DOI:
10.1039/D1SC00803J
(Edge Article)
Chem. Sci., 2021,
12, 10605-10612
Oxygen atom transfer promoted nitrate to nitric oxide transformation: a step-wise reduction of nitrate → nitrite → nitric oxide†
Received
9th February 2021
, Accepted 1st July 2021
First published on 2nd July 2021
Abstract
Nitrate reductases (NRs) are molybdoenzymes that reduce nitrate (NO3−) to nitrite (NO2−) in both mammals and plants. In mammals, the salival microbes take part in the generation of the NO2− from NO3−, which further produces nitric oxide (NO) either in acid-induced NO2− reduction or in the presence of nitrite reductases (NiRs). Here, we report a new approach of VCl3 (V3+ ion source) induced step-wise reduction of NO3− in a CoII-nitrato complex, [(12-TMC)CoII(NO3−)]+ (2,{CoII–NO3−}), to a CoIII–nitrosyl complex, [(12-TMC)CoIII(NO)]2+ (4,{CoNO}8), bearing an N-tetramethylated cyclam (TMC) ligand. The VCl3 inspired reduction of NO3− to NO is believed to occur in two consecutive oxygen atom transfer (OAT) reactions, i.e., OAT-1 = NO3− → NO2− (r1) and OAT-2 = NO2− → NO (r2). In these OAT reactions, VCl3 functions as an O-atom abstracting species, and the reaction of 2 with VCl3 produces a CoIII-nitrosyl ({CoNO}8) with VV-Oxo ({VV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O}3+) species, via a proposed CoII-nitrito (3, {CoII–NO2−}) intermediate species. Further, in a separate experiment, we explored the reaction of isolated complex 3 with VCl3, which showed the generation of 4 with VV-Oxo, validating our proposed reaction sequences of OAT reactions. We ensured and characterized 3 using VCl3 as a limiting reagent, as the second-order rate constant of OAT-2 (k2/) is found to be ∼1420 times faster than that of the OAT-1 (k2) reaction. Binding constant (Kb) calculations also support our proposition of NO3− to NO transformation in two successive OAT reactions, as Kb(CoII–NO2−) is higher than Kb(CoII–NO3−), hence the reaction moves in the forward direction (OAT-1). However, Kb(CoII–NO2−) is comparable to Kb{CoNO}8, and therefore sequenced the second OAT reaction (OAT-2). Mechanistic investigations of these reactions using 15N-labeled-15NO3− and 15NO2− revealed that the N-atom in the {CoNO}8 is derived from NO3− ligand. This work highlights the first-ever report of VCl3 induced step-wise NO3− reduction (NRs activity) followed by the OAT induced NO2− reduction and then the generation of Co-nitrosyl species {CoNO}8.
O}3+) species, via a proposed CoII-nitrito (3, {CoII–NO2−}) intermediate species. Further, in a separate experiment, we explored the reaction of isolated complex 3 with VCl3, which showed the generation of 4 with VV-Oxo, validating our proposed reaction sequences of OAT reactions. We ensured and characterized 3 using VCl3 as a limiting reagent, as the second-order rate constant of OAT-2 (k2/) is found to be ∼1420 times faster than that of the OAT-1 (k2) reaction. Binding constant (Kb) calculations also support our proposition of NO3− to NO transformation in two successive OAT reactions, as Kb(CoII–NO2−) is higher than Kb(CoII–NO3−), hence the reaction moves in the forward direction (OAT-1). However, Kb(CoII–NO2−) is comparable to Kb{CoNO}8, and therefore sequenced the second OAT reaction (OAT-2). Mechanistic investigations of these reactions using 15N-labeled-15NO3− and 15NO2− revealed that the N-atom in the {CoNO}8 is derived from NO3− ligand. This work highlights the first-ever report of VCl3 induced step-wise NO3− reduction (NRs activity) followed by the OAT induced NO2− reduction and then the generation of Co-nitrosyl species {CoNO}8.
Introduction
The mechanism of microbial denitrification is still one of the most mysterious subjects, despite being explored in detail in both in vivo and in vitro systems. Based on the extensive research and literature available, denitrification has been well accepted to be a four-step reductive process of nitrate (NO3−) to dinitrogen (N2) conversion {NO3− → NO2− → NO → N2O → N2}, through a series of intermediate gaseous nitrogen oxide products.1 In mammals and bacteria, inorganic NO3− and nitrite (NO2−) serve as a fundamental storage material of NO for its bio-physiological processes.2,3 However, in humans, an excessive amount of NO3− has been discovered to cause gastric cancer and other disorders.4 To maintain an optimal NO3− level in the bio-system, commensal bacteria in the human oral cavity play a vital role in converting NO3− to NO2− (eqn (1)).2 In bacteria, molybdenum-based nitrate reductase (NRs) enzymes generate NO2−via an OAT reaction from NO3− anion.5 At the bio-physiological level, NO2− serves as a pool of NO and can easily be transformed to NO, either (i) in non-enzymatic acid-catalyzed NO2− reduction in the stomach6,7 or (ii) by Fe and Cu based nitrite reductase (NiRs) enzymes catalyzed reactions (eqn (2)).8,9| | | NO3− + MoIV → NO2− + MoVIO | (1) |
| | | M–NO2− + 2H+ + e− → M–NO + H2O | (2) |
| | | M–NO2 + R2S → M–NO + R2SO | (3) |
| | | M–NO2 + RSH → M–NO + R(O)SH | (4) |
| | | M–NO2 + PPh3 → M–NO + OPPh3 | (5) |
| | | M–NO2 + Sub + hν → M–NO + (O)Prod | (6) |
NO is a gaseous secondary messenger in animals, plants, fungi, and bacteria. In plants, NO is involved in different physiological processes, such as plant growth & development, metabolism, aging, defense against pathogens, biotic and abiotic trauma.10 However, NO regulates various physiological processes in mammals.11 For instance, NO inadequacy possibly will aggravate the pathogenic effects associated with atherosclerosis, diabetic hypertension, etc.12 Also, the immune response of NO towards the harmful pathogens is related to the oxidized NO species,13i.e., peroxynitrite (PN, ONOO−)13a,14 or/nitrogen dioxide (˙NO2).15,16 Hence, balanced production of NO is required to maintain a normal homeostatic bio-physiological condition. In bio-systems, enzymes, i.e., NiRs8,9,17 and endothelial nitric oxide synthases (eNOSs)17,18 are available for NO generation. The NOSs enzymes catalyze the biosynthesis of NO from L-arginine.18
In the case of NO overproduction, nitric oxide dioxygenase (NODs) generates NO3− in the reaction of the iron–dioxygen adduct with NO via a proposed PN intermediate,19 as explored in other biomimetic systems.19d,20 Also, there are various reports on NO2− formation in nitric oxide monooxygenation (NOM) reaction from metal-nitrosyls in the presence of O2, O2˙− and OH−.19a,20b,21 Oxidized species of NO (NO3− & NO2−) may also generate via different oxidative processes (vide supra). However, NO3− to NO2− transformation and NO3−/NO2− to NO conversion (vise versa) are critical steps of the denitrification process.1
In an attempt to mimic the denitrification process, R. H. Holm and co-workers reported NRs activity, molybdenum, and tungsten-based catalyst for catalytic reduction of NO3− to NO2− with metal-Oxo species.22 Similarly, S. Sarkar and co-workers have reported MoIV mediated reduction of NO3− to NO2− to mimic NRs activity.23 Eunsuk Kim proposed the reduction of NO3− to NO2− using the Lewis acid Sc3+ to activate the OAT reaction from NO3− to Mo metal center.24 On the other hand, nitrite reduction chemistry is explored widely; Ford and co-workers examined the acid-induced reduction of NO2− to N2O in a Fe–porphyrin complex.25 Warren group described the NiR by using the thiol group.26 Recently, we have reported acid-induced nitrite reduction to NO.27 However, the conversion of NO3− to NO using a single catalyst is barely explored; Yunho Lee and co-workers testified Ni catalyzed the transformation of inorganic NO3− to N2via NO2− intermediate using the carbon monoxide (CO) as oxophilic species.28
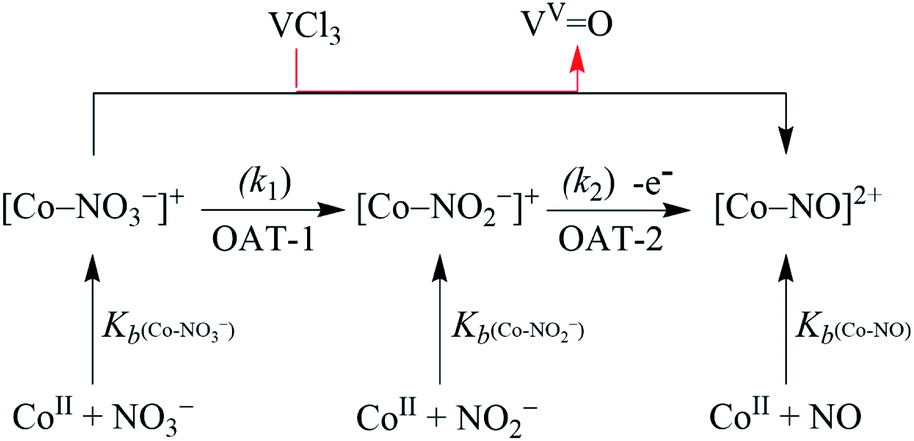
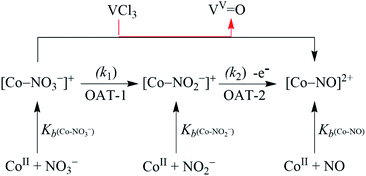
In biological systems3,5a and biomimetic22,23,27,29/or catalytic reactions,24 the approach towards converting NO3− to NO is usually a two-enzymes/-catalysts-induced two-step process in two different reactions (i.e., NO3− reduction followed by NO2− reduction).5a However, only a few reports simultaneously carry out both the NO3− and NO2− reduction using a single metal center in a biomimetic system. Here, our eagerness is to mimic the NRs enzymatic reaction followed by the NO2− reduction, sponsored by the same reagent (i.e., single metal-induced two-step NO3− to NO conversion). Herein, we report the NO3− reduction chemistry of a CoII–NO3− complex, [(12-TMC)CoII(NO3)]+ (2), bearing a 12-TMC ligand (12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane) via the two consecutive oxygen atom transfer (OAT) reactions using VCl3 as an oxophilic compound (Scheme 1, reaction II & III). Complex 2 reacts with VCl3 to form corresponding CoIII-nitrosyl complex, [(12-TMC)CoIII(NO)]2+ (ref. 19d, 20b, 21a, 27 and 30) (4), with VV-Oxo ({VVO}3+) species, which further decomposes to V2O5, via the formation of a presumed CoII–nitrito (3,{CoII–NO2−) intermediate in MeOH or H2O at 298 K (Scheme 1, reaction II). Interpretation of various spectral measurements, we have confirmed the generation of 3 with VV-Oxo by the transfer of one O-atom from NO3− moiety of 2 to VCl3 (OAT-1). Further, we observed the generation of 4 with VV-Oxo from 3 upon reaction with VCl3 (OAT-2), under similar reaction conditions, showing the similar OAT induced NO2− reduction reactivity as reported in the case of PPh3 and sulphur based compounds (thiols and thioethers) (eqn (3)–(6)).26,31 Combining these two OAT reactions, we can predict the reaction sequences, i.e., the first OAT from NO3− moiety of 2 to VCl3 and the generation of 3, {OAT-1 = CoII–NO3− + VCl3 → CoII–NO2− + VOCl3 (r1)}. Subsequently, the second OAT from NO2− moiety of 3 to another VCl3 moiety and the generation of 4 {OAT-2 = CoII–NO2− + VCl3 → {CoNO}8 + VOCl3 (r2)}. Mechanistic investigation using 15N-labeled-15NO3− & 15NO2− confirmed clearly that the N-atom in the {CoNO}8 is derived from NO3− anion of 2. To the best of our knowledge, the present work reports the first example of VCl3 induced conversion of CoII–NO3− to {CoNO}8 in two successive OAT reactions, demonstrating a new mechanistic approach for one-metal induced NO3− to NO2− reduction (NRs activity) followed by NO2− to NO conversion (OAT induced NO2− reduction).
 |
| | Scheme 1 | |
Results and discussion
Preparation of CoII–nitrate complex, [(12-TMC)CoII(NO3−)]+ (2)
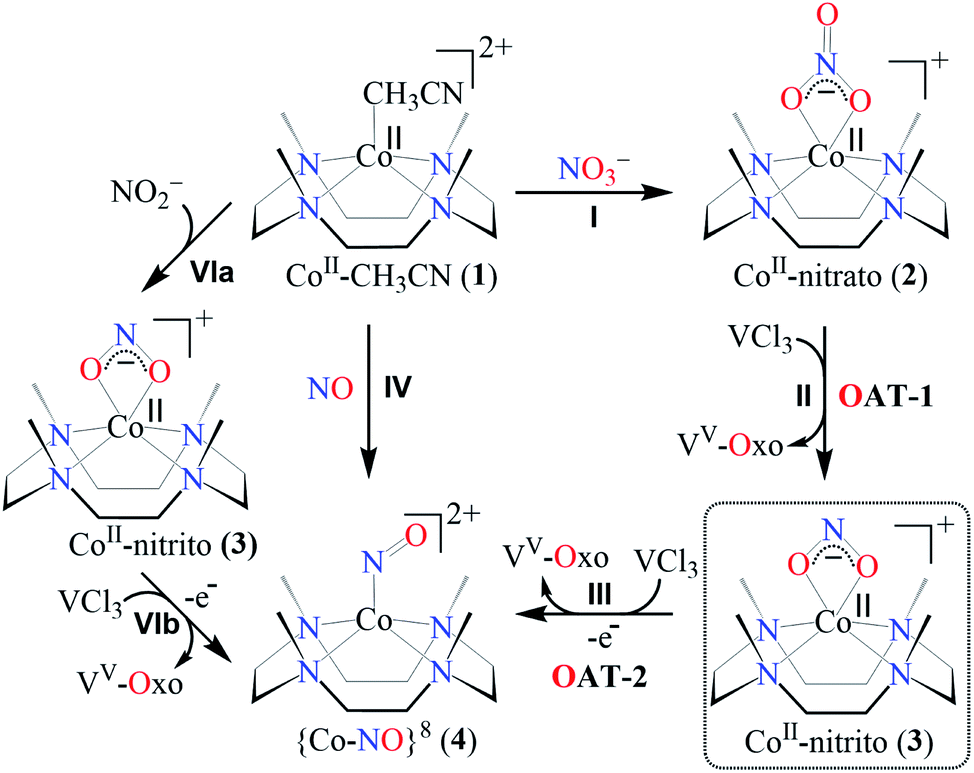
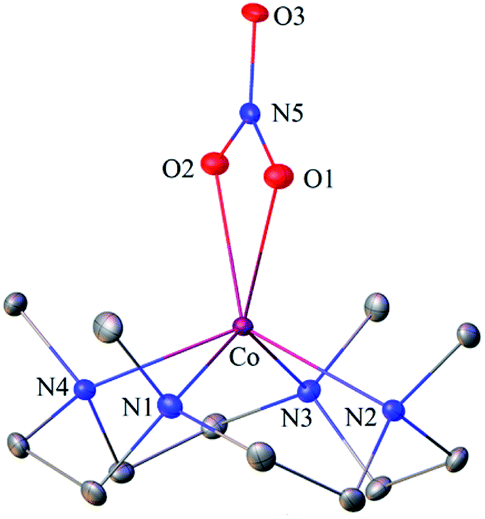
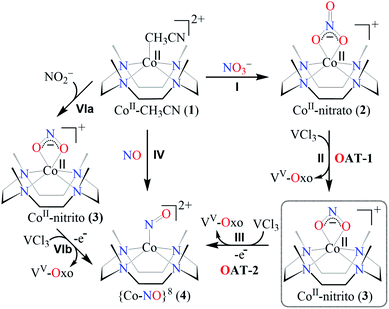
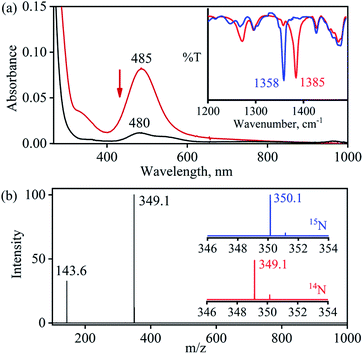
The primary CoII–nitrate complex, [(12-TMC)CoII(NO3−)]+ (2), was prepared by reacting CoII-complex, [(12-TMC)CoII(NCCH3)]2+ (1), with 1 equivalent of NaNO3 in H2O/CH3CN (Scheme 1, the reaction I; also see ESI† and Experimental section (ES)). Further, 2 was characterized by various spectroscopic techniques, including the single-crystal X-ray structure determination. UV-vis absorption band of 1 (λmax = 485 nm) changed to a new band (λmax = 480 nm, ε = 25 M−1 cm−1) upon addition of 1 equivalent NaNO3 in CH3CN at RT, suggesting the formation of 2 (Fig. 1a). FT-IR spectrum of 2 showed a characteristic peak for CoII-bound NO3− anion at 1384 cm−1 and shifted to 1358 cm−1 when exchanged with 15N-labeled-NO3− (15N16O3−) (Fig. 1a; ESI, Fig. S1†). A wide range 1H-NMR spectrum of 2 showed fairly clean paramagnetic proton-signals (Figure S2a†), suggesting a magnetically active Co-center. The spin state of 2 was determined by calculating the magnetic moment of the CoII metal-center by the Evans' method and found to be 4.46 BM, suggesting a high spin CoII-ion in complex 2 (ES, ESI, Fig. S2b†). Electrospray ionization mass spectrum (ESI-MS) of 2 showed a prominent peak at m/z 349.1, which shifted to m/z 350.1 when prepared with 15N-labeled Na15NO3, and their mass and isotope distribution pattern corresponds to [(12-TMC)CoII(14NO3)]+ (calc. m/z 349.1) and [(12-TMC)CoII(15NO3)]+ (calc. m/z 350.1), respectively (Fig. 1a; ESI, Fig. S3†). In addition to the above experimental characterization, 2 was structurally characterized by single-crystal X-ray crystallography. Complex 2 has a six-coordinate distorted octahedral geometry around the CoII-center, possessing O, O/-chelated bi-dentate NO3− anion (Fig. 2; ESI, ES, Fig. S4, Tables T1 and T2†).
 |
| | Fig. 1 (a) UV-vis spectra of 1 (0.50 mM, brick red line) and 2 (0.50 mM, black line) in CH3CN under Ar at 298 K. Inset: IR spectra of 2-14NO3− (red line) and 2-15NO3− (blue line) in KBr. (b) ESI-MS spectra of 2. The peak at 349.1 is assigned to [(12TMC)CoII(NO3)]+ (calcd m/z 349.1). Inset: isotopic distribution pattern for 2-14NO3− (red line) and 2-15NO3− (blue line). | |
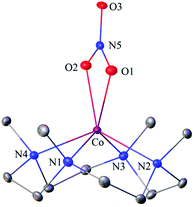 |
| | Fig. 2 Displacement ellipsoid plot (20% probability) of 2 at 100 K. Disorder C-atoms of TMC ring, anion and H-atoms have been removed for clarity. | |
OAT reaction of CoII–nitrate complex (2)
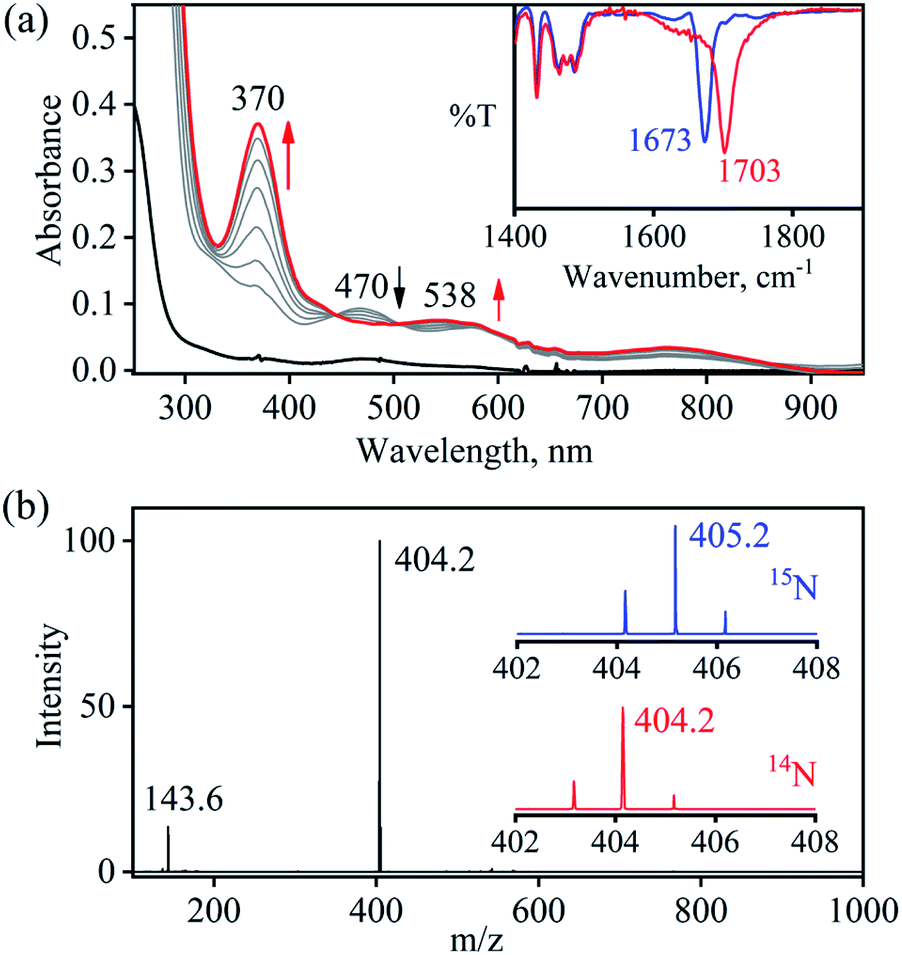
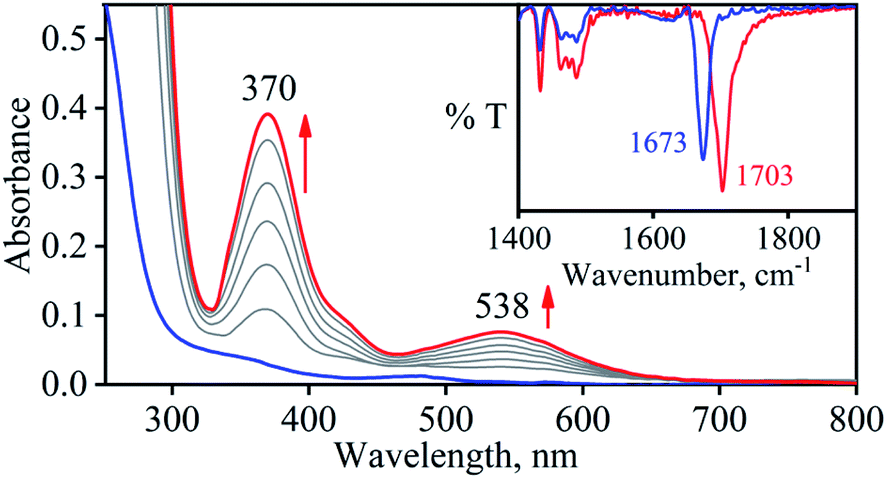
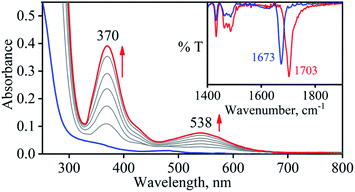
So as to understand the NO3− reduction chemistry of 2, we explored its reaction with VCl3 to mimic the OAT based NRs enzymatic reaction. We observed a visible color change from pink to wine-red in the reaction of 2 with VCl3 and a new absorption band (at 370 nm) formed, which is corresponding to the characteristic absorption band of a CoIII-nitrosyl ({CoNO}8, 4) (Fig. 3a and 6a).19d,20b,21a,27,30,32 Astonishingly, 2 upon reaction with VCl3 generated corresponding CoIII-nitrosyl complex 4, ({CoNO}8),19d,20b,21a,27,30 with VV-Oxo species in both aqueous/or methanol medium at 298 K (Scheme 1; reaction II). It is important to note that 2 did not show any spectral changes in the absence of VCl3, suggesting that 2 is highly stable in H2O/or MeOH and at 298 K (ESI, ES, and Fig. S5†). Finally, the product of NO3− reduction, formed in the reaction of 2 and VCl3, was established to be {CoNO}8 (4) based on various spectroscopic (UV-vis, FT-IR, ESI-MS, NMR) and structural characterization (vide infra).19d,20b,21a,27,30,32-33 The FT-IR spectrum of 2 showed a peak at 1384 cm−1, characteristic to the CoII bound NO3− stretching frequency which shifted to 1703 cm−1 when 2 was reacted with VCl3, which is characteristic of NO stretching frequency of {CoNO}8 (4).19d,20b,21a,27,30 The peak at 1703 cm−1 shifted to 1673 cm−1 when 4 was prepared by reacting 15N-labeled-NO3− (CoII–15NO3−) with VCl3, evidently suggesting the formation of {Co15NO}8 (inset: Fig. 3a; ESI and Fig. S6†). The shifting of NO stretching frequency (Δ = 30 cm−1) indicates that N-atom in NO ligand is derived from CoII–NO3−. The ESI-MS spectrum of 4 showed a prominent peak at m/z 404.2, [(12-TMC)CoIII(NO)(BF4)]+ (calcd m/z 404.2), and shifted to 405.2, [(12-TMC)CoIII(15NO)(BF4)]+ (calcd m/z 405.2) when the reaction was performed with CoII–15NO3− (Fig. 3b; ESI, Fig. S7†); suggests clearly that NO moiety in 4 is derived from NO3− moiety. The 1H-NMR spectrum of 4 showed the peaks for the protons of 12-TMC ligand frameworks, confirming a low spin diamagnetic CoIII center (d6, S = 0) in complex 4 (ESI, Fig. S8†).32,33 Further, we have calculated the yield of 4 from NMR spectra using benzene as an internal standard and found to be 90 ± 3% (ESI, Fig. S9†). As a final point, the exact conformation of 4 was provided by its single-crystal X-ray crystallographic analysis (ESI, ES, Fig. S10, Tables T1 and T2†) and comparable with previously reported CoIII–NO−/M–NO− having sp2 hybridized N-atom.20b,21a,27,30,34 The lone pair present on N-atom is responsible for the significant bending of the CoIII–NO− moiety, with Co(1)–N(5)–O(1) bond angles of 128.52 (18)° for 4 and, therefore, further consistent with the assignment of 4 as {CoNO}8 species.
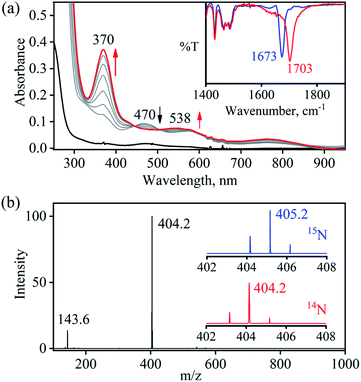 |
| | Fig. 3 (a) UV-vis spectral changes of 2 (0.50 mM, black line) upon addition of VCl3 (2.2 equiv.) in H2O at 298 K. Black line (2) changed to a red line (4) upon addition of VCl3. Inset: IR spectra 4-14NO (blue line) and 4-15NO (red line) in KBr. (b) ESI-MS spectra of 4. The peak at 404.2 is assigned to [(12TMC)CoIII(NO)(BF4)]+ (calcd m/z 404.2). Inset: isotopic distribution pattern for 4-14NO (red line) and 4-15NO (blue line). | |
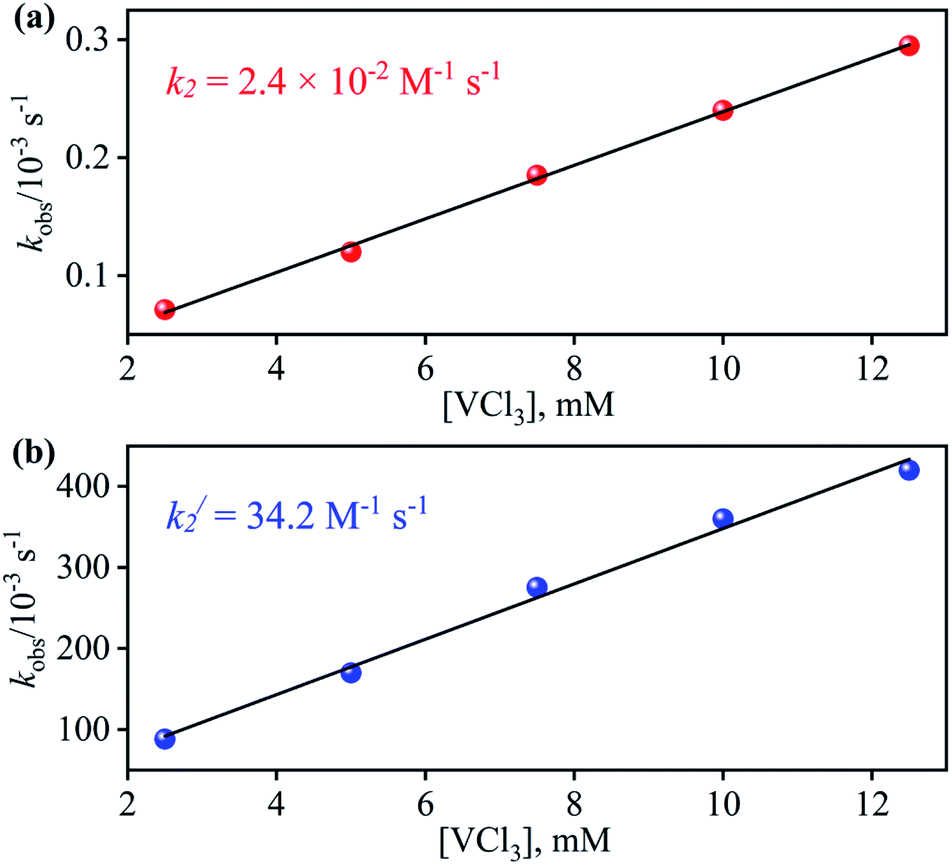
From the final spectrum (black line in Fig. 3a), we have calculated the amount of 4 (90 ± 2%) by comparing its ε (M−1 cm−1) value at 370 nm, since VV-Oxo and VCl3 species does not show any absorption at 370 nm. This value is also in good agreement with the yield calculated from NMR spectroscopy (ESI, Fig. S9†). Further, we had also determined the isolated yield of the formation of 4 and found it to be 90 (±2)%, depicting clearly VCl3 induced NO3− to NO transformation. The reduction of NO3− was observed to be slow; however, it enhanced with an increase in VCl3 amount, suggesting that the NO3− to NO transformation follows the second-order reaction. Upon adding 10 equivalents of VCl3 to the solution of 2 (0.5 mM), the UV-visible band at 370 nm starts forming with a pseudo-first-order rate constant, kobs = 1.2 × 10−1 s−1, and showed the isosbestic points at 418 and 497 nm (Fig. 3a). Upon increasing the concentration of VCl3, the pseudo-first-order rate constants increased proportionally, allowing us to determine a second-order rate constant (k2) of 2.4 × 10−2 M−1 s−1 (Fig. 4a) for the reaction of 2 with the various equivalents of VCl3 (5, 10, 15, 20, 25).
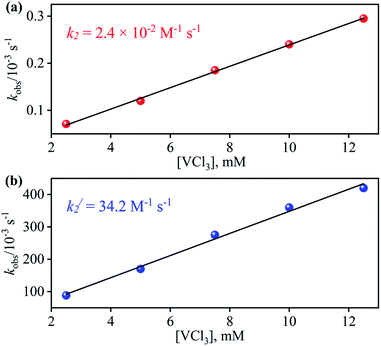 |
| | Fig. 4 Plot of kobsversus the concentration of VCl3 to determine the second order rate constant in the OAT reaction of (a) 2 (b) 3 in H2O at 298 K. | |
Confirming VV-Oxo generation in NO3− reduction reaction via OAT
In order to authenticate our proposition of OAT promoted NO3− to NO reduction, we should observe the generation of VV-Oxo species during this transformation. In this regard, we have confirmed the conversion of VCl3 to VV-Oxo species in the NO3− reduction reaction by 51V-NMR. We observed the characteristic peaks of VV-Oxo species in the 51V-NMR spectrum for the reaction mixture obtained after the completion of the reaction of 2 (4 mM) with VCl3 (8 mM) in CD3OD, at −365, −525, and −598 assignable to VOCl3, VOCl(OMe2)2 and VO(OMe2)3, respectively, as reported previously (ESI and Fig. S11†).35 The observation of VV-Oxo species in the VCl3 promoted NO3− reduction reaction indisputably illustrates that the VCl3 sponsored NO3− to NO conversion should proceed via the two consecutive OAT reactions, where OAT-1 mimics the NRs enzymatic reaction,5b,36 while OAT-2 mimics the phosphorus or sulphur induced OAT transfer reactions (Schemes 1 and 2).26,31
 |
| | Scheme 2 | |
Mechanistic investigation of NO3− reduction
In the biological system, the conversion of NO3− to NO proceeds via a common NO2− intermediate in two consecutive steps (vide supra). In this report, it is dreadfully clear that the formation of NO from NO3− could only be accomplished via the VCl3 induced two consecutive OAT reactions (vide supra), i.e., OAT-1 & OAT-2. Hence, the conversion of NO3− to NO is likely to proceed via a CoII–NO2− intermediate (3). Although we were unable to isolate the intermediate 3; however, we were able to show its generation by using VCl3 as a limiting reagent (ES). In the reaction of 2 with 1.0-fold of VCl3, we observed the generation of 4 with CoII–NO3− and CoII–NO2− and confirmed with various spectroscopic measurements. The FT-IR spectrum of the above reaction mixture showed the characteristic peaks for CoII–NO3− (at 1385 cm−1), CoII–NO2− (at 1272 cm−1) and {Co14NO}8 (at 1703 cm−1), those shifted to 1358 cm−1, 1245 cm−1 and 1673 cm−1 when 15N-labeled-nitrate complex (3-15NO3−) reacted with VCl3, respectively (ESI, Fig. S12a and b†). Also, we have recorded the ESI-MS spectrum of the reaction mixture, which showed the prominent peaks at m/z 404.2, [(12-TMC)CoIII(NO)(BF4)]+ (calcd m/z 404.2), 333.1, [(12-TMC)CoII(NO2−)]+ (calcd m/z 333.1) and [(12-TMC)CoII(NO3−)]+ (calcd m/z 349.1), those shifted to 405.2, [(12-TMC)CoIII(15NO)(BF4)]+ (calcd m/z 405.2), 334.1, [(12-TMC)CoII(15NO2−)]+ (calcd m/z 334.1) and [(12-TMC)CoII(15NO3−)]+ (calcd m/z 350.1), when the reaction was performed with CoII–15NO3− and VCl3, respectively (ESI, Fig. S12c and d†). Further, when we reacted 2 with 1.5-fold of VCl3, we observed the generation of 4 with CoII–NO2− and confirmed by FT-IR and ESI-MS measurements. The FT-IR spectrum showed the characteristic peaks for CoII–NO2− (at 1272 cm−1) and {Co(14NO)8 (at 1703 cm−1), and shifted to 1245 cm−1 (CoII–15NO2−) and 1673 cm−1 ({Co15NO}8) when using 15N-labeled-nitrate complex (3-15NO3−) (SI, Fig. S13a and b†). The ESI-MS spectrum, for the above reaction mixture, showed the prominent peaks at m/z 404.2, [(12-TMC)CoIII(NO)(BF4)]+ (calcd m/z 404.2), and 333.1, [(12-TMC)CoII(NO2−)]+ (calcd m/z 333.2), and shifted to 405.2, [(12-TMC)CoIII(15NO)(BF4)]+ (calcd m/z 405.2) and 333.1, [(12-TMC)CoII(15NO2−)]+ (calcd m/z 334.1), when using CoII–15NO3− as starting reacting material in OAT reaction, correspondingly (ESI, Fig. S13d†). Together, the FT-IR and ESI-MS spectra confirmed that VCl3 induced reduction of NO3− to NO is going through a CoII–NO2− (3) intermediate. Furthermore, as described above, when 2 reacted with 2.2-fold of VCl3, we had observed the generation of only complex 4 with nearly 90 ± 2% yield (vide supra).
With the intention of further validate our concept of CoII–NO2− species formation in the NO3− reduction reaction, in a control experiment, we explored the VCl3 induced transformation of CoII–NO2− to one-electron oxidized {CoNO}8 species;37 possibly by the release of one electron which usually gets solvated as observed in other cases,38 and trailed the fate of OAT reaction (Fig. 5). For this reaction, the initial CoII–NO2− complex was prepared by following the reported literature (Scheme 1, reaction VIa).21a,27 Upon addition of one fold VCl3 to a solution of 3 in MeOH/H2O at RT, the color of the reaction solution immediately changed from light pink to wine red, suggesting the generation of {CoNO}8 (4),37 and its characteristic absorption band (at 370 nm) appeared in ∼1 minute as shown in Fig. 5 and 6b (Scheme 1, reaction VIb). Also, we have calculated the yield of 4 by comparing its ε (M−1 cm−1) value at 370 nm and found it to be (>95 ± 2%). Further, spectral titration data confirmed that the stoichiometric ratio of 3 with VCl3 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI, Fig. S14†). Furthermore, the final product (4) was confirmed by the FT-IR and 1H-NMR spectroscopy (ESI, Fig. S15 and S16†).19d,20b,21a,27,30 Also, the generation of VV-Oxo species was confirmed by the 51V-NMR (ESI, Fig. S17†). Speedy conversion of 3 to 4 in the presence of VCl3 justifies our inability to isolate intermediate 3 in the VCl3 induced conversion of 2 to 4 and supports our supposition of two consecutive OAT reactions in reducing NO3− to NO. We have also determined the second-order rate constant for the reduction of NO2− to NO to understand the reaction mechanism and NO formation fate. The second-order rate constant (k2/) for NO2− reduction was determined by plotting the pseudo-first-order rate constant against various equivalents of VCl3 (5, 10, 15, 20, and 25) and found to be 34.2 M−1 s−1 (Fig. 4b). Our efforts to isolate the CoII–NO2− intermediate in the NO3− reduction reaction is unsuccessful due to the high reactivity of CoII–NO2− with VCl3 (OAT-2), which is ∼1420 times faster than the second-order rate constant of VCl3 induced NO3− to NO reduction (k2 = 2.4 × 10−2 M−1 s−1).
:1 (ESI, Fig. S14†). Furthermore, the final product (4) was confirmed by the FT-IR and 1H-NMR spectroscopy (ESI, Fig. S15 and S16†).19d,20b,21a,27,30 Also, the generation of VV-Oxo species was confirmed by the 51V-NMR (ESI, Fig. S17†). Speedy conversion of 3 to 4 in the presence of VCl3 justifies our inability to isolate intermediate 3 in the VCl3 induced conversion of 2 to 4 and supports our supposition of two consecutive OAT reactions in reducing NO3− to NO. We have also determined the second-order rate constant for the reduction of NO2− to NO to understand the reaction mechanism and NO formation fate. The second-order rate constant (k2/) for NO2− reduction was determined by plotting the pseudo-first-order rate constant against various equivalents of VCl3 (5, 10, 15, 20, and 25) and found to be 34.2 M−1 s−1 (Fig. 4b). Our efforts to isolate the CoII–NO2− intermediate in the NO3− reduction reaction is unsuccessful due to the high reactivity of CoII–NO2− with VCl3 (OAT-2), which is ∼1420 times faster than the second-order rate constant of VCl3 induced NO3− to NO reduction (k2 = 2.4 × 10−2 M−1 s−1).
 |
| | Fig. 5 UV-vis spectral changes of 3 (0.50 mM, blue line) upon addition of VCl3 (1 equiv.) in H2O at 298 K. Blue line (3) changed to red line (4) upon addition of VCl3. Inset: IR spectra 4-14NO (blue line) and 4-15NO (red line) in KBr. | |
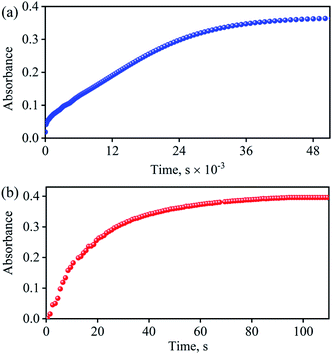 |
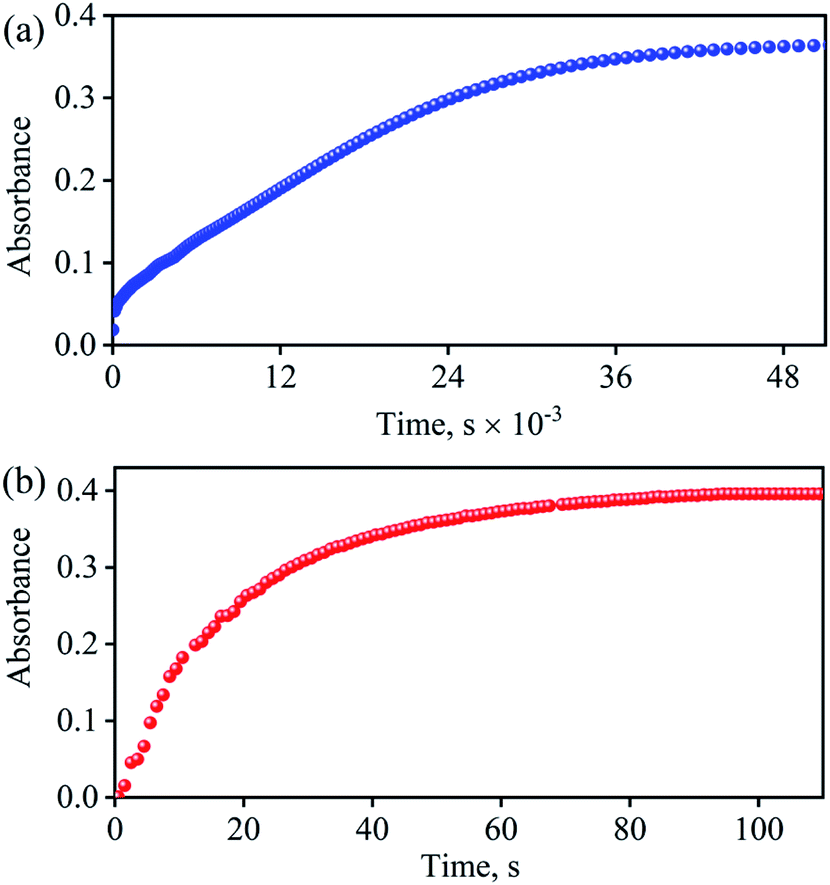
| | Fig. 6 (a) Time course of the formation of 4 (blue circles) monitored at 370 nm upon addition VCl3 (2.2 equiv.) to a solution of 2 (0.5 mM) in H2O at 298 K. (b) Time course of the formation of 4 (Red circles) monitored at 370 nm upon addition VCl3 (1 equiv.) to a solution of 3 (0.5 mM) in H2O at 298 K. | |
Spectroscopic and kinetic measurements undeniably confirmed that the reaction of 3 with VCl3 generates 4 ({CoNO}8) plus VV-Oxo species with a second-order rate constant (k2/ = 34.2 M−1 s−1), suggesting a rapid conversion (Fig. 6b). However, the transformation of 2 to 4 was observed to be a prolonged reaction, based on the spectral measurements (vide infra), in two sequential OAT reactions with a second-order rate-constant (k2 = 2.4 × 10−2 M−1 s−1) via a CoII–NO2− intermediate. This comparison of rate constants (k2 ⋘ k2/) suggests that the formation of NO2− from NO3− is a rate-determining step in the NO3− to NO reduction chemistry. Kinetic measurements (vide supra) confirmed clearly that the first step of the reaction (Scheme 1, pathway II) is the slowest step of the reaction; hence, a rate-determining step. Therefore, the overall second-order rate constant (k2 = 2.4 × 10−2 M−1 s−1) is equal to the rate constant of the conversion of 2 to 3 (Fig. 4a and 6a). Additionally, the binding constants (Kb(CoII–NO3−), Kb(CoII–NO2−), & Kb{(CoNO)8) for the generation of different species, CoII–NO3−, CoII–NO2− & {CoNO}8 in the reaction of [CoII(CH3CN)(12TMC)]2+ with NO3−, NO2− & NO, were determined by using Benesi–Hildebrand equation39 and found to be 2.3 × 102 M−1, 2.5 × 103 M−1 & 2.4 × 103 M−1 (ESI, ES, and Fig. S18†), respectively (Scheme 2). Structural parameters and ambiphilic nature of O-atom in coordinated NO3− & NO2−species, in their respective complexes,28 and the binding constants calculations further support our proposal of two OAT reactions with different reaction rates (r2 ⋙ r1). The Kb(CoII–NO2−) is higher than that of Kb(CoII–NO3−); hence the reaction moves in the forward direction once the NO2− generates from NO3− in OAT-1. However, the abstraction of the first non-coordinated O-atom from NO3− using VCl3 is somewhat hard due to its less electrophilic nature and more bond strength (bond length(N–O) = 1.215 Å) compare to other more electrophilic Co2+-coordinated O-atoms (bond length(N–O) = 1.267 Å & 1.269 Å); therefore showed a slower rate of OAT-1 (r1) than OAT-2 (r2) reaction, as proposed theoretically in CO induced Ni–NO3− reduction chemistry, suggesting the slow rate of NO3− to NO2− than NO2− to NO.28 Kim and co-workers reported that the alteration of O-atom's electrophilic behavior in NO3− species, induced by the Sc3+ metal (Lewis acid) binding, showed the NO3− to NO2− reduction, which was not observed in the absence of Sc3+ ion, suggesting the O-atom activation upon Sc3+ binding. Discussion on the OAT chemistry28 and metal-induced activation of O-atom24 (vide supra) undoubtedly support our supposition of a faster OAT-2 than the OAT-1, as non-co- ordinated O-atom of NO3− is difficult to abstract by VCl3 than Co2+-co-ordinated O-atoms of nitrite moiety.24,28 Recent reports on Lewis acid induced OAT reactions showed an increase in the oxidizing power of M–oxygen adducts and their OAT reaction rate,40 which coincides with the activation of N2 by Lewis acid.28,41 Further, the difference in the rates of the NO3− & NO2− reduction were supported by the inert and labile behavior of Co-complexes. High spin CoII-complexes, 2 and 3, are labile (d7, S = 3/2);42 hence the conversion of 2 to 3 was found to be slow as there is not much change in the CFSE; however, the conversion of labile 3 to an inert 4 (d6, S = 0) found to be very fast as there is a huge change in the CFSE, in order to achieve more stable inert electronic configuration.19d,43 These results verify our theory of step-wise conversion of NO3− to NO2−, which further reduces to NO in the presence of VCl3 in two consecutive OAT reactions.
Conclusion
Investigation of insights into the mechanistic aspects of the NO3− & NO2− reduction process became a most significant research area in modern-day chemistry as it deals with the biological and environmental aspects.1 Reduction of NO3− to NO via NO2− intermediate species are key steps in biological NO generation (salival NRs followed by NiRs in mammalian system)5,44 and also for the denitrification process (biogeochemical systems).1,45 Reduction of NO3− to NO using a single metal complex is still a challenge to the scientific community as two different enzymes play the catalytic role in each step in the biological system.2,3 In this report, for the very first time, we have shown the direct reduction of NO3− in a CoII–nitrato complex, [(12TMC)CoII(NO3−)]+ (2), to a Co–nitrosyl complex {CoNO}8 (4), in the presence of an oxophilic reagent (VCl3). Mechanistic investigation suggests that the reaction proceeds via a CoII–NO2− (3) species, as observed in the case of biological NRs enzymatic chemistry,5 in two consecutive OAT reactions. Kinetic measurements suggest that the VCl3 induced reduction of NO3− to NO2− is a rate-determining step (k2 = 2.4 × 10−2, OAT-1), mimicking the salival molybdate NRs enzymatic reaction.5 In the second step (OAT-2), a speedy reduction process (k2/ = 34.2), NO2− further reduces to NO in the presence of one-fold VCl3. Isolation of 3 was difficult due to the fast conversion of 3 to 4; however, we could characterize it with various spectroscopic techniques. The results observed in VCl3 induced NO3− reduction to NO in two consecutive OAT reactions are found to be in good agreement with our proposed concept. The results are explained in the light of the bond strength and the electrophilic behavior of O-atoms of NO3−/NO2− ligands and based on the inert & labile nature of Co-complexes. Due to high bond strength and less electrophilic character of metal-unbound O-atom of NO3−, it showed a slower OAT reaction (r1); in contrast, O-atoms of NO2− moiety is activated due to their binding with CoII-center, and the OAT from NO2− to VCl3 found to be very fast (r2), as observed in Ni–NO3− reduction chemistry.28 Also, the conversion of high spin 2 to 3 (d7, S = 3/2) is slow due to a very less change in the CFSE compare to the transformation of high spin 3 to a low spin 4 (d6, S = 0) with much change in the CFSE, additionally support our chemistry. Furthermore, direct generation of 4 from 3 supports our proposition that CoII–NO2− involved as an intermediate species in NO3− to NO transformation. Tracking the reactions using 15N-labeled-15NO3− and 15NO2− evidently suggests that the N-atom in the {CoNO}8 species is derived from NO3− moiety. N–O bond activation20b,21a,27,30,34a,34c of coordinated NO3− in 2 generates 4,19d hence implying that the OAT reaction of 2 in the presence of VCl3 generates the Co–nitrosyl species. This work highlights the first-ever report of VCl3 encouraged the reduction of NO3− to NO2− (NRs activity, OAT-1) followed by another OAT induced NO2− to NO transformation (OAT-2). In nature, both the reduction process needs two different enzymes for converting NO3− to NO; hence, the proposed OAT reagent (VCl3), which is capable of doing the same in a one-shot, has border significance with respect to biological as well as environmental systems.
Author contributions
PKK & Kulbir discovered/conceptualized the initial project. Kulbir, SD, MG, PB, & MY carried out the different experiments and gathered the data. PKK, SG & TD helped in interpreting the experimental results. Kulbir and SD write the first draft of the article. PKK & TD have corrected the manuscript, finalized the final draft, and guided during the revision. PKK followed and guided the whole project work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid (Grant No. EEQ/2016/000466) from SERB-DST. KK, SD, MY, PB, and SG thank IISER Tirupati for financial assistance and for providing the research facility. SCS thanks DST-FIST for the single crystal facility at PU. Special thanks to Prof. K. Vijayamohanan Pillai (IISER Tirupati) for fruitful discussion and support.
References
- P. Tavares, A. S. Pereira, J. J. Moura and I. Moura, J. Inorg. Biochem., 2006, 100, 2087–2100 CrossRef CAS PubMed.
- E. Weitzberg and J. O. Lundberg, Nitric Oxide, 1998, 2, 1–7 CrossRef CAS PubMed.
- J. O. Lundberg and M. Govoni, Free Radical Biol. Med., 2004, 37, 395–400 CrossRef CAS PubMed.
-
(a) L. Ma, L. Hu, X. Feng and S. Wang, Aging Dis., 2018, 9, 938–945 CrossRef PubMed;
(b) G. M. McKnight, C. W. Duncan, C. Leifert and M. H. Golden, Br. J. Nutr., 1999, 81, 349–358 CrossRef CAS PubMed;
(c) M. J. Hill, G. Hawksworth and G. Tattersall, Br. J. Cancer, 1973, 28, 562–567 CrossRef CAS PubMed;
(d) L. Juhasz, M. J. Hill and G. Nagy, IARC Sci. Publ., 1980, 619–623 CAS;
(e) O. M. Jensen, Ecotoxicol. Environ. Saf., 1982, 6, 258–267 CrossRef CAS PubMed;
(f) C. J. Johnson and B. C. Kross, Am. J. Ind. Med., 1990, 18, 449–456 CrossRef CAS PubMed.
-
(a) L. I. Hochstein and G. A. Tomlinson, Annu. Rev. Microbiol., 1988, 42, 231–261 CrossRef CAS PubMed;
(b) W. H. Campbell, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1999, 50, 277–303 CrossRef CAS PubMed.
- N. Benjamin, F. O'Driscoll, H. Dougall, C. Duncan, L. Smith, M. Golden and H. McKenzie, Nature, 1994, 368, 502 CrossRef CAS PubMed.
- J. O. Lundberg, E. Weitzberg, J. M. Lundberg and K. Alving, Gut, 1994, 35, 1543–1546 CrossRef CAS PubMed.
- B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. Murphy, Science, 2004, 304, 867–870 CrossRef CAS PubMed.
- R. B. S. Nabi, R. Tayade, A. Hussain, K. P. Kulkarni, Q. M. Imran, B.-G. Mun and B.-W. Yun, Environ. Exp. Bot., 2019, 161, 120–133 CrossRef CAS.
-
(a) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS;
(b) L. J. Ignarro, Angew. Chem., Int. Ed., 1999, 38, 1882–1892 CrossRef CAS;
(c)
L. J. Ignarro, Nitric oxide: biology and pathobiology, Academic press, 2000 Search PubMed;
(d) G. B. Richter-Addo, P. Legzdins and J. Burstyn, Chem. Rev., 2002, 102, 857–860 CrossRef CAS PubMed;
(e) I. M. Wasser, S. de Vries, P. Moënne-Loccoz, I. Schröder and K. D. Karlin, Chem. Rev., 2002, 102, 1201–1234 CrossRef CAS PubMed.
- F. Vargas, J. M. Moreno, R. Wangensteen, I. Rodriguez-Gomez and J. Garcia-Estan, Eur. J. Endocrinol., 2007, 156, 1–12 CAS.
-
(a) P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed;
(b) R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed;
(c) B. Kalyanaraman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11527–11528 CrossRef CAS PubMed;
(d) P. C. Dedon and S. R. Tannenbaum, Arch. Biochem. Biophys., 2004, 423, 12–22 CrossRef CAS PubMed.
-
(a) R. E. Huie and S. Padmaja, Free Radical Res. Commun., 1993, 18, 195–199 CrossRef CAS PubMed;
(b) C. Prolo, M. N. Alvarez and R. Radi, Biofactors, 2014, 40, 215–225 CrossRef CAS PubMed.
-
(a) W. C. Nottingham and J. R. Sutter, Int. J. Chem. Kinet., 1986, 18, 1289–1302 CrossRef CAS;
(b) C. H. Lim, P. C. Dedon and W. M. Deen, Chem. Res. Toxicol., 2008, 21, 2134–2147 Search PubMed.
-
(a) D. D. Thomas, L. A. Ridnour, J. S. Isenberg, W. Flores-Santana, C. H. Switzer, S. Donzelli, P. Hussain, C. Vecoli, N. Paolocci, S. Ambs, C. A. Colton, C. C. Harris, D. D. Roberts and D. A. Wink, Free Radical Biol. Med., 2008, 45, 18–31 CrossRef CAS PubMed;
(b) D. L. Granger and J. B. Hibbs Jr, Trends Microbiol., 1996, 4, 46–47 CrossRef CAS PubMed;
(c) C. F. Nathan and J. B. Hibbs Jr, Curr. Opin. Immunol., 1991, 3, 65–70 CrossRef CAS.
-
N. Lehnert, T. C. Berto, M. G. I. Galinato and L. E. Goodrich, in Handbook of Porphyrin Science, ed. K. Kadish, K. Smith and R. Guilard, World Scientific Publishing, Singapore, 2011, p. 1 Search PubMed.
-
(a) T. B. McCall, N. K. Boughton-Smith, R. M. Palmer, B. J. Whittle and S. Moncada, Biochem. J., 1989, 261, 293–296 CrossRef CAS PubMed;
(b) R. G. Knowles and S. Moncada, Biochem. J., 1994, 298(Pt 2), 249–258 CrossRef CAS PubMed.
-
(a) P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed;
(b) M. P. Schopfer, B. Mondal, D. H. Lee, A. A. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 11304–11305 CrossRef CAS PubMed;
(c) M. P. Doyle and J. W. Hoekstra, J. Inorg. Biochem., 1981, 14, 351–358 CrossRef CAS PubMed;
(d) M. Yenuganti, S. Das, Kulbir, S. Ghosh, P. Bhardwaj, S. S. Pawar, S. C. Sahoo and P. Kumar, Inorg. Chem. Front., 2020, 7, 4872–4882 RSC.
-
(a) S. G. Clarkson and F. Basolo, Inorg. Chem., 1973, 12, 1528–1534 CrossRef CAS;
(b) P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed;
(c) K. Gogoi, S. Saha, B. Mondal, H. Deka, S. Ghosh and B. Mondal, Inorg. Chem., 2017, 56, 14438–14445 CrossRef CAS PubMed;
(d) G. Y. Park, S. Deepalatha, S. C. Puiu, D. H. Lee, B. Mondal, A. A. Narducci Sarjeant, D. del Rio, M. Y. Pau, E. I. Solomon and K. D. Karlin, J. Biol. Inorg Chem., 2009, 14, 1301–1311 CrossRef CAS PubMed;
(e) A. Yokoyama, K. B. Cho, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2013, 135, 14900–14903 CrossRef CAS PubMed;
(f) S. Hong, P. Kumar, K. B. Cho, Y. M. Lee, K. D. Karlin and W. Nam, Angew. Chem., Int. Ed. Engl., 2016, 55, 12403–12407 CrossRef CAS PubMed;
(g) A. Yokoyama, J. E. Han, K. D. Karlin and W. Nam, Chem. Commun., 2014, 50, 1742–1744 RSC;
(h) A. Kalita, P. Kumar and B. Mondal, Chem Commun., 2012, 48, 4636–4638 RSC.
-
(a) S. Das, Kulbir, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2020, 11, 5037–5042 RSC;
(b) F. Roncaroli, L. M. Baraldo, L. D. Slep and J. A. Olabe, Inorg. Chem., 2002, 41, 1930–1939 CrossRef CAS PubMed;
(c) A. Kalita, P. Kumar, R. C. Deka and B. Mondal, Chem Commun., 2012, 48, 1251–1253 RSC.
- J. Jiang and R. H. Holm, Inorg. Chem., 2005, 44, 1068–1072 CrossRef CAS.
- A. Majumdar, K. Pal and S. Sarkar, J. Am. Chem. Soc., 2006, 128, 4196–4197 CrossRef CAS PubMed.
- L. T. Elrod and E. Kim, Inorg. Chem., 2018, 57, 2594–2602 CrossRef CAS PubMed.
- C. Khin, J. Heinecke and P. C. Ford, J. Am. Chem. Soc., 2008, 130, 13830–13831 CrossRef CAS PubMed.
- S. Kundu, W. Y. Kim, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 1045–1048 CrossRef CAS PubMed.
- M. A. Puthiyaveetil Yoosaf, S. Ghosh, Y. Narayan, M. Yadav, S. C. Sahoo and P. Kumar, Dalton Trans., 2019, 48, 13916–13920 RSC.
- J. Gwak, S. Ahn, M. H. Baik and Y. Lee, Chem. Sci., 2019, 10, 4767–4774 RSC.
- J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1994, 116, 5475–5476 CrossRef CAS.
- P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed.
-
(a) L. Cheng, M. A. Khan, G. B. Richter-Addo and D. R. Powell, Chem Commun., 2000, 2301–2302, 10.1039/b006775j;
(b) A. K. Patra, R. K. Afshar, J. M. Rowland, M. M. Olmstead and P. K. Mascharak, Angew. Chem., Int. Ed., 2003, 42, 4517–4521 CrossRef CAS PubMed;
(c) B. C. Sanders, S. M. Hassan and T. C. Harrop, J. Am. Chem. Soc., 2014, 136, 10230–10233 CrossRef CAS PubMed;
(d) T. S. Kurtikyan, A. A. Hovhannisyan, A. V. Iretskii and P. C. Ford, Inorg. Chem., 2009, 48, 11236–11241 CrossRef CAS PubMed.
- P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed.
- P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed.
-
(a) C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12023–12031 CrossRef CAS PubMed;
(b) M. R. Walter, S. P. Dzul, A. V. Rodrigues, T. L. Stemmler, J. Telser, J. Conradie, A. Ghosh and T. C. Harrop, J. Am. Chem. Soc., 2016, 138, 12459–12471 CrossRef CAS PubMed;
(c) Y. Guo, J. R. Stroka, B. Kandemir, C. E. Dickerson and K. L. Bren, J. Am. Chem. Soc., 2018, 140, 16888–16892 CrossRef CAS;
(d) C. H. Chuang, W. F. Liaw and C. H. Hung, Angew. Chem., Int. Ed., 2016, 55, 5190–5194 CrossRef CAS PubMed.
-
(a) O. W. Howarth, Prog. Nucl. Magn. Reson. Spectrosc., 1990, 22, 453–485 CrossRef CAS;
(b) D. Rehder, Bull. Magn. Reson., 1982, 4, 33–83 CAS.
- W. H. Campbell, Cell. Mol. Life Sci., 2001, 58, 194–204 CrossRef CAS PubMed.
- The product of VCl3 induced OAT-2 reaction is {CoNO}8 not {CoNO}9, suggesting a missing electron in the Co–NO2− reduction reaction. One electron loss has been shown in the Scheme 1 and 2 for better understanding, which probably solvated in presence of excess solvent. As a tiny entity, it is very challenging to track an electron in the system. As there is a missing electron in the overall NO2− reduction reaction, we did not describe a more detailed mechanism and based on the product analysis, we proposed the OAT transfer from NO2− moiety.
-
(a) K. R. Siefermann, Y. Liu, E. Lugovoy, O. Link, M. Faubel, U. Buck, B. Winter and B. Abel, Nat. Chem., 2010, 2, 274–279 CrossRef CAS PubMed;
(b) C. R. Wang, J. Nguyen and Q. B. Lu, J. Am. Chem. Soc., 2009, 131, 11320–11322 CrossRef CAS PubMed;
(c) L. Sanche, Nature, 2009, 461, 358–359 CrossRef CAS PubMed;
(d) V. Petrosyan, M. E. Niyazymbetov, é. V. Ul'yanov and U. Niyazymbetov, Division of chemical science, 1989, 38, 1548–1551 Search PubMed.
-
(a) S. Goswami, D. Sen, N. K. Das, H. K. Fun and C. K. Quah, Chem Commun., 2011, 47, 9101–9103 RSC;
(b) J. Chen, H. Yoon, Y. M. Lee, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Chem. Sci., 2015, 6, 3624–3632 RSC;
(c) Y. M. Lee, M. Yoo, H. Yoon, X. X. Li, W. Nam and S. Fukuzumi, Chem Commun., 2017, 53, 9352–9355 RSC.
- S. Fukuzumi, K. Ohkubo, Y.-M. Lee and W. Nam, Chem. -Eur. J., 2015, 21, 17548–17559 CrossRef CAS PubMed.
- J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS PubMed.
- H. Taube, Chem. Rev., 1952, 50, 69–126 CrossRef CAS.
-
G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, 5th edn, 2014 Search PubMed.
- B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed.
- A. Tilstra, Y. C. El-Khaled, F. Roth, N. Radecker, C. Pogoreutz, C. R. Voolstra and C. Wild, Sci. Rep., 2019, 9, 19460 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For the Experimental details. CCDC 2058406 and 2058407. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00803j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sandip
Das
a,
Sandip
Das