the ammonium templates 52+ and 6+ in the formation of the ethoxy and propoxy prismarenes. In fact, we previously showed that the cation 52+ acted as a template for the synthesis of the methylated pentamer PrS[5]Me (47%, Table 1, entry 2).19 Differently, starting from 4b, under the conditions in Scheme 1 and in the presence of 52+·2I−, the hexamer PrS[6]Et was preferentially formed in 35% yield after 22 h (thermodynamic conditions), while the pentamer PrS[5]Et was obtained in 10% yield (Table 1, entry 6). HPLC analysis of the reaction mixture (Scheme 1) (ESI†) showed that after 60 min the hexamer was formed faster than PrS[5]Et, which was not detected in the reaction mixture. After 70 min, PrS[6]Et started to decrease slightly, while PrS[5]Et increased.
Starting from propoxy derivative 4c, the hexamer was still the kinetically favored product, while after 3 h the concentration of the pentamer PrS[5]nPr started to increase, and after 22 h an equilibrium value of 1/1 was reached (Table 1, entry 8). By comparison between the data in Table 1 (entry 2)19 and the new results (entries 6 and 8), we can conclude that the cation 52+ is surely less effective as a thermodynamic template in the synthesis of ethoxy and propoxy pentamers. In detail, the thermodynamic effect of the host–guest complexation invoked for the synthesis of PrS[5]Me,19 is overcome by the intramolecular “self-templating” effect of the ethyl and propyl chains. At this point, we investigated the role of 6+ in the synthesis of PrS[6]Et starting from 4b in 1,2-DCE (Scheme 1).
As previously reported,196+ is an effective template for the synthesis of PrS[6]Me (Table 1, entry 3).19 We monitored the progress of the reaction, and in the presence of 6+·I− after 45 min the yield of PrS[6]Et was <10%, significantly lower than that obtained after the same time (75%, Table 1, entry 4) in the absence of 6+. After 22 h (Table 1, entry 7), the yield of PrS[6]Et reached a value of 60%. Analogous results were observed starting from 4c in 1,2-DCE in the presence of 6+·I− that gave PrS[6]nPr in 55% yield after 22 h (Table 1, entry 9), while after 90 min the yield of PrS[6]nPr was <10%, once again significantly lower than that obtained after the same time in the absence of 6+ (65%, Table 1, entry 5). From the analysis of these data, it emerges that, in contrast to the clear cation templating effect on the formation of the methylated-analogue PrS[6]Me (Table 1, entry 3),19 the ammonium cation 6+ has a negative influence on the kinetics of the formation of prismarene hexamers PrS[6]Et and PrS[6]nPr. This is demonstrated by lower yields over longer reaction times. In other words, kinetically and thermodynamically self-templating by the Et and nPr alkyl chains in the synthesis of prismarene hexamers is verified by the higher yield (65–75%) of the product and a shorter reaction time, in the absence of cations. With these results in hand, we investigated the nature of the self-templating effect from the alkyl chains by detailed X-ray, DFT, and 2D NMR studies.
Structural properties of ethoxy- and propoxy-prismarenes in the solid state: the role of the alkyl chains
The structure assumed by prismarenes were studied experimentally, both in solution and in the solid state, by single crystal X-ray diffraction (Fig. 2) and NMR techniques (ESI†), respectively, and theoretically by DFT calculations (Fig. 3 and ESI†).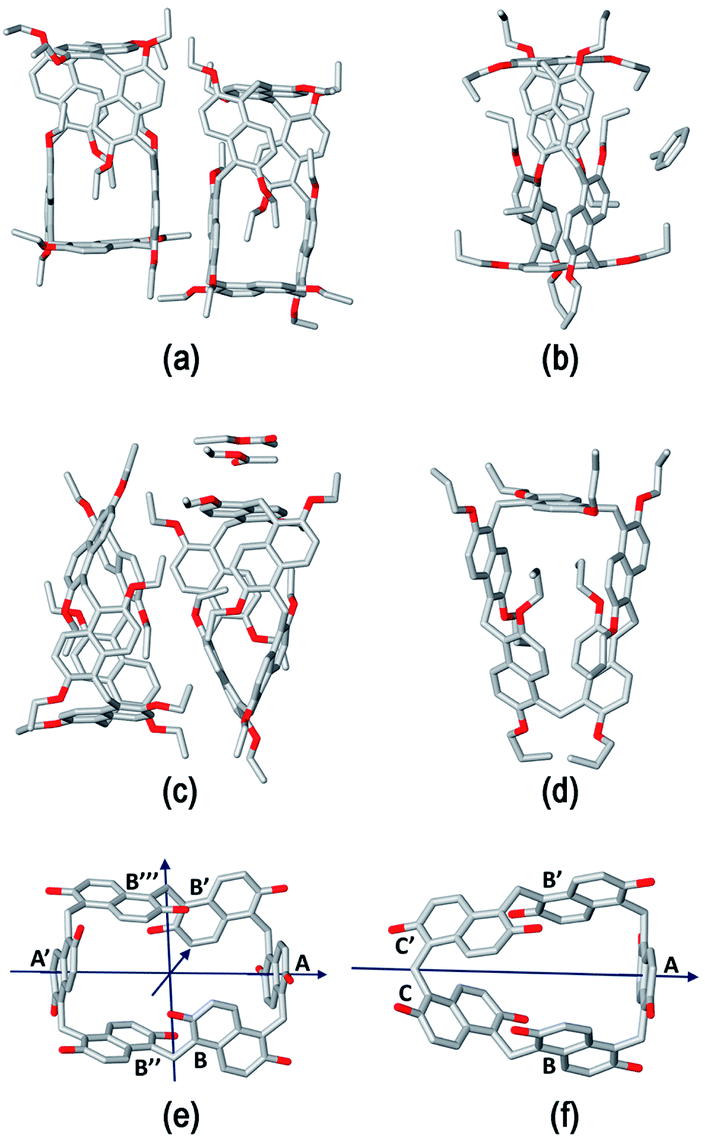 | ||
| Fig. 2 Stick representation of the asymmetric units of (a) PrS[6]Et, (b) PrS[6]nPr, (c) PrS[5]Et, and (d) PrS[5]nPr. Hydrogen atoms and one orientation of the disordered alkyl groups and a toluene molecule are omitted for clarity. For PrS[6]nPr, the entire molecule generated by a crystallographic two-fold axis is shown. (e) and (f) Stick representation of (e) the D2 symmetric PrS[6]R and (f) the C2 symmetric PrS[5]R scaffolds. The pseudo-two-fold symmetry axes are represented with arrows. For each molecule, the same letter indicates symmetry equivalent naphthalene rings. The alkyl groups are omitted for clarity. | ||
 | ||
| Fig. 3 Different views of the DFT optimized structure of Pr[6]nPr. Top views of the CPK (a) and tube (b) structure. Side views (c) and (d). In blue are the propoxy chains inward oriented and filling the cavity, in green are the propoxy chains outside oriented, and in yellow are the propoxy chains on the parallel naphthalene rings. | ||
Detailed structural models of the series PrS[6]R and PrS[5]R (R = Et and n-Pr) were obtained by single crystal X-ray diffraction experiments (Fig. 2 and ESI†) using synchrotron radiation and cryo-cooling techniques (see the ESI†). All four centrosymmetric crystal structures are composed of a racemic mixture of inherently24–27 chiral macrocyclic molecules, in which the naphthalene moieties are 1,5 connected, with the alkoxy groups at the 2,6-positions (Fig. 2). The ethyl derivatives, PrS[6]Et (Fig. 2a) and PrS[5]Et (Fig. 2c), show two crystallographic independent molecules in the asymmetric units, while the propyl derivatives, PrS[6]nPr (Fig. 2b) and PrS[5]nPr (Fig. 2d), contain a half molecule which lies on a two-fold crystallographic axis and one entire macrocyclic molecule, respectively. The prism[6]arene derivatives show a common dissymmetric conformation for the independent molecules, with a pseudo-D2 point symmetry of the macro-ring (Fig. 2e).
The DFT-optimized PrS[6]nPr molecule (Fig. 3) is very similar to the corresponding experimental solid-state structure as demonstrated by a rmsd value of 0.34 Å (ESI†) obtained by superimposing the two macrocyclic models (excluding the mobile alkyl chains).
The macro-ring is folded into a nearly square cuboid-shape (Fig. 2a, e and 3a) with the side-faces defined by the six naphthalene planar groups. Each aromatic planar group makes an angle of about 90° with respect to the contiguous naphthalene moieties (see ESI, Table S6†). Two opposite naphthalene rings (A and A′ of Fig. 2e at a 12.0 Å distance) define the square faces of the prism with about 6.5 Å side lengths. A twofold symmetry axis passes through the centers of these naphthalene rings. Thus, these two naphthalene units show a canting angle θ value of about 90° (see ESI, Table S5†).28 A second orthogonal twofold axis passes through two opposite methylene bridges of naphthalene moieties located on two edges of elongated faces of the parallelepiped. These naphthalene moieties show supplementary canting angles of ca. 40°/140° for the B/B′′ and B′′′/B′ couples (Fig. 2e and Table S5†). The third orthogonal twofold axis passes through the two open edges of the prism (Fig. 2e). The four inward oriented alkyl chains of the naphthalene moieties located on the elongated faces of the prism fill the cavity of the macrocycle (Fig. 3). These chains, organized in couples oriented in an antiparallel fashion, establish C–H⋯π interactions with the aromatic walls and various inter-chain van der Waals interactions (Fig. 3).
In detail, four strong C–H⋯π interactions are present with a C–H⋯πcentroid distance of 2.9 Å and a C–H⋯πcentroid angle of 148°. Clearly, this self-filling of the central cavity of the hexamer (Fig. 3) plays a crucial role in the thermodynamic stabilization of the hexameric skeleton, and it is relevant with respect to the conformational properties in solution and recognition abilities within the family PrS[6]nPr, PrS[6]Et, and PrS[6]Me. In addition, the inward oriented alkyl chains enhance the rigidity of the cuboid scaffold, as evidenced by NMR studies (vide infra). All three independent molecules of the prism[5]arene derivatives show a pseudo-C2 point symmetry of the macro-ring and therefore a similar dissymmetric conformation (Fig. 2c, d and f). This pseudo-symmetry was also observed in the three pseudo-polymorphic crystal forms of PrS[5]Me.19
The superimposition of the DFT-optimized PrS[5]nPr molecule with the solid-state structure shows a rmsd value of 0.07 Å. The pseudo-twofold symmetry axis passes through the methylene bridge connecting the C and C′ rings and the barycenter of the opposite naphthalene moiety (A in Fig. 2f). The canting angles reported in Table S5† reflect this situation. The dihedral angles of the B/B′ and C′/C couples (see Fig. 2f) are supplementary angles (ca. 70°/110° for B/B′ and ca. 40°/140° for C′/C), while the A angle is ca. 90°. In this way, two adjacent naphthalene rings (ESI†) are tilted in a way to give a self-filling of the cavity with their chains (ESI†). To gain insight into the structural features of the potential cavities of the prism[n]arenes (n = 5 and 6), the surfaces and volumes of the ideal prismatic solids enclosed by the aromatic walls were evaluated (ESI† shows the comparison with the analogous pillararene and pagodarene17). The geometrical parameters used for these calculations were obtained from the CCDC database as mean values of distances and angles observed in the X-ray structures containing the relevant monomeric unit (see Fig. S107† for details).
The prism[6]arene shows a cavity opening of 52.4 Å2 almost twice that (27.3 Å2) of the prism[5]arene, and, analogously, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume cavity ratio was calculated for the two prisms (490 Å3/255 Å3). Another important geometric feature is the internal potential contact surface areas (A) derived from the total area of the rectangular prism faces. The ratio between the contact surface areas of prism[6]arene and prism[5]arene is 252 Å2/186 Å2 = 1.35.
:1 volume cavity ratio was calculated for the two prisms (490 Å3/255 Å3). Another important geometric feature is the internal potential contact surface areas (A) derived from the total area of the rectangular prism faces. The ratio between the contact surface areas of prism[6]arene and prism[5]arene is 252 Å2/186 Å2 = 1.35.
Conformational properties of ethoxy- and propoxy-prismarenes in solution: the role of the alkyl chains
The 1D and 2D NMR spectra (243 K, 600 MHz) of PrS[6]nPr (Fig. S20† and 4a) in CD2Cl2 are consistent with the D2 symmetry of the cuboid structure found in the solid state.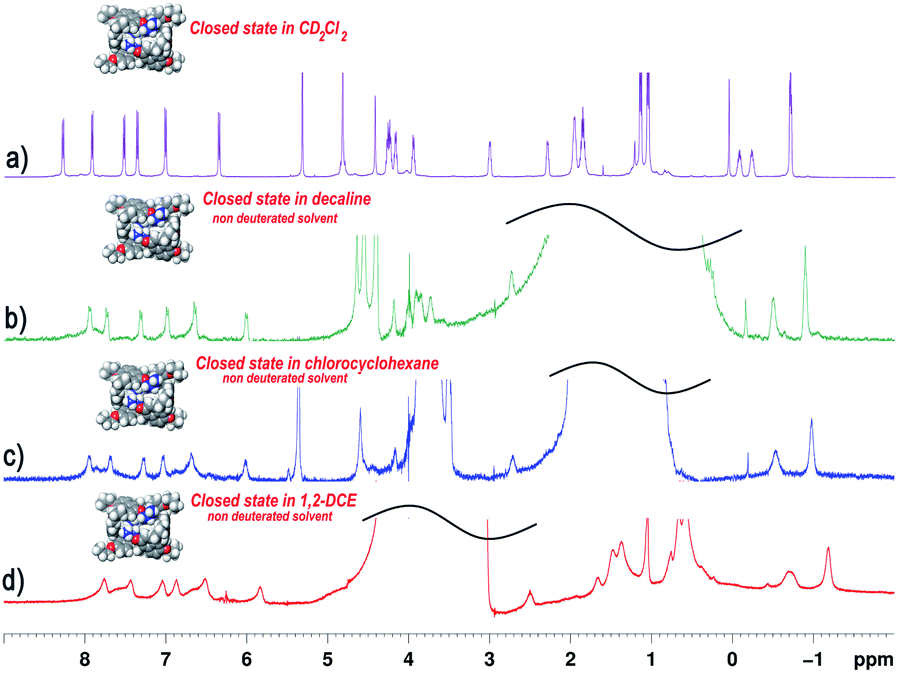 | ||
| Fig. 4 1H NMR spectra of Pr[6]nPr in: (a) CD2Cl2 at 243 K (600 MHz); (b) non-deuterated decaline (298 K); (c) non-deuterated chlorocyclohexane (298 K); (d) non-deuterated 1,2-dichloroethane (298 K). The spectra are consistent with the D2 symmetry of the cuboid structure of the prism[6]arene. | ||
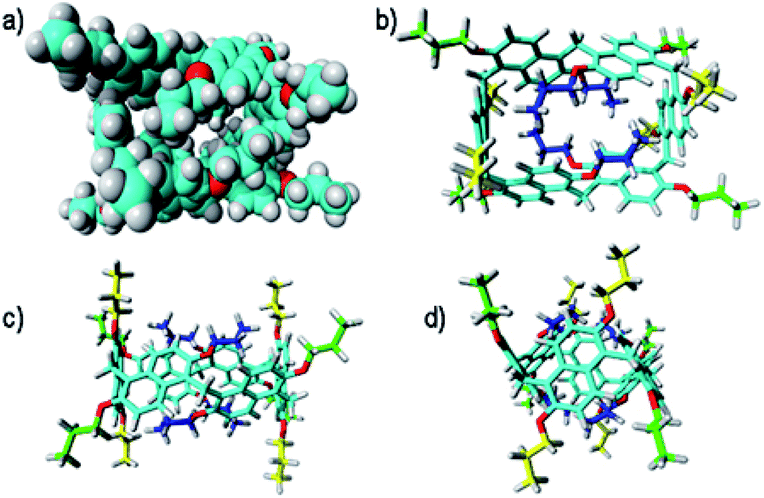
In fact, a close inspection of the 1H NMR spectrum of PrS[6]nPr revealed the presence of 3 aromatic AX/AB systems (COSY) at 8.28/7.35 (9.6 Hz), 7.91/7.01 (9.6 Hz), and 7.52/6.35 (9.6 Hz) ppm (Fig. S29c and e†), and two singlets at 4.82 and 4.42 ppm in a 2:1 ratio, attributable to the methylene-bridged groups. In addition, the OCH2 groups showed three diastereotopic AB systems at 4.25/4.23, 4.17/3.95, and 3.00/2.29 ppm (Fig. S29c, d and f†). The presence of diastereotopic resonances for the OCH2 groups of PrS[6]nPr (Fig. 4a and S29c, d, f†) is due to the planar chirality of the macrocycle, in agreement with the considerations previously reported by Ogoshi and coworkers24 for pillar[5]arenes. Thus, this 1D and 2D NMR analysis indicates that in solution PrS[6]nPr also adopts a conformation with a pseudo-D2 point symmetry. The presence of shielded 1H NMR signals at negative chemical shift values (Fig. 4a), attributable to the terminal methylene and methyl groups of the propyl-chains (CH3: −0.6 ppm, 12H, ESI†), clearly corroborates the finding that PrS[6]nPr also adopts in solution the cuboid structure depicted in Fig. 3a and 5a, in which there is a folding of the propyl chains inside the cavity. The 1D and 2D NMR spectra of PrS[6]Et show analogous features that confirm its cuboid structure in solution. A broad singlet is present in the 1H NMR spectrum of PrS[6]Et in CD2Cl2 at 298 K at −1.12 ppm (12H), attributable to the methyl groups belonging to the four ethyl groups folded inside the cavity. Impressively, these OCH2CH3 methyl groups experienced an up-field shift of △δ = δmacrocycle − δmonomer-4b = −2.74 ppm, significantly larger than that shown by the methyl groups of PrS[6]nPr of −1.67 ppm. This is evident from the close inspection of the X-ray structures in Fig. 5a and d. In Fig. 5b, the terminal methyl group of the chains of PrS[6]nPr are outward oriented, while the four ethyl groups of PrS[6]Et are entirely included inside the cavity of the macrocycle. The CPK visualization of the ethyl chains in Fig. 5d clearly reveals that they are close-packed inside the cavity, and fill the inner space more efficiently than the propyl chains of PrS[6]nPr in Fig. 5b. To quantify the differences in the self-filling of PrS[6]Et and PrS[6]nPr, the free volume of each enclosed internal cavity was evaluated from accessible surface area calculations (see the ESI† and Fig. 5). The smaller cavity volume of 49 Å3 obtained for PrS[6]Et with respect to 77 Å3 for PrS[6]nPr indicates the more efficient packing of this ethyl derivative (Fig. 5c and f).
 | ||
| Fig. 5 Self-filling of the cavities of prism[6]arenes as obtained by X-ray structures. (a) and (b) Different views of PrS[6]nPr. (d) and (e) Different views of PrS[6]Et. (c) and (f) Self-filling of the cuboid cavity. Cross sections of the PrS[6]Et (f) and PrS[6]nPr (c) molecules. An enclosed internal cavity was obtained from accessible surface area calculations using a 0.4 Å probe. The free cavity volume is 49 Å3 and 77 Å3 for PrS[6]Et and PrS[6]nPr, respectively. | ||
These results support the hypothesis that the nature of the alkyl chains plays a crucial role in the thermodynamically controlled cyclization of the hexamer. The cyclization process of the prism[6]arene hexamer is thermodynamically controlled by the alkyl chains on the rims and by their ability to fill the internal cavity of the macrocycle. In particular, the ethyl chains show a better steric complementarity with the internal cavity of the prism[6]arene and fill it in a more effective way (90%) than the propyl chains (84%). Consequently, PrS[6]Et is obtained in higher yields (about 75%) than PrS[6]nPr independent of the used solvent. Longer butyl and pentyl chains are less effective at self-filling the internal cavity and consequently the yield of the hexamer is lower.
Analogously, no evidence of the hexamer was detected in the reaction mixture starting from 2,6-dimethoxynaphthalene 4a; in this case, the methyl groups is too small to produce an effective filling of the internal cavity of the hexamer. Consequently, the equilibrium of formation of the methoxy-prismarenes is shifted toward the confused prism[5]arene c-PrS[5]Me, which shows a more effective self-filling of the internal cavity. When the 52+ or 6+ cation is present, the filling of the PrS[5]Me or PrS[6]Me cavity with the formation of the host–guest complex stabilizes the pentamer or hexamer respectively, which remains the preferential product over time.
On these bases, we can invoke an intramolecular thermodynamic self-templating effect in which the formation of the prism[6]arene is not driven by an external templating agent (e.g.: solvent) but by “intramolecular self-templating”29 of the alkyl chains. In other words, the self-filling of the internal cavity of PrS[6]Et or PrS[6]nPr stabilizes their cuboid structure driving the equilibrium toward their formation. This represents a rare example of a documented thermodynamically controlled cyclization process driven through an intramolecular self-template effect.
Ogoshi reported that 1,2-DCE and chlorocyclohexane are thermodynamic template solvents in the synthesis of pillar[5]arenes.22 In detail, DCE fills the pillar[5]arene cavity and its equilibrium of formation is driven by the thermodynamic effect of the complexation, solvent@host.22 The same authors clearly showed that 1,2-DCE didn't play a template role in the synthesis of the pillar[6]arene.22 In an attempt to examine if the solvents in Table 1 play a thermodynamic templating role for the selective formation of prism[6]arenes, by an endo-cavity inclusion process (solvent@PrS[6]R complexation), we performed 1H NMR experiments following a standard procedure already reported for pillararenes.22 The cuboid structure of the prism[6]arene shows an inaccessible closed-cavity (closed state, Fig. 5a, b and d, e) and consequently a conformational change of the prism[6]arene should be expected during an eventual endo-cavity inclusion of the solvent (vide infra).
The 1H NMR spectra of PrS[6]nPr in Fig. 4b–d were acquired in non-deuterated solvents such as chloro-cyclohexane, decaline, and 1,2-DCE. As is evident, the 1H NMR spectra in these solvents agree with the D2-symmetry of the cuboid structure. This result clearly indicated that in the presence of these solvents, the prism[6]arene remains in a closed state (Fig. 4) and consequently the inclusion of solvent molecules inside the cavity doesn't occur. To confirm this assumption, 1H NMR experiments were performed in which PrS[6]nPr and PrS[6]Et were titrated with 1 equiv. of solvent such as cyclohexane, chloro-cyclohexane, toluene, decaline, and 1,2-DCE and in no case was the shift of the solvent signals observed (ESI†), confirming that the host cuboid structure was retained. From these results, it can be inferred that these solvents don't act as a thermodynamic template for prism[6]arene formation. These results are consistent with the finding that, independent of the nature and size of the solvent, the ethoxy- and propoxy-hexamers are always kinetically and thermodynamically favored with respect to the pentamer.
In order to investigate whether the solvent 1,2-DCE plays a role in the stabilization of the transition states (TSs) for the macrocyclization steps of the intermediate carbocation of PrS[6]Et, a preliminary DFT study (ESI†) was performed. The transition state holding the solvent molecule inside the cavity (ESI†) is predicted to lie above the one holding the ethyl group inside the cavity by 3.4 kcal mol−1 in terms of internal energy and 2.3 kcal mol−1 in terms of Gibbs free energy. Based on the reasonable hypothesis that a late transition state is involved in the rate-determining step, DFT results suggest that the ethyl group could indeed fill the cavity, thus acting as an internal kinetic templating agent.
Molecular recognition properties of ethoxy- and propoxy-prism[n]arenes: the role of the alkyl chains
On the basis of these conformational aspects, a question arises as to whether PrS[6]nPr and PrS[6]Et are also capable of adopting a conformation with an open-cavity (open state) in the presence of appropriate guests. Due to the presence of π-electron rich cavities,19 it is worth investigating the recognition abilities of PrS[6]nPr and PrS[6]Et hosts toward cationic ammonium guests (Scheme 2). The complexation of the N,N,N′,N′-tetramethylpiperazonium 72+ cation (Scheme 2) showed very interesting features. The addition of 1 equiv. of 72+ as a barfate salt (BArF−)30,31 to a solution of PrS[6]nPr in CD2Cl2 caused significant changes in its 1H NMR spectrum (Fig. 6), indicative of the formation of a 72+@PrS[6]nPr complex with the cation hosted inside the cavity of PrS[6]nPr. | ||
| Scheme 2 Schematic complexation equilibrium of PrS[n]R with ammonium guests 52+–10+ as barfate salts. | ||
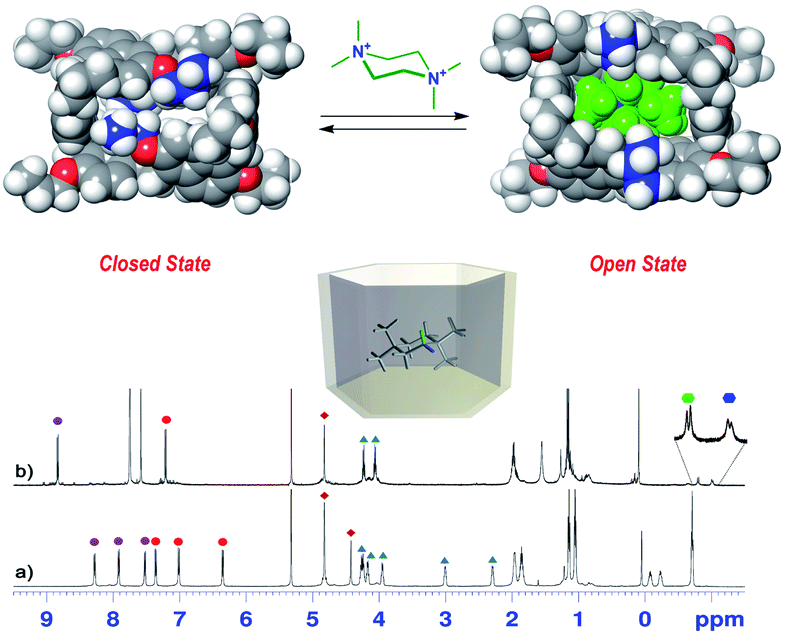 | ||
| Fig. 6
1H NMR spectra of (a) PrS[6]nPr in CD2Cl2 at 243 K (600 MHz) and (b) a 1:1 mixture of PrS[6]nPr and 72+·(BArF)2 (3 mM). The correspondence between the marked signals (red and purple circles for aromatic H-atoms and blue triangles for OCH2 groups) indicates the conformational change from a closed state to an open state upon complexation with 72+. The blue and green hexagons indicate the diastereotopic methylene H atoms of 72+ shielded inside the macrocycle. (Top) DFT optimized structure of PrS[6]nPr in the closed state and of its complex with 72+ (72+@PrS[6]nPr). | ||
Furthermore, in the presence of 72+, a more symmetric conformation of PrS[6]nPr emerged in the 1D and 2D NMR spectra (ESI†). In fact, the 1H NMR signals of the macrocycle in the 72+@PrS[6]nPr complex (Fig. 6b) are in agreement with an average D6 symmetry.32 The most plausible rationalization of this behavior is that the introduction of the guest into the macrocycle cavity forces the cuboid scaffold to open. This assumption was corroborated by DFT calculations (Fig. 6 top). The optimized structure (ESI†) of the complex 72+@PrS[6]nPr shows an opening of the cuboid scaffold (open state in Fig. 6), with the guest 72+ that occupies the central cavity in which the average plane of the cyclohexane ring is tilted by 20° with respect to the mean plane of the host methylene bridges. An association constant value of 1.2 × 108 M−1 (Table 2, 298 K, CD2Cl2, see the ESI†) was determined for the 72+@PrS[6]nPr complex by a series of competition experiments (ESI†).33,34 The host–guest complex is stabilized by ion-dipole (N+⋯OR), cation⋯π, C–H⋯π, and van der Waals interactions.
| 52+ | 6+ | 72+ | 82+ | 92+ | |
|---|---|---|---|---|---|
| a An intricate mixture of complexes was present at the equilibrium due to the presence of macrocyclic hosts in different conformations. | |||||
| PrS[5]Me | 3.9 × 107 | 90 | 1.8 × 107 | 6.6 × 106 | 5.8 × 103 |
| PrS[5]Et | 1.4 × 108 | 50 | 2.8 × 108 | 2.9 × 106 | 1.0 × 105 |
| PrS[5]nPr | 1.7 × 108 | 100 | 1.4 × 109 | 6.3 × 107 | 4.8 × 105 |
| PrS[6]Me | 50 | 2700 | —a | 4200 | 3700 |
| PrS[6]Et | 790 | 2745 | 1.0 × 108 | 420 | —a |
| PrS[6]nPr | 400 | 340 | 1.2 × 108 | 370 | —a |
Regarding the C–H⋯π interactions, Natural Bond Orbital (NBO)35,36 (ESI†) and Non-Covalent Interactions (NCI) calculations (ESI†) revealed that a significant contribution to the stabilization energy of the 72+@PrS[6]nPr complex was provided by the four equatorial C–H groups of the guest and the aromatic naphthalene walls (26%, see RDG based NCI analysis in the ESI†), while the axial C–H groups give a negligible contribution (3.0%). Finally, the +NCH3 group also establishes cationic C–H⋯π interactions with the aromatic walls of the host, as confirmed by RDG-based NCI analysis (ESI†).
An analogous conformational change from a closed to an open conformation was also revealed for the PrS[6]Et host after endo-cavity complexation with 72+. Attempts to obtain crystals of 72+@PrS[6]nPr or 72+@PrS[6]Et complexes were unsuccessful. However diffracting crystals of 6+@PrS[6]Me·BArF19 were obtained and the X-ray structure of this complex (Fig. 7a) provides interesting structural details regarding the open state of the prism[6]arene skeleton. The symmetric PrS[6]Me host lies on the crystallographic two-fold axis passing through the center of two opposite naphthalene rings of the host. The internal guest results in an opening of the cuboid arrangement towards the formation of a hexagonal prism. This is reflected by the supplementary canting angles of the B/B′′ and B′′′/B′ couples, which change from ca. 40°/140° of free PrS[6]R to ca. 60°/120° in the complex (Table S7†), closer to the ideal angle of 90° of a regular hexagonal prism.
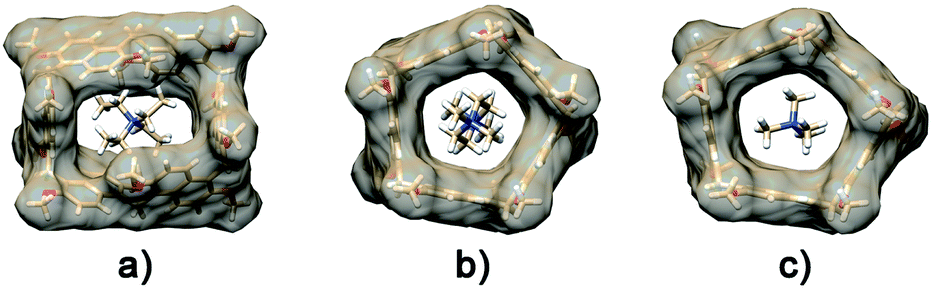 | ||
| Fig. 7 X-ray structural models of the host–guest complexes: (a) 6+@PrS[6]Me, (b) 72+@PrS[5]Me, and (c) 10+@PrS[5]Me. The transparent van der Waals surface of the host is shown to illustrate the effective prismarene cavities. Counter ions, solvents, and disordered guest atoms are omitted for clarity. | ||
Furthermore, the interior dihedral angles between the naphthalene planes increase from an averaged value of 90° observed in free PrS[6]R molecules to an averaged value of 105°, closer to the ideal angle of 120° of a regular hexagonal prism. A close inspection of the solid state structure of the 6+@PrS[6]Me·BArF complex reveals the important role of cationic C–H⋯π interactions in the host–guest complexation. Analogously, PrS[5]nPr undergoes a conformational change from a closed to an open state upon endo-cavity complexation with the cationic guest 72+. When PrS[5]nPr was mixed with N,N,N′,N′-tetramethylpiperazonium salt 72+·(BArF)2, in CD2Cl2 at room temperature, drastic changes were observed in the 1H NMR spectrum (Fig. 8). The presence of upfield shifted signals of 72+ methylene H atoms to negative values (Fig. 8b) is indicative of the inclusion of 72+ inside the cavity, shielded by the PrS[5]nPr walls. An AB system is observed at −0.82/−1.04 ppm (J = 11.5 Hz). Furthermore, the Δδ value between the aromatic doublets experiences a variation from 1.18 (Δδ = 8.07 − 6.89 = 1.18) for the free host (Fig. 8a) to 1.62 ppm (Δδ = 8.82 − 7.20 = 1.62) (Fig. 8b) upon complexation. This result clearly indicates an opening of the folded structure of PrS[5]nPr (closed state in Fig. 8) that upon complexation with 72+ adopts the open state in Fig. 8 in which the PrS[5]nPr host shows a D5 symmetry.
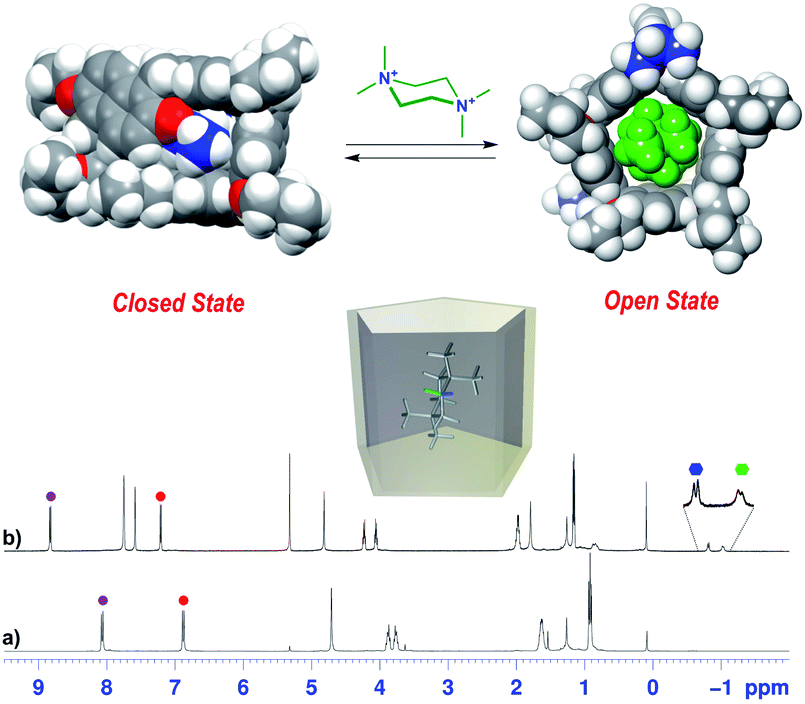 | ||
| Fig. 8
1H NMR spectra of (a) PrS[5]nPr in CD2Cl2 at 298 K (600 MHz) and (b) a 1:1 mixture of PrS[5]nPr and 72+·(BArF)2 (3 mM). The circles indicate the signals of aromatic H atoms, while the hexagons indicate the diastereotopic methylene H atoms of 72+ shielded inside the macrocycle. The correspondence between the marked signals (red and purple circles for aromatic H atoms) indicates the conformational change from a closed state to an open state upon complexation with 72+. The blue and green hexagons indicate the diastereotopic methylene H atoms of 72+ shielded inside the macrocycle. (Top) DFT optimized structure of PrS[5]nPr in the closed state and of its complex with 72+ (72+@PrS[5]nPr). | ||
In this conformation, the aromatic walls adopt canting angle values of about 90° and therefore define a regular pentagonal prism (Fig. 8).37 Thus, in the open state the aromatic H atoms move away from the cavity and experience a down-field shift (Fig. 8b) with respect to the closed state (Fig. 8a). The attempts to obtain crystals of 72+@PrS[5]nPr or 72+@PrS[5]Et complexes were also unsuccessful in this case. However diffracting crystals of 72+@PrS[5]Me·BArF and 10+@PrS[5]Me·BArF were obtained and the X-ray structures of these complexes (Fig. 7b and c) provide structural details regarding the open state of the prism[5]arene skeleton (see also the DFT optimized structure of the 72+@PrS[5]nPr complex in Fig. 8 and the ESI†). The crystal structure of 72+@PrS[5]Me·(BArF)2 (Fig. 7b) shows the guest located at the center of the prismatic host with the chair's plane perpendicular to the mean plane of methylene bridges of the macrocycle (85.8°). The pseudo-two-fold symmetry axis of 72+, orthogonal to the chair's plane, forms an angle of about 50° with a crystallographic two-fold axis, thereby resulting in a two-position statistical disorder of the 72+ ion.
The X-ray structure of the 72+@PrS[5]Me complex reveals a high steric complementarity between the PrS[5]Me cavity and the cationic 72+ guest that gives rise to relevant C–H⋯π interactions. The host resembles an almost regular pentagon prism with canting angles of 90.0° ± 3.2° and interior dihedral angles ranging from 104.6° to 110.5°, with an average angle of 107.9°, very close to the ideal 108° angle (Fig. 7b). Similarly, the crystal structure of 10+@PrS[5]Me·BArF also evidences an opening of the prismatic host in both crystallographic independent host–guest complexes (see Tables S7 and S8†) (Fig. 7c). The macrocycle adopts an almost regular pentagon prism with canting angles of 90.0° ± 3.3° and interior dihedral angles ranging from 102.1° to 113.7°, with an average angle of 108°, equal to the ideal angle (Fig. 7c).
The tetramethylammonium guests are well-centered in the PrS[5]Me cavity to give the important cationic C–H⋯π interactions (Fig. 7c). By a series of NMR competition experiments, an association constant value of 1.4 × 109 M−1 (298 K, CD2Cl2) was determined for the 72+@PrS[5]nPr complex (Table 2). This was confirmed by fluorescence titration (Kass = 1.7 × 109 M−1, see the ESI†). Interestingly, NBO analysis (ESI†) conducted on 72+@PrS[5]nPr indicates that both axial and equatorial C–H⋯π interactions play a significant role in the stabilization of the complex. In a similar way, PrS[5]nPr shows high affinity for dication N,N′-dihexyl-DABCO 52+ (ESI†). In fact, the 1H NMR spectrum of PrS[5]nPr shows significant changes upon addition of 52+ as a barfate salt (ESI†) and is indicative of the formation of pseudorotaxane 52+@PrS[5]nPr. The association constant values for the formation of 52+@PrS[5]R complexes (Table 2) were determined by competition experiments (ESI†). Values of 1.7 × 108 M−1 and 1.4 × 108 M−1 were obtained for PrS[5]nPr and PrS[5]Et, respectively.
Comparison among the affinities of the PrS[5]R hosts toward the N,N′-dialkyl-DABCO 52+ and 82+ cations in Table 2 indicates that the longer alkyl chains of axle 52+ play a crucial role in the stabilization of pseudorotaxanes 52+@PrS[5]R.
Conclusions
Prism[6]arenes bearing ethyl or propyl chains on their rims are obtained in high yields and in short reaction times, independent of the nature and size of the solvent. Differently, in the presence of small methoxy groups, the hexamer PrS[6]Me is not formed in any solvent (CyHex, toluene, or 1,2-DCE), and analogously the PrS[6]Pentyl derivative is formed in a very low yield. The reaction progress, monitored by HPLC, revealed that PrS[6]R (R = nPr and Et) are the favored products, both kinetically and thermodynamically. Differently, as previously reported for methylated prismarenes,19 the pentamer PrS[5]Me is the kinetically favored product, while its confused isomer c-PrS[5]Me prevails under thermodynamic conditions. Based on these results, we have invoked a kinetically and thermodynamically self-templating effect by the Et and nPr alkyl chains in the synthesis of prismarene hexamers. Insights into this effect were obtained by detailed X-ray and 1D and 2D NMR investigations supported by DFT calculations. These studies clearly showed that PrS[6]Et and PrS[6]nPr are folded into a square cuboid-shape with D2 symmetry. In this conformation, four inward oriented alkyl chains of the naphthalene moieties, located on the elongated faces of the prism, fill the cavity of the macrocycle, and establish C–H⋯π interactions with the aromatic walls and various inter-chain van der Waals interactions. 1D and 2D NMR studies show that the cuboid structure of the hexamers is also maintained in solutions of different solvents, such as 1,2-DCE, CH2Cl2, decaline, and chloro-cyclohexane. On these bases, we propose that the synthesis of the ethyl and propyl prism[6]arenes occurs under an intramolecular thermodynamic self-templating effect in which the formation of the prism[6]arene is not driven by an external templating agent (e.g., solvent) but by intramolecular self-templating of the alkyl chains. In other words, the self-filling of the internal cavity of PrS[6]Et or PrS[6]nPr stabilizes their cuboid structure driving the equilibrium toward their formation.The cuboid structure of the prism[6]arene shows an inaccessible closed cavity, and molecular recognition studies, both in solution and in the solid state, show that the introduction of guests into the macrocycle cavity forces the cuboid scaffold to open, through an induced-fit mechanism, which originates from the conformational flexibility of the prismarene macrocycle. An analogous conformational change from a closed to an open state occurs during the endo-cavity complexation process of the pentamer.
Thus, we here have described a rare example of a documented cyclization process kinetically and thermodynamically templated by intramolecular interactions. The in-depth investigation of the self-filling effects of the alkyl chains on the synthesis of prism[6]arenes, and on their conformational properties, could pave the way to the synthesis of macrocycles exploiting this novel thermodynamically controlled intramolecularly self-templated cyclization process, different from those currently known, based on the external effect of solvents or guests.
Data availability
CCDC 2025594 (for PrS[5]Et); 2025589 (for PrS[5]nPr); 2025583 (for PrS[6]Et); 2025585 (for PrS[6]nPr); 2050614 (for 7·PrS[5]Me·(BArF)2); 2050617 (for 3·PrS[5]Me·BArF); 2050618 (for 6·PrS[6]Me·BArF) contain the supplementary crystallographic data for this paper.Author contributions
P. D. S.: DFT calculations; R. D. R., L. D. M., C. C. and C. P.: synthesis; S. G. and N. H.: X-ray studies; C. T., N. C. I. and N. B. O. calculations; A. C.: TS DFT calculations; C. G. and S. G.: conceptualization, data analysis and manuscript writing; P. N.: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Regione Campania (POR CAMPANIA FESR 2007/2013 O.O.2.1, CUP B46D14002660009) for the FT-ICR mass spectrometer facilities and the “Centro di Tecnologie Integrate per la Salute” (CITIS) (project PONa3_00138) for the 600 MHz NMR facilities. Financial support from the University of Salerno (FARB) is acknowledged.Notes and references
- F. Diederich, P. J. Stang and R. R. Tykwinski, Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- C. Gaeta and D.-X. Wang, Front. Chem., 2020, 8, 128, DOI:10.3389/fchem.2020.00128.
- J. M. Benyus, Biomimicry: Innovation Inspired by Nature, Harper Perennial, 2002 Search PubMed.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 Search PubMed.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 Search PubMed.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 Search PubMed.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, The Royal Society of Chemistry, Cambridge, 1994 Search PubMed.
- P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and Beyond, Springer Int Publishing, 2016 Search PubMed.
- T. Ogoshi, Pillararenes, The Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- H. Yao and W. Jiang, Naphthol-Based Macrocycles, in Handbook of Macrocyclic Supramolecular Assembly, Springer, Singapore, 2019 Search PubMed.
- A. H. Tran, D. O. Miller and P. E. Georghiou, J. Org. Chem., 2005, 70, 1115–1121 Search PubMed.
- F. Jia, Z. He, L.-P. Yang, Z.-S. Pan, M. Yi, R.-W. Jiang and W. Jiang, Chem. Sci., 2015, 6, 6731–6738 Search PubMed.
- L.-P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 Search PubMed.
- F. Jia, H. Hupatz, L.-P. Yang, H. V. Schröder, D.-H. Li, S. Xin, D. Lentz, F. Witte, X. Xie, B. Paulus, C. A. Schalley and W. Jiang, J. Am. Chem. Soc., 2019, 141, 4468–4473 Search PubMed.
- F. Jia, H. V. Schröder, L.-P. Yang, C. von Essen, S. Sobottka, B. Sarkar, K. Rissanen, W. Jiang and C. A. Schalley, J. Am. Chem. Soc., 2020, 142, 3306–3310 Search PubMed.
- X. Wang, F. Jia, L.-P. Yang, H. Zhou and W. Jiang, Chem. Soc. Rev., 2020, 49, 4176–4188 Search PubMed.
- X.-N. Han, Y. Han and C.-F. Chen, J. Am. Chem. Soc., 2020, 142, 8262–8269 Search PubMed.
- R. Del Regno, P. Della Sala, A. Spinella, C. Talotta, D. Iannone, S. Geremia, N. Hickey, P. Neri and C. Gaeta, Org. Lett., 2020, 22, 6166–6170 Search PubMed.
- P. Della Sala, R. Del Regno, C. Talotta, A. Capobianco, N. Hickey, S. Geremia, M. De Rosa, A. Spinella, A. Soriente, A. Spinella, P. Neri and C. Gaeta, J. Am. Chem. Soc., 2020, 142, 1752–1756 Search PubMed.
- L.-P. Yang and W. Jiang, Angew. Chem., Int. Ed., 2020, 7, 15794–15796 Search PubMed.
- (a) R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 Search PubMed , and references cited therein.; (b) As reported by Sanders and Stoddart: “There is an important distinction to be made between kinetic and thermodynamic templating in relation to macrocycle formation. In kinetic templating, the template binds a linear species, which brings the two chain ends into proximity, thus resulting in an increase in the rate of ring formation for a particular macrocycle. By contrast, in thermodynamic templating, the template binds to the macrocycle itself and forms a complex which stabilizes the macrocycle. This situation results in an equilibrium involving a range of different macrocycles becoming biased toward the formation of one particular macrocycle”, S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 Search PubMed.
- T. Ogoshi, N. Ueshima, T. Akutsu, D. Yamafuji, T. Furuta, F. Sakakibara and T.-a. Yamagishi, Chem. Commun., 2014, 50, 5774–5777 Search PubMed.
- (a) X.-H. Tian, X. Hao, T.-L. Liang and C.-F. Chen, Chem. Commun., 2009, 6771–6773 Search PubMed; (b) T. Boinski and A. Szumma, Tetrahedron, 2012, 68, 9419–9422 Search PubMed; (c) P. Della Sala, C. Gaeta, W. Navarra, C. Talotta, M. De Rosa, G. Brancatelli, S. Geremia, F. Capitelli and P. Neri, J. Org. Chem., 2016, 81, 5726–5731 Search PubMed.
- T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima and T.-a. Yamagishi, Org. Lett., 2011, 13, 1264–1266 Search PubMed.
- N. L. Strutt, H. Zhang and J. F. Stoddart, Chem. Commun., 2014, 50, 7455–7458 Search PubMed.
- T. Ogoshi, K. Kitajima, T. Aoki, S. Fujinami, T. a. Yamagishi and Y. Nakamoto, J. Org. Chem., 2010, 75, 3268–3273 Search PubMed.
- K. Du, P. Demay-Drouhard, K. Samanta, S. Li, T. U. Thikekar, H. Wang, M. Guo, B. van Lagen, H. Zuilhof and A. C. H. Sue, J. Org. Chem., 2020, 85, 11368–11374 Search PubMed.
- The canting angle is defined as the dihedral angle between the naphthalene plane and the mean plane of the bridging methylene carbon atoms of the prism[n]arene molecule.
- Very recently, Bunchuay and co-workers reported an interesting example of self-template assisted synthesis of the decatosylate pillar[5]arene. In this example, the p-toluenesulfonate group acts as an effective intermolecular template in the macrocyclisation reaction. By X-ray studies, the authors showed that the self-templated mechanism was due to an intermolecular inclusion of the tosylate chain into the cavity of a neighbouring pillararene macrocycle forming a self-assembled 1D supramolecular polymer that was retained both in solution and in the solid state: A. Ruengsuk, K. Khamphaijan, P. Pananusorn, A. Docker, J. Tantirungrotechai, M. Sukwattanasinitt, D. J. Harding and T. Bunchuaya, Chem. Commun., 2020, 56, 8739–8742 Search PubMed . In our case, close inspection of the solid state packing of PrS[6]nPr L. Tu, H. B. Wu and X. W. D. Lou, Acc. Chem. Res., 2017, 50, 293–301 Search PubMed and PrS[6]Et (Fig. S106†) did not show any intermolecular inclusion of alkyl chains into the cavity of neighbouring macrocycles (see. S106†). Thus, PrS[6]nPr and PrS[6]Et adopt a cuboid structure in which the central cavity (see Fig. 6 and 8) of the macrocycle is closed exclusively by intramolecular inclusion (self-inclusion) of alkyl chains, which thermodynamically drives their formation. Self-templated synthesis is well-documented in material chemistry, but also in these instances intermolecular processes occur, see: Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 Search PubMed; Q. Zhang, W. Wang, J. Goebl and Y. Yin, Nanotoday, 2009, 4, 494–507 Search PubMed; H. Li, A. M. Evans, W. R. Dichtel and J.-L. Bredas, ACS Materials Lett, 2021, 3, 398–405 Search PubMed; P. Wang, Y. Aihua, Z. Wang, X. Shen, H. Chen and H. Zhou, Nanoscale, 2021, 13, 1988–1996 Search PubMed; Y.-Y. Liu, X.-C. Li, S. Wang, T. Cheng, H. Yang, C. Liu, Y. Gong, W.-Y. Lai and W. Huang, Nat. Commun., 2020, 11, 5561–6669 Search PubMed . In addition, some self-templated syntheses of catenanes have been reported and were based on intermolecular effects: J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int., Ed., 2016, 55, 8938–8943 Search PubMed; J. E. M. Lewis, Org. Biomol. Chem., 2019, 17, 2442–2447 Search PubMed.
- C. Gaeta, C. Talotta, F. Farina, G. Campi, M. Camalli and P. Neri, Chem.–Eur. J., 2012, 18, 1219–1230 Search PubMed.
- C. Gaeta, C. Talotta and P. Neri, Chem. Commun., 2014, 50, 9917–9920 Search PubMed.
- The presence of the N,N,N′,N′-tetramethylpiperazonium guest 72+ inside the PrS[6]nPr cavity should lower the symmetry of the macrocycle compared to the D6-one. In detail, the observed D6 symmetry of PrS[6]nPr in the complex with 72+ can be explained on the basis of the fast tumbling (with respect to the NMR time scale, 600 MHz) of the piperazinium guest inside the cavity of the macrocycle. In fact, upon lowering the temperature a broadening was observed for the AB system of the guest shielded at negative values of chemical shifts at −0.80 ppm and −1.02 ppm (J = 11.4 Hz). Below 263 K the AB system of the guest coalesced, and new signals appeared at negative values of chemical shifts, indicative of a slowing down of the tumbling motion of 72+.
- K. Hirose, Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2007, pp. 17–54 Search PubMed.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 20, 988–993 Search PubMed.
- F. Weinhold and C. R. Landis, Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 Search PubMed.
- This is an example of an induced fit complex, in which the host structure undergoes a conformational adaptation around the guest. For an analogous phenomenon in which the chemical shift of aromatic protons of a calixarene host shifted as a function of the canting angle of the aromatic rings, see: T. Pierro, C. Gaeta, F. Troisi and P. Neri, Tetrahedron Lett., 2009, 50, 350–353 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. Detailed synthetic procedures, 1D and 2D NMR spectra of prismarenes and their complexes, HR mass spectra, details on stability constant determination, DFT calculation details. X-ray figures, and tables of crystal data: CCDC 2025583, 2025585, 2025589, 2025594, 2050614, 2050617 and 2050618. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02199k |
| ‡ P. D. S. and R. D. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |