
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeepReac+: deep active learning for quantitative modeling of organic chemical reactions†
Yukang
Gong
,
Dongyu
Xue
,
Guohui
Chuai
,
Jing
Yu
 * and
Qi
Liu
*
* and
Qi
Liu
*
Department of Ophthalmology, Shanghai Tenth People's Hospital, Bioinformatics Department, School of Life Sciences and Technology, Tongji University, Shanghai, 200072, China. E-mail: dryujing@aliyun.com; qiliu@tongji.edu.cn
First published on 9th October 2021
Abstract
Various computational methods have been developed for quantitative modeling of organic chemical reactions; however, the lack of universality as well as the requirement of large amounts of experimental data limit their broad applications. Here, we present DeepReac+, an efficient and universal computational framework for prediction of chemical reaction outcomes and identification of optimal reaction conditions based on deep active learning. Under this framework, DeepReac is designed as a graph-neural-network-based model, which directly takes 2D molecular structures as inputs and automatically adapts to different prediction tasks. In addition, carefully-designed active learning strategies are incorporated to substantially reduce the number of necessary experiments for model training. We demonstrate the universality and high efficiency of DeepReac+ by achieving the state-of-the-art results with a minimum of labeled data on three diverse chemical reaction datasets in several scenarios. Collectively, DeepReac+ has great potential and utility in the development of AI-aided chemical synthesis. DeepReac+ is freely accessible at https://github.com/bm2-lab/DeepReac.
Introduction
Synthetic organic chemistry is a cornerstone of several disciplines and industries,1–3 such as chemical biology, materials science and the pharmaceutical industry. Due to the complex and nonlinear nature of organic chemistry, organic synthesis is frequently described as an art that must be routinely practiced for several years to be mastered.4,5 As a key aspect of the synthetic methodology, the optimization of reaction conditions is often driven by chemical intuition, which can be biased by personal preferences and chemical education.6–8 Within a modern chemistry setting, the reaction performance, including yield and selectivity (chemo-, regio-, diastereo- and enantioselectivity), can be controlled by dozens of variables, such as the leaving groups, (co-)catalysts, temperature, solvents, and additives. The combinatorial fabrication of these factors can produce a large reaction space to be explored, which is known as combinatorial explosion, making it impractical and challenging for chemists to assess all available options to identify the optimal reaction conditions. A growing number of researchers and companies have recognized this issue, and various systematic synthesis techniques have been developed with the aid of high-throughput experimentation9 and flow chemistry.10,11While such synthesis techniques enable standardization and parallelization, an exhaustive search of the entire reaction space is impossible as it is too large. Artificial intelligence technologies such as various machine learning models, which have been successfully applied in similar scenarios, including virtual screening,12–16 material discovery,17–20 molecular design,21–24 and synthesis planning,25 were rapidly introduced to predict potential reaction outcomes before experimentation.26–32 However, two limitations exist for current computational approaches: (1) there is a lack of universality and generalization in the modeling of various sorts of chemical reactions. For different reaction mechanisms or prediction tasks, researchers have to design different customized reaction descriptors based on certain scientific hypotheses. These handcrafted descriptors are subject to a limited application scope as well as the bias of specific designers. There is no guarantee that enough task-related information will be considered and represented well, which is a well-known bottleneck of traditional machine learning. A recent report shared the same concern, in which a universal version of molecular fingerprints was developed to achieve state-of-the-art predictive performance in three different chemical reaction prediction tasks.33 Although the combined molecular fingerprints contain more structural information, the feature dimension of a single molecule is up to 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 374. For a complex organic reaction containing multiple components, i.e. 4 components, the feature dimension of the reaction can reach tens of thousands. In addition, the random forest model which achieved state-of-the-art predictive performances has 5000–10000 decision trees. The above two points make the approach require high demanding computing resources. (2) Another challenge is that current methods still need a large amount of data to train the model for high predictive performance. To reduce the cost and accelerate the optimization process, active strategies need to be comprehensively investigated to selectively explore the reaction space with reduced cost and time. As an efficient optimization algorithm, Bayesian optimization has been used in reaction condition optimization to reduce the number of experiments.34 It typically consists of two major steps: (1) construct a surrogate to the underlying black box function; (2) propose new parameter points for querying the function based on this probabilistic approximation. Although Bayesian optimization is a powerful tool, its application might be limited due to its surrogate models. While several different models have been proposed to be the surrogate, including random forests,35 Gaussian processes36,37 and Bayesian neural networks,34,38 not all models can meet the requirement and serve readily as a surrogate. Moreover, just as the above-mentioned point, the effect could be compromised without carefully designed descriptors.
374. For a complex organic reaction containing multiple components, i.e. 4 components, the feature dimension of the reaction can reach tens of thousands. In addition, the random forest model which achieved state-of-the-art predictive performances has 5000–10000 decision trees. The above two points make the approach require high demanding computing resources. (2) Another challenge is that current methods still need a large amount of data to train the model for high predictive performance. To reduce the cost and accelerate the optimization process, active strategies need to be comprehensively investigated to selectively explore the reaction space with reduced cost and time. As an efficient optimization algorithm, Bayesian optimization has been used in reaction condition optimization to reduce the number of experiments.34 It typically consists of two major steps: (1) construct a surrogate to the underlying black box function; (2) propose new parameter points for querying the function based on this probabilistic approximation. Although Bayesian optimization is a powerful tool, its application might be limited due to its surrogate models. While several different models have been proposed to be the surrogate, including random forests,35 Gaussian processes36,37 and Bayesian neural networks,34,38 not all models can meet the requirement and serve readily as a surrogate. Moreover, just as the above-mentioned point, the effect could be compromised without carefully designed descriptors.
The first chemical reaction representation issue could potentially be addressed by applying graph neural networks (GNNs). With the rapid development of deep learning techniques in recent years, GNNs have recently become one of the main research focuses in view of their powerful ability to model graph-structured data.39–41 Graph structures prevail in society and nature, especially in biology and chemistry, where GNNs have been applied and have made remarkable achievements.42–47 Due to their capacity to directly model molecular structures, GNNs have obtained better performance on different prediction tasks, including biological activity, toxicity and quantum chemical properties, than traditional shallow-learning-based approaches that use manually designed descriptors derived from molecular structures.48–53 Thus, we hypothesize that GNNs could also be applied well to the quantitative modeling of reaction outcomes. Such a model would directly take the (2D) Lewis structure of the reaction components as input and automatically extract task-related information in a representation learning way, which is expected to achieve a universal and generalized prediction performance for different types of chemical reactions. Regarding the second issue, active learning54,55 seems to be another promising solution besides Bayesian optimization. Instead of training a learning algorithm with large amounts of data to build static models, active learning can yield similarly competitive performance using only a fraction of the training data selected by a certain strategy, and it has been applied in several fields, including drug discovery,56–59 material design,60–63 molecular dynamics,64,65 and protein production optimization.66 It is noteworthy that not all data are equally valuable to the model, and similar training entries may be so redundant or uninformative that it is cost inefficient to label them.67 This is of great significance in chemical reaction modeling scenarios, which can actively select valuable samples to conduct experiments to save time and costs. Given a certain sampling strategy, active learning enables a model to selectively explore the sample space and select certain informative training data that largely improve the model performance.
We therefore present DeepReac+ (Fig. 1), which is an efficient and universal computational framework for the prediction of chemical reaction outcomes and selection of optimized reaction conditions, which particularly addresses the two aforementioned issues. It should be noted that for clarity, DeepReac+ is referred to as a computational framework, which contains the deep learning model DeepReac specifically designed for chemical reaction representation learning, and then an active learning strategy is applied to improve the model performance and save costs. The main contributions of DeepReac+ are as follows: (a) under the framework of DeepReac+, DeepReac is designed as an efficient graph-neural-network-based representation learning model for chemical reaction outcome prediction, where the 2D structures of molecules can serve as inputs for feature representation learning and subsequent prediction with a universal and generalized prediction ability. Such a model can handle any reaction performance prediction tasks, including those for yield and stereoselectivity. For some reaction components which are inappropriate or even impossible to be presented by a graph structure, we apply a mechanism-agnostic embedding strategy, which further broadens the application scope of DeepReac. (b) An active learning strategy is proposed to explore the chemical reaction space efficiently. Such a strategy helps to substantially save costs and time in reaction outcome prediction and optimal reaction condition searching by reducing the number of necessary experiments for model training. Unlike the traditional uncertainty-based sampling strategy applied in active learning, two novel sampling strategies are presented based on the representation of the reaction space for reaction outcome prediction, i.e., diversity-based sampling and adversary-based sampling. While the former is novel in the context of reaction outcome prediction, the latter is a generally new sampling strategy in the wider context of cheminformatics. In addition, two other sampling strategies, i.e., greed-based sampling and balance-based sampling, are proposed for optimal reaction condition searching. Finally, the performance of DeepReac+ is evaluated comprehensively based on three recently reported chemical reaction datasets covering different reaction mechanisms, predictive targets and synthesis platforms. The efficiencies of the proposed sampling strategies are also investigated and validated to (a) substantially improve the chemical reaction outcome prediction performance and (b) rapidly and precisely identify the optimal reaction conditions.
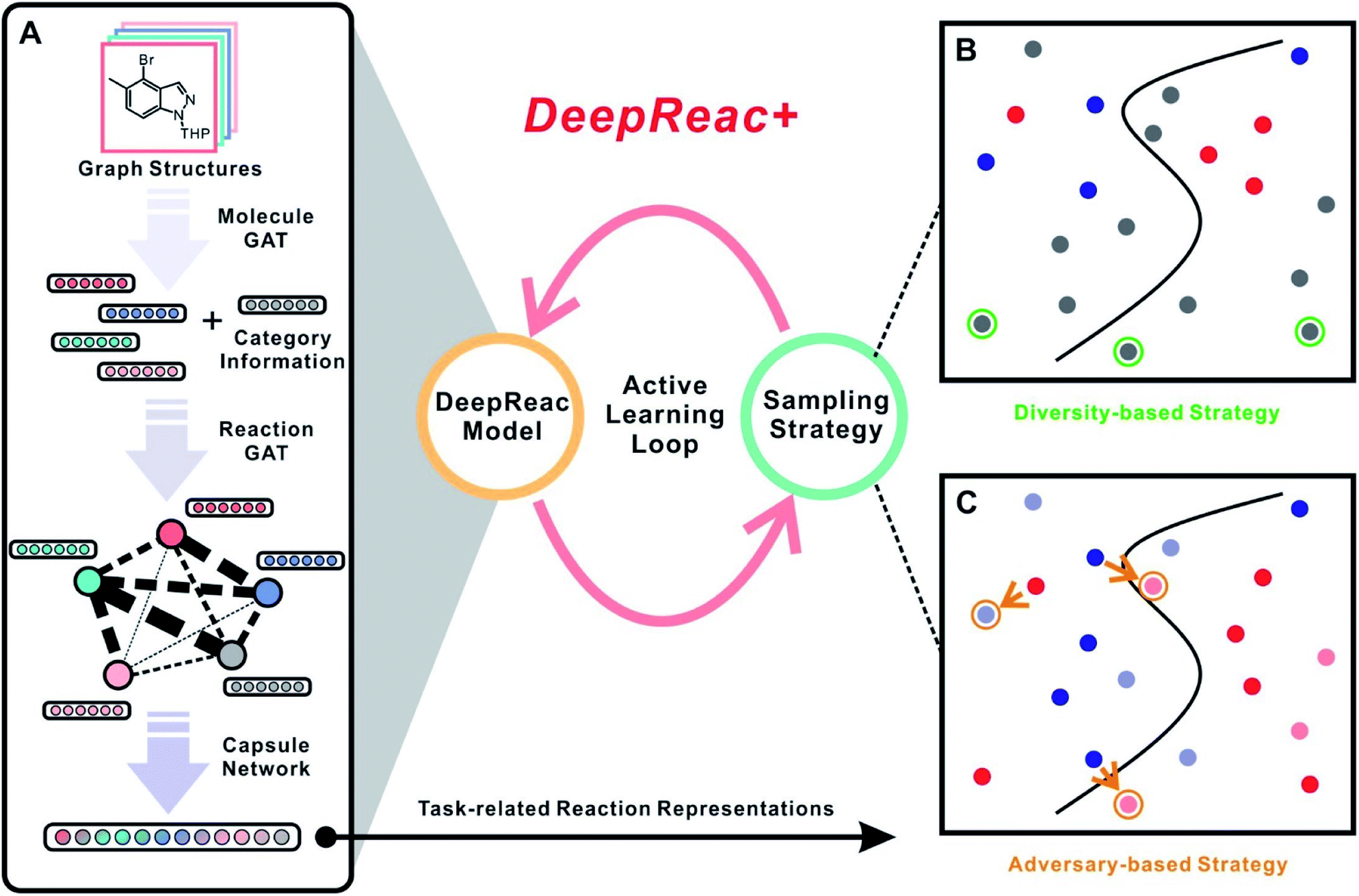 | ||
| Fig. 1 Schematic workflow of the DeepReac+ framework. (A) Architecture of the DeepReac model. The Lewis structures of organic components are used directly as inputs and encoded as feature vectors by the Molecule GAT module. If there are some components which can't be represented by graphs, they are encoded as feature vectors by an embedding layer. Then, a reaction graph is constructed, and all the feature vectors are fed into the Reaction GAT module to model the interactions among the reaction components. Finally, a Capsule module is used to aggregate all the information to produce task-related reaction representations that will be used by sampling strategies. (B) Illustration of the diversity-based sampling strategy. The blue and red circles indicate labeled data points belonging to two different classes, while the gray circles indicate unlabeled data points. The unlabeled data points marked by green circles are candidates according to the diversity-based sampling strategy. (C) Illustration of the adversary-based sampling strategy. The blue and red circles indicate labeled data points belonging to two different classes, while the light circles indicate unlabeled data points. The unlabeled data points marked by orange circles are candidates according to the adversary-based sampling strategy. The orange arrows indicate adversarial samples of the labeled data points. Note that these two illustrations use the classification problem as an example just for clarity and to make the strategies easier to be understood. The ideas behind them are also applied easily to the regression problem as reported in this paper. GAT, graph attention network. | ||
Results and discussion
The general framework of DeepReac+
DeepReac+ consists of two main parts: a deep-learning model DeepReac for chemical reaction representation learning as well as outcome prediction and an active sampler for experimental design. The architecture of the DeepReac model is shown in Fig. 1A, whose core module is a GNN. To the best of our knowledge, this is the first application to utilize a GNN for quantitative modeling of chemical reaction outcomes. A probable obstacle is that the size of the current reaction outcome datasets can only reach a few thousand data points, which encourages overfitting during the training of a deep learning model. To reduce the risk of overfitting, DeepReac is carefully designed by making the inductive bias conform to the underlying and universal principles of chemical reactions.68 The following model design principles are considered: (a) for most organic reactions, especially complex reactions, intertwined interactions within multiple components have a decisive impact on the reaction outcome. For example, a transition-metal-catalyzed organic reaction consists of several elementary reactions that connect to each other to form a catalytic cycle.69 The molecules in each step influence each other and the resulting intermediates of each step ultimately determine the performance of the entire reaction. Therefore, the interactions between reaction components should be modeled explicitly. (b) Moreover, since interaction patterns are flexible with regard to different reaction mechanisms and predictive targets, DeepReac should be able to focus on specific interactions related to the tasks of interest in an adaptative way.To satisfy the above principles, the graph attention network (GAT) is designed as a core building block in DeepReac.70–72 This graph-based attention network structure explicitly enables the model to leverage rich information by aggregating and propagating information through the attention mechanism, which focuses on the task-related part of the graph. The nodes attend to their neighbor's features and dynamically learn the edge-weight proportions with neighbors according to their importance, which enables the GAT to generalize to unseen graph structures. As shown in Fig. 1A, GAT modules are deployed in two steps: in the first step, the GAT module is utilized to encode 2D molecular structures of the reaction components as feature vectors, which is denoted as Molecule GAT; in the second step, each component is treated as a virtual node and connected to form a reaction graph whose node features come from the first step; each edge represents an interaction between two linked reaction components. The second GAT module is then utilized to deliver messages between the reaction components, which is denoted as the Reaction GAT. The weight of each edge can be learned according to the specific tasks. In addition, some reaction conditions are inappropriate or even impossible to be presented by the graph structure, including inorganic additives, special reaction media and so on. In these cases, mechanism-agnostic embeddings will be used to represent them and they are introduced into Reaction GAT along with the outputs of Molecule GAT, which means that we only need the types of the inorganic components.
Furthermore, to avoid the loss of reaction information and alleviate the need for training data, we introduce the Capsule module to aggregate the feature vectors of the reaction components after message passing. Unlike most deep learning architectures, capsule networks73 have achieved outstanding performance for small-sample learning in the fields of life sciences.74–76 As the core element, the capsule is a new type of neuron that encapsulates more information than common pooling operations by computing a small vector of highly informative outputs rather than taking only a scalar output. The dynamic routing mechanism, which can be viewed as a parallel attention mechanism, allows the network to attend to some internal capsules related to prediction. Therefore, we introduce a capsule layer as the output module to learn a task-related representation of the entire reaction (Fig. 1A). Eventually, the resulting reaction features are used to perform regression tasks, and they play a crucial role in the active learning framework.
Representation-learning-based active learning strategies presented in DeepReac+
Another key point designed in DeepReac+ is the active learning strategy, which can be applied to select the training data and substantially reduce the number of experiments to be conducted. With a well-designed sampling strategy, DeepReac is able to achieve satisfactory prediction performance rapidly through iterative retraining after every inclusion of a small number of selected experiments (Fig. 1). As a core of active learning, the sampling strategy is designed to distinguish more valuable data from other data. Traditional strategies of active learning are uncertainty-based strategies,77 which are often called “curious”, i.e., the unlabeled data are predicted, and those with a lower prediction confidence have priority in being labeled. However, deep learning models tend to be so overconfident about their predictions that the corresponding uncertainty estimation is very difficult and unreliable.78 Thus, due to their powerful representation learning ability, we designed two representation-based sampling strategies, i.e., diversity-based sampling and adversary-based sampling (Fig. 1B and C). With the reaction features automatically learned by DeepReac, we can determine the similarity between experiments with regard to specific tasks, which lays the foundation for the two strategies. In the diversity-based strategy, unlabeled data that have the least similarity to the labeled data should be labeled first (Fig. 1B). The intuition behind this strategy is that diverse data can provide the model with a global view of the reaction space and improve its generalization capability. In fact, the distance to available data in the latent space can be used empirically as a measure of uncertainty,79 which means the diversity-based strategy belongs to uncertainty-based strategies in a broad sense. However, in order to distinguish it from other methods of uncertainty estimation, “diversity-based” is used to describe this strategy. Adversarial samples, which were recently proposed in the machine learning community,80–82 generally mean that a small perturbation in the sample can cause prediction failure. This phenomenon is common in chemistry, as a minor transformation of the molecular structure, including substituted atoms and reversed chiral centers, can cause significant changes in the properties. Thus, the adversary-based strategy designed in DeepReac+ indicates that unlabeled data should be labeled first if there is a large difference between its prediction and the ground-truth value of highly similar labeled data (Fig. 1C). The intuition behind this strategy is that seeing these experimental data on the “cliffs” in the reactivity landscape can make the model robust.Benchmark datasets
We chose three datasets (Scheme 1) to test our DeepReac+ framework. To validate the versatility of our solution, these three datasets cover different kinds of reactions and predictive targets.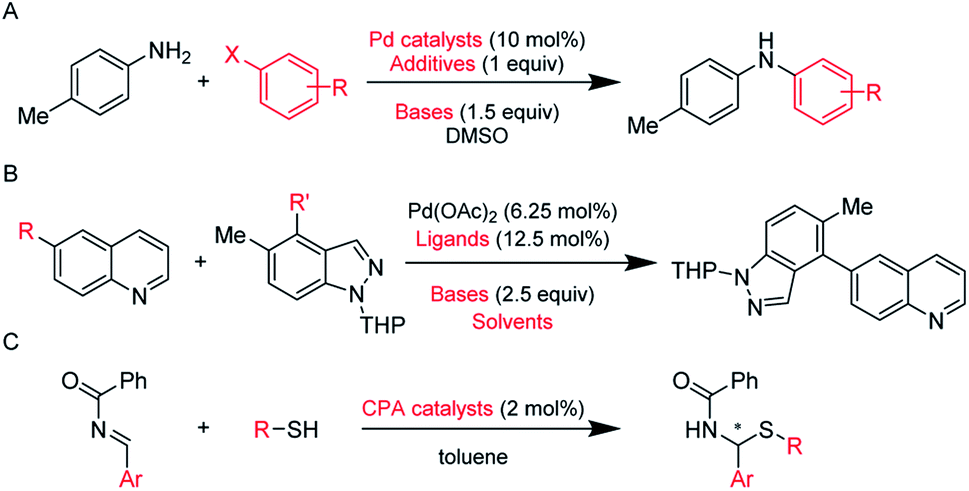 | ||
| Scheme 1 Datasets of different reaction types. (A) C–N cross coupling reactions of 4-methylaniline with various aryl halides by Doyle et al.9 The predictive target is the yield. (B) Suzuki–Miyaura cross-coupling reactions of various aryl boronic acids with various electrophiles by Sach et al.11 The predictive target is the yield. (C) Asymmetric N,S-acetal formation using CPA catalysts by Denmark et al.83 The predictive target is the enantioselectivity. The reaction variables are highlighted in red. The details of data preprocessing can be found in the ESI.† | ||
Chemical reaction outcome prediction by DeepReac without an active learning strategy
We first tested the predictive performance of DeepReac in a normal setting and compared it with the following baseline models: (1) mean; (2) median; (3) the best models reported in the original studies; (4) multiple fingerprint feature (MFF) combined with the random forest model, which achieved the state-of-the-art performance on Dataset A & C.33 To make a statistically valid comparison, five-fold cross-validation was conducted on the three datasets. As shown in Table 1, DeepReac achieved better performance in all tasks. It should be noted that the only inputs needed for DeepReac are 2D molecular structures or types of reaction components, regardless of the reaction mechanisms and predictive targets, while elaborately designed and calculated descriptors are required to meet certain hypotheses related to specific tasks. For example, the design of reaction descriptors of Dataset A takes account of the *C-3 nuclear magnetic resonance (NMR) shift (where the asterisk indicates a shared atom), lowest unoccupied molecular orbital (LUMO) energy, and *O-1 and *C-5 electrostatic charges of isoxazole additives. It's clear that the same calculation method can't be directly used on Dataset B or C which don't even have isoxazole additives as the reaction component. Therefore, the advantage clearly implies that DeepReac can be utilized effectively in a variety of predictive tasks involving organic reactions with its universal representation learning ability. While the MFF is also a universal representation of reactions, its application may be hindered by the requirement of high demanding computing resources. For instance, it spent about 24 hours for a round of cross validation on Dataset A when DeepReac only spent about 4 hours with the same computational device.| Dataset A | Dataset B | Dataset C | ||||
|---|---|---|---|---|---|---|
| RMSE | R 2 | RMSE | R 2 | MAE | R 2 | |
| a Because the validation method is different from the original studies, we retrained these models and tested. Note that the retained models have a slightly lower prediction performance than these methods reported originally. b The R2 values for the mean and median models turn out to be all negative, which are not meaningful, so they were omitted. c Since MFF didn't indicate how to encode inorganic compounds which are included in Dataset B, we didn't train the MFF + RF model on this dataset. d The values correspond to mean ± standard deviation of the CV results. The best results are given in bold. RMSE, root-mean-square error. MAE, mean absolute error, in kcal mol−1. R2, coefficient of determination. MFF, multiple fingerprint feature. RF, random forest. See also Fig. S12–S26. | ||||||
| Mean | 0.273 ± 0.002 | —b | 0.290 ± 0.004 | —b | 0.558 ± 0.035 | —b |
| Median | 0.276 ± 0.003 | —b | 0.303 ± 0.006 | —b | 0.557 ± 0.036 | —b |
| Previous work9,83,84a | 0.073 ± 0.004 | 0.919 ± 0.010 | 0.180 ± 0.004 | 0.354 ± 0.034 | 0.186 ± 0.010 | 0.822 ± 0.020 |
| MFF + RF33a | 0.071 ± 0.004 | 0.924 ± 0.009 | —c | —c | 0.132 ± 0.010 | 0.912 ± 0.012 |
| DeepReac | 0.053 ± 0.004 | 0.960 ± 0.006 | 0.088 ± 0.006 | 0.901 ± 0.013 | 0.096 ± 0.018 | 0.956 ± 0.012 |
| DeepReac_noG | 0.134 ± 0.011 | 0.674 ± 0.067 | 0.171 ± 0.008 | 0.467 ± 0.072 | 0.178 ± 0.021 | 0.852 ± 0.026 |
| DeepReac_noC | 0.061 ± 0.003 | 0.949 ± 0.005 | 0.096 ± 0.001 | 0.884 ± 0.003 | 0.185 ± 0.011 | 0.847 ± 0.025 |
| DeepReac_noGC | 0.150 ± 0.004 | 0.568 ± 0.007 | 0.200 ± 0.004 | 0.114 ± 0.068 | 0.198 ± 0.014 | 0.837 ± 0.017 |
Additionally, we conducted an ablation study to validate the necessity of the modules of DeepReac, especially the Reaction GAT module and Capsule module. We designed three ablation test scenarios: (1) DeepReac_noG: DeepReac without the Reaction GAT module; (2) DeepReac_noC: DeepReac without the Capsule module; and (3) DeepReac_noGC: DeepReac without both the Reaction GAT module and Capsule module. These tests were then performed on the three datasets. Various degrees of decline in the predictive performance were observed for all ablated models, especially for the model lacking both modules (Table 1), which means that a simple concatenation of the molecular feature is not enough for the representation of the reaction. Absence of the Capsule module didn't cause a dramatic decline on Dataset A & B but on Dataset C, implying the effect of the capsule network on small-sample learning. This result indicates that both the Reaction GAT module and the Capsule module are likely to fit the inductive bias and guarantee the predictive performance of DeepReac.
Chemical reaction outcome prediction by DeepReac with an active learning strategy
After validating the predictive ability of DeepReac, the overall effect of DeepReac+, i.e., the DeepReac model with an active learning strategy, was examined. We tested two sampling strategies based on the learned reaction representation, including diversity-based sampling and adversary-based sampling, by running simulations on the three datasets (the detailed process is described in the Methods section of the ESI†). In addition, a random strategy was used as the baseline. Since the initial 10% training set is too small to perform meaningful hyperparameter optimization, the same hyperparameters that achieve the best performance in most of the splits on the three datasets were used to perform simulations on all the three datasets for consistency. During simulation, the predictive performance of DeepReac was recorded after each retraining with a few selected data points. The results of 30 simulations on the three datasets are summarized in Fig. 2A–C. For each dataset, using only approximately 30–50% of the data, the well retrained DeepReac achieved the same predictive performance as that obtained using 70% training data without active learning, which is shown as a dashed line. Compared with the random sampling strategy, the two active learning strategies can obtain a model with similarly good predictive performance by using much less data on all three datasets. The adversary-based strategy is slightly superior to the diversity-based strategy.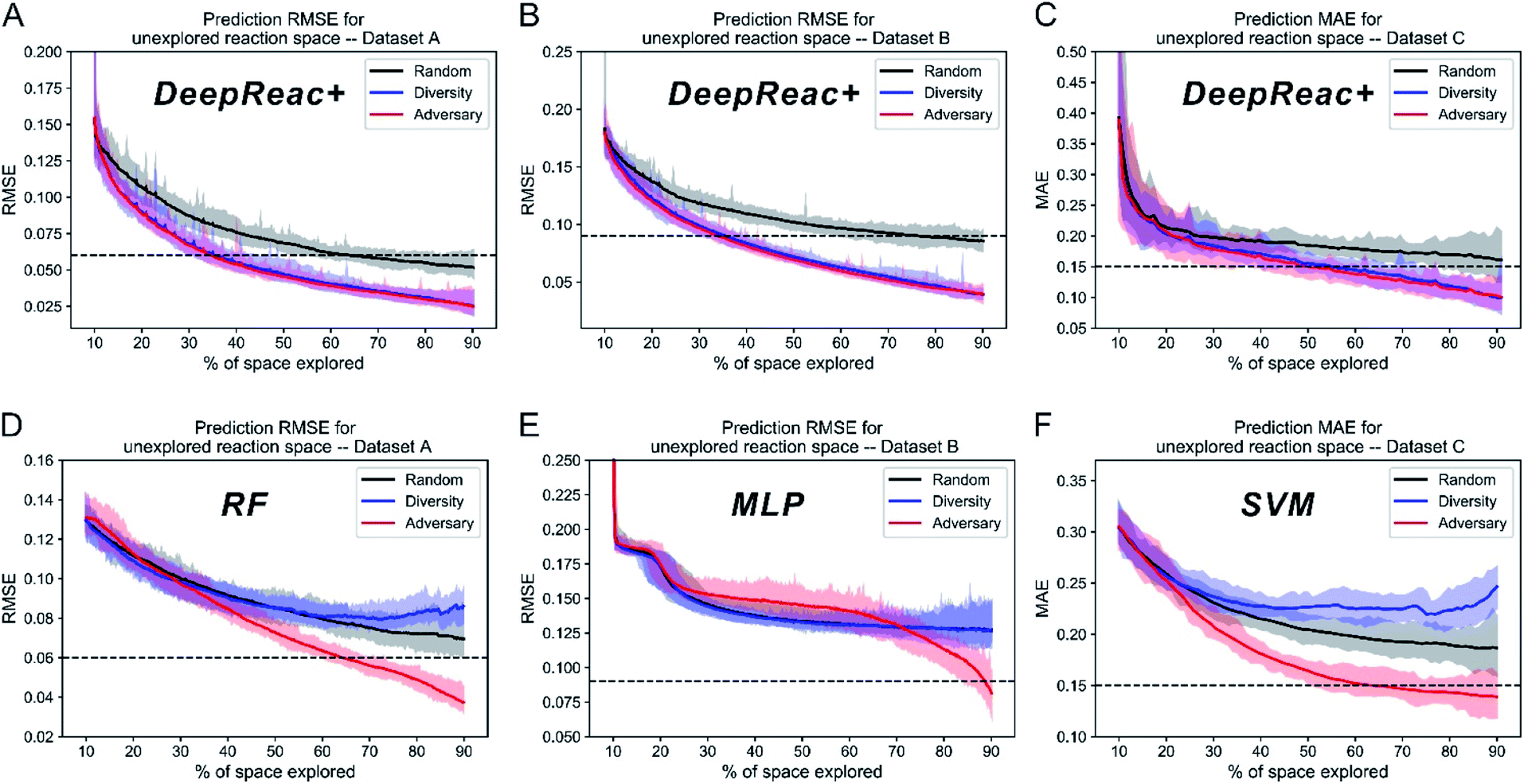 | ||
| Fig. 2 Simulation results of three sampling strategies with DeepReac and other models on three benchmark datasets in library mode. The aggregated results from 30 simulations show the average RMSE/MAE of DeepReac on Dataset A (A), Dataset B (B) and Dataset C (C) versus the fraction of the chemical space explored; the filled areas around the curves are defined by the maximum and minimum values. The black line indicates the random sampling strategy, the blue line indicates the diversity-based sampling strategy, and the red line indicates the adversary-based sampling strategy. The horizontal dashed black line indicates the model performance achieved using 70% training data without active learning. Since the hyperparameters used during simulation don't perform best on Dataset C, the MAE here is larger than that obtained during cross validation. The aggregated results from 30 simulations showing the average RMSE/MAE of RF/MLP/SVM on Dataset A (D), Dataset B (E) and Dataset C (F) versus the fraction of the chemical space explored; the filled areas around the curves are defined by the maximum and minimum values. The black line indicates the random sampling strategy, the blue line indicates the diversity-based sampling strategy, and the red line indicates the adversary-based sampling strategy. The horizontal dashed black line indicates the model performance achieved using 70% training data without active learning. RMSE, root-mean-square error. MAE, mean absolute error, in kcal mol−1. RF, random forest. MLP, multilayer perceptron. SVM, support vector machine. | ||
Since various synthesis platforms can conduct different number of experiments in one batch, we also tested the impact of the number of candidates of each iteration on the effect of sampling strategies. Taking the adversary-based strategy as an example, we chose 10, 50 and 96 as the number of candidates respectively and performed the same simulation. The results are summarized in Fig. S1† and they indicate that the choice of the number of candidates do not affect the upward tendency of model performance.
To vividly demonstrate the capacity of the representation learning of DeepReac and the difference between the sampling strategies, we used the t-SNE (t-distributed stochastic neighbor embedding) technique,85 which is a technique for dimensionality reduction that is particularly well suited for the visualization of high-dimensional data, to visualize the reaction features learned at different stages with each sampling strategy. Taking Dataset A as an example, the reaction features were extracted to be visualized when 10% and 30% of the data were used to train the model, and the corresponding candidates according to each sampling strategy were marked (Fig. 3). The same 10% of the data were used to pretrain the model so the preferences of the different sampling strategies could be clearly compared. From the perspective of representation learning, along with the improvement in the predictive performance, the distribution of the samples based on the representation learned by DeepReac becomes increasingly regular, which means that the samples with similar labels have similar representations. A comparison of the candidates chosen by different sampling strategies shows a certain degree of preference: the samples chosen by the random strategy are distributed over the whole reaction space (Fig. 3A and B), while those chosen by the diversity and adversary-based sampling strategies tend to focus on some specific areas (Fig. 3C–F). Similar results were observed on the other two datasets (Fig. S2 and S3†) and confirmed further by corresponding principal component analysis (PCA) which preserves distances in the latent space vectors (Fig. S4–S6†).
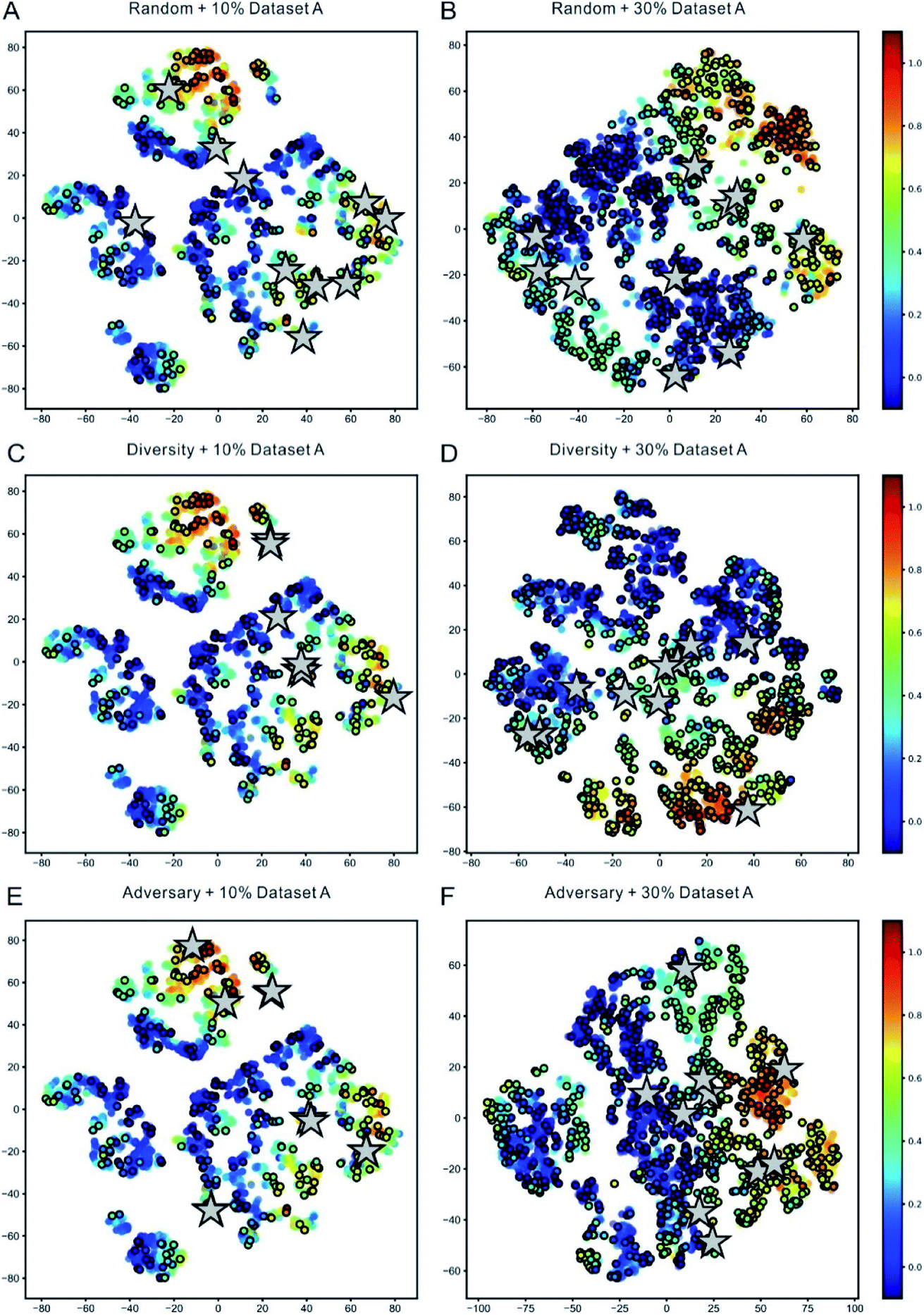 | ||
| Fig. 3 Visualization of representation learning for the DeepReac model with three sampling strategies on Dataset A. t-Distributed stochastic neighbor embedding (t-SNE) of all reaction representations outputted by the Capsule module in the DeepReac model trained with 10% data. The data points are colored according to the true yield, and the labeled points are indicated by black edges. The candidates selected by the random sampling strategy (A), diversity-based sampling strategy (C) and adversary-based sampling strategy (E) are marked as grey stars. t-SNE of all reaction presentations outputted by the Capsule module in the DeepReac model trained with 30% data in the active learning setting. The data points are colored according to the true yield, and the labeled points are indicated by black edges. The candidates selected by the random sampling strategy (B), diversity-based sampling strategy (D) and adversary-based sampling strategy (F) are marked as grey stars. See also Fig. S2–S6.† | ||
Comparison between DeepReac+ and traditional machine learning models with active learning strategies in chemical reaction outcome prediction
For a more objective and comprehensive comparison, we also tested the two active learning strategies with traditional machine learning models, including the random forest (RF), multilayer perceptron (MLP) and support vector machine (SVM) on Datasets A, B and C, respectively. Since the MFF approach consumes a lot of computing resources, i.e. one training process spends several hours on our servers, and simulation requires dozens or hundreds of rounds of retraining, it's not practical to use the MFF to perform simulation. The customized descriptors (Datasets A & C) as well as one-hot encoding (Dataset B) were used as the reaction features, and the simulation process mentioned above was conducted on all three datasets (Fig. 2D–F). According to the results, the diversity-based strategy is inferior to the random strategy on Datasets A and C (Fig. 2D and F), while there is no difference on Dataset B (Fig. 2E), indicating that the effect of the diversity-based strategy is representation dependent. Surprisingly, unlike the diversity-based strategy, the adversary-based strategy is almost representation independent as well as model independent since it performed best at all times except in the early stage on Dataset B (Fig. 2E). It should be noted that after repeated selective sampling by active learning strategies, the remaining small amount of data will have a strongly non-random distribution. Compared with DeepReac+, traditional machine learning models seem to be affected and behave strangely with a certain active learning strategy, i.e. declining performance with more training data. A possible reason is that the reaction representations are learnt adaptively by DeepReac+, while descriptors are pre-defined which could cause limited predictive ability on non-random data distribution. By combining all the simulation results into a summary (Table 2), it is clearly concluded that our proposed DeepReac model equipped with an adversary-based sampling strategy can take a single form of input to fit different reaction mechanisms and predictive targets and can achieve better performance with fewer samples, simultaneously exhibiting versatility and efficiency.| Sampling strategy | Random | Diversity | Adversary | |
|---|---|---|---|---|
| a The best results are given in bold. The criteria of model performance on the three benchmark datasets are shown in parentheses. RMSE, root-mean-square error. MAE, mean absolute error, in kcal mol−1. RF, random forest. MLP, multilayer perceptron. SVM, support vector machine. | ||||
| Dataset A expert annotated (RMSE < 0.06) | DeepReac | 64.3% | 35.3% | 34.8% |
| RF | >90.0% | >90.0% | 64.3% | |
| Dataset B expert annotated (RMSE < 0.09) | DeepReac | 76.6% | 35.6% | 34.0% |
| MLP | >90.0% | >90.0% | 88.8% | |
| Dataset C expert annotated (MAE < 0.15) | DeepReac | >90.0% | 55.5% | 50.9% |
| SVM | >90.0% | >90.0% | 64.8% | |
Performance of DeepReac+ and traditional machine learning models with active learning strategies on unseen reaction components
The above simulation can be named library mode, which means that for a certain type of reaction, reaction components, including substrates and reagents, to be screened have been defined in advance. According to the results, active learning achieves a good predictive ability on the whole library using only a small fraction of data. However, sometimes substrates or reagents are not easily available due to high cost or other reasons. Thus, we also tested whether active learning can enable the model to achieve generalized predictive ability when reactions contain components unseen in the library. For consistency, (co-)catalysts, which three datasets have in common, are chosen as the reference. According to (co-)catalysts, all datasets are divided into 4 sets since Dataset A contains only 4 catalysts (Table S1†). Three of them make up a library for screening and the remaining one works as a validation set which can't be sampled by active learning strategies. For convenience, we named this simulation as “catalyst-unknown mode”. The corresponding results are summarized in Fig. 4A–C and S7–S9.† Both diversity-based sampling and adversary-based sampling have slight or even no advantage over random sampling depending on different data splitting. This indicates that active learning strategies work better in library mode and have limited power to boost the generalized predictive ability of DeepReac. In other words, active learning strategies can help the model to generalize its predictive ability on unseen combinations rather than unseen reaction components. We also tested the two active learning strategies with traditional machine learning models in catalyst-unknown mode. The corresponding results are summarized in Fig. 4D–F and S7–S9.† There was no difference between the effects of sampling strategies. In addition, SVM outperformed DeepReac on Dataset C whose size is the smallest, indicating that the generalization ability of the deep learning model will be compromised by small datasets.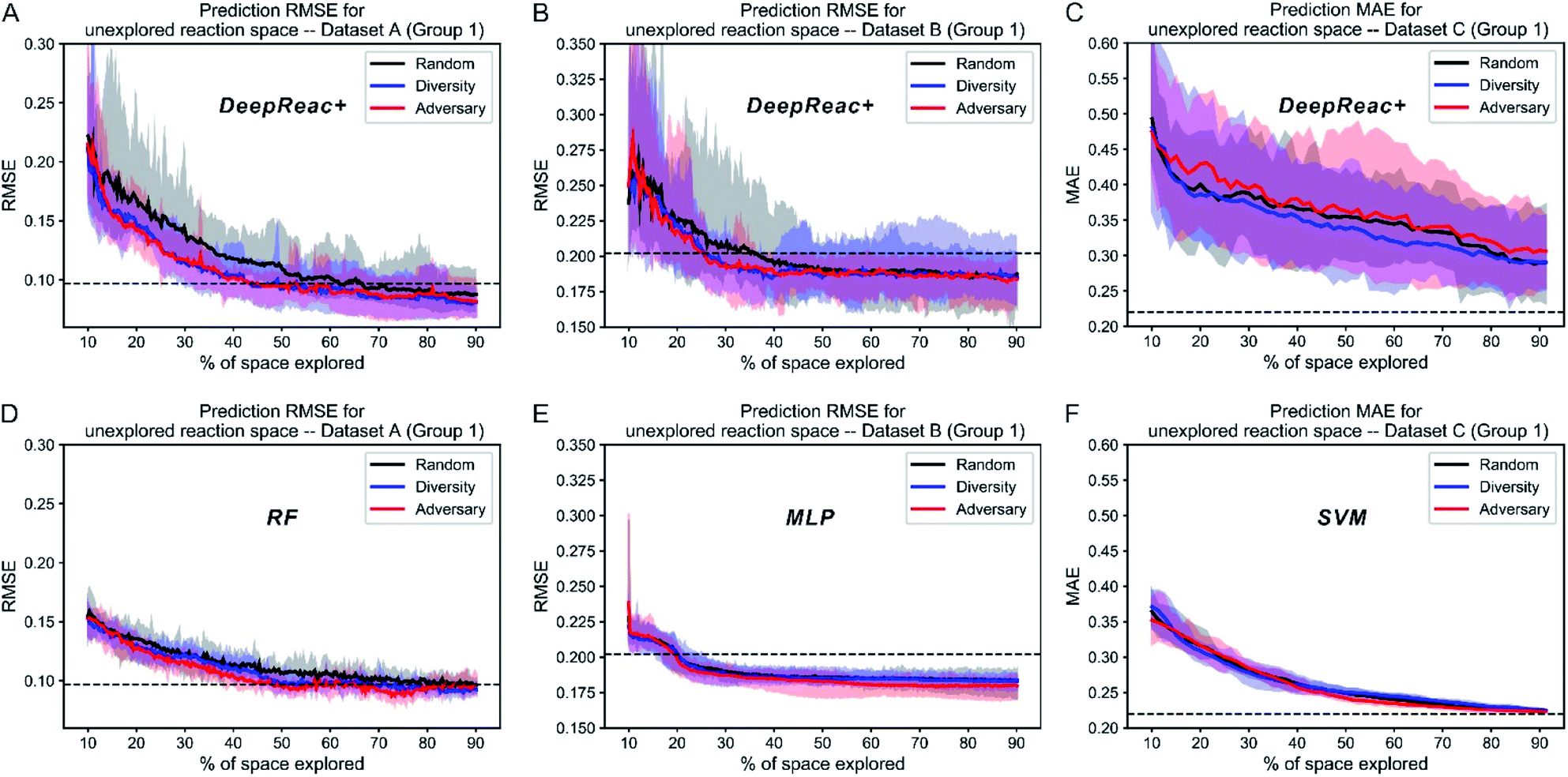 | ||
| Fig. 4 Simulation results of three sampling strategies with DeepReac and other models on Group 1 of three benchmark datasets in catalyst-unknown mode. The aggregated results from 30 simulations show the average RMSE/MAE of DeepReac on Dataset A (A), Dataset B (B) and Dataset C (C) versus the fraction of the chemical space explored; the filled areas around the curves are defined by the maximum and minimum values. The black line indicates the random sampling strategy, the blue line indicates the diversity-based sampling strategy, and the red line indicates the adversary-based sampling strategy. The horizontal dashed black line indicates the best model performance achieved using all training data without active learning. The aggregated results from 30 simulations showing the average RMSE/MAE of RF/MLP/SVM on Dataset A (D), Dataset B (E) and Dataset C (F) versus the fraction of the chemical space explored; the filled areas around the curves are defined by the maximum and minimum values. The black line indicates the random sampling strategy, the blue line indicates the diversity-based sampling strategy, and the red line indicates the adversary-based sampling strategy. The horizontal dashed black line indicates the best model performance achieved using all training data without active learning. RMSE, root-mean-square error. MAE, mean absolute error, in kcal mol−1. RF, random forest. MLP, multilayer perceptron. SVM, support vector machine. See also Fig. S7–S9.† | ||
Identification of the optimal reaction conditions and starting materials by DeepReac+
Identifying the optimal reaction conditions is always a crucial goal of chemical synthesis. As an industry standard, design of experiments (DOE) has been applied successfully to optimize the reaction conditions including solvent, temperature, catalyst loading, etc.86–88 The response surface model is often used with pre-defined optimal designs, i.e. fractional factorial designs, to assist and guide experimenters during experimentation planning. However, this approach may not be implemented readily for certain complex situations such as Dataset A which has 4 factors with 15, 23, 4 and 3 levels, respectively, according to the terms of DOE. On the other hand, the exploitation strategy, also known as the “greedy” strategy, has also been used to achieve the goal,59 which means that the sample predicted to be optimal should be labeled first. However, this strategy is likely to cause low predictive performance for the model, which in turn compromises the sample selection process.55 We here propose a balance-based strategy in which not only the adversarial samples but also samples predicted to be high yield or stereoselective have a high priority in being labeled. To make the simulation more practical, our goal is to optimize the yield of specific products. Dataset A has five products in total and each has 990 reactions whereas Dataset B has only one product. It should be noted that the same product can be obtained by different starting materials which only differ in the leaving group. They will therefore be optimized at the same time. In view of the number of products in two datasets, we designed two different scenarios to do simulation, respectively:(1) in Dataset A, we assume that the experimental data of one product is used as historical data to identify the optimal reaction conditions of another. To be specific, Dataset A is divided into 5 subsets according to the products (Table S2†). One of them works as the pretraining set to train an initial DeepReac model which then searches the optimal reaction conditions iteratively in another subset with a certain sampling strategy. Each subset in turn works as the pretraining set.
(2) in Dataset B where only one product is involved, we assume that there is no historical data, thus optimizing the reaction conditions from scratch. To be specific, we randomly select 96 experimental data at first to train an initial DeepReac model, and search the optimal reaction conditions in the remaining dataset with a certain sampling strategy. In each iteration, 96 experimental data will be sampled. Thirty simulations were conducted to test the effect of various sampling strategies. For each scenario, we performed a statistical analysis of the target value distribution of the candidates selected by the different sampling strategies during the first five iterative rounds (Fig. 5). It is clearly shown that both the greed-based and balance-based strategies can recognize more conditions that have high yields in the early stage of iteration than the other strategies. However, from the perspective of improving the predictive performance, the greed-based strategy is the worst, which is in accordance with a previous report, and the balance-based strategy is compromised but still acceptable (Fig. S10†).
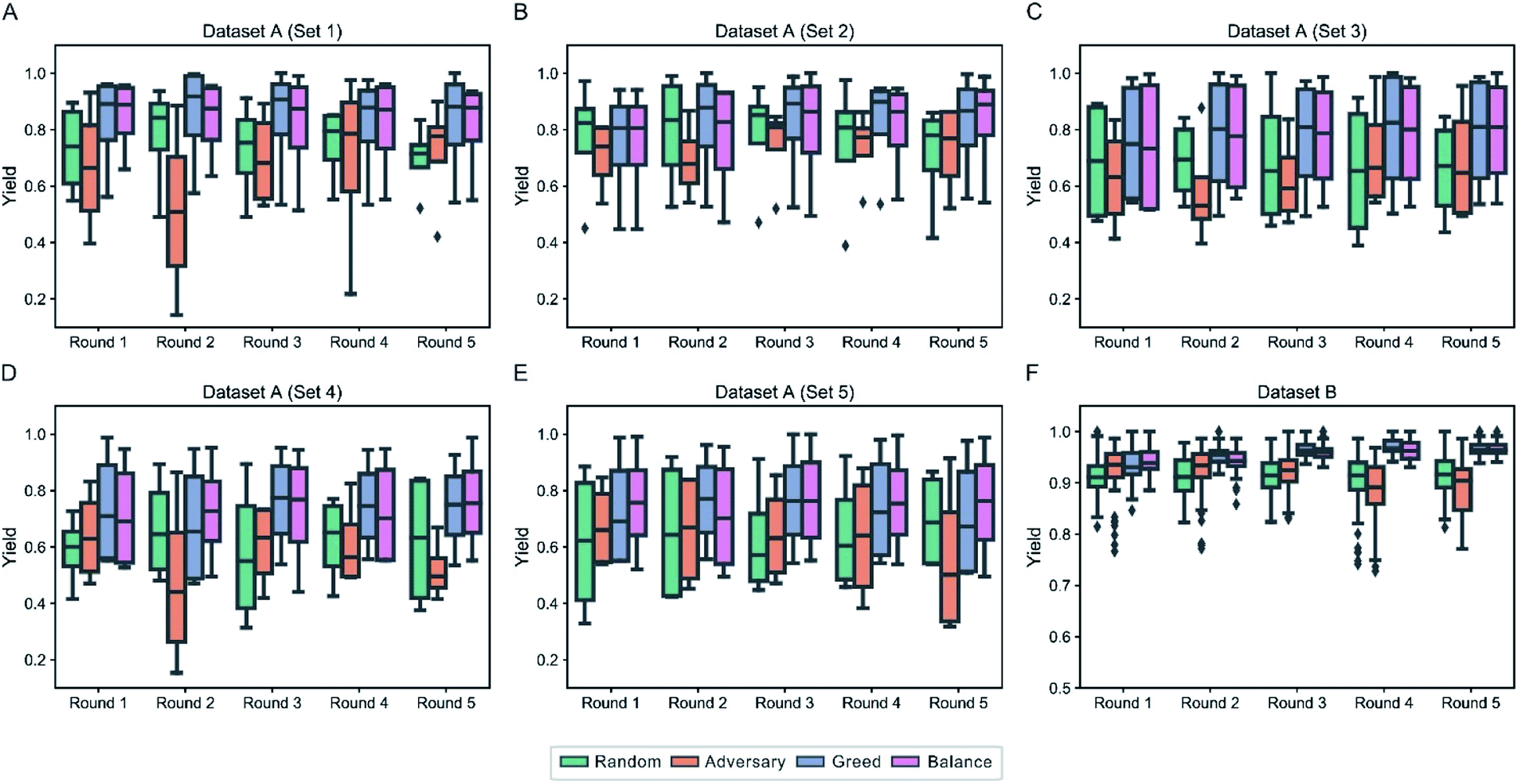 | ||
| Fig. 5 Statistical analysis of the ground-truth distribution of the candidates of four sampling strategies in the first 5 rounds of iteration on Dataset A & B. Each box plot shows the ground-truth distribution of the candidates of a certain sampling strategy in a round of iteration, which are colored according to the strategy type. The quartiles are shown, and the outliers are marked as square points. The results of the first 5 rounds of iteration are summarized for 5 subsets of Dataset A (A–E) and Dataset B (F). | ||
In theory, it's also feasible to identify reaction conditions that generate specific products with high stereoselectivity. However, each product in Dataset C only has 25 experimental data which is too few to obtain meaningful results. As an alternative we did the simulation in library mode which means that the goal of the optimization wasn't set to aim at specific products. The statistical analysis of the target value distribution of the candidates during the first five iterative rounds (Fig. S11†) shows similar results. Although it was less practical, it demonstrated the broad application of active learning strategies.
Discussions
So far, we have tested DeepReac+ in various situations and the advantages of the computational framework have been indicated. The form of input is universal and the model can adapt rapidly to various types of reactions and predictive targets with competitive performance. The number of new reactions keeps growing rapidly. Not all reaction mechanisms have been rigorously studied, which makes the design of descriptors more difficult. DeepReac+ doesn't have such limitation and can rapidly boost predictive performance as well as identify the optimal reaction conditions with proper active learning strategies, which is helpful to reduce the manpower and resources. Recently automated experimentation platforms equipped with robotics have been designed and implemented to boost productivity and reproducibility as well as liberate the scientific workforce from repetitive tasks.84,89–93 The universality and high efficiency of DeepReac+ gives us reasons to believe that it can be embedded in such automated reaction systems and play an important role. Nevertheless, there are also some limitations of our proposed model, which remain to be addressed in the future:(1) We need to train corresponding models for different reaction mechanisms. Namely, a model trained with data of one type of reaction can't be used to predict the outcome of another type of reaction. It's well known that the same group can have a different role in different reaction mechanisms. Hence, a model with “genuine” generalized predictive ability is required to recognize both types of reaction mechanisms and roles of various molecular structures under specific reaction mechanism. The attempt has been made but not successful by training on patent data.94
(2) For the unseen reaction components that can't be represented by the graph structure, our model has to be retrained since they are only represented as one-hot encoding. It also limits the application of our model on some reaction types whose most components cannot trivially be expressed in terms of graphs. That would involve many types of inorganic reactions or reactions on surfaces or within materials. Hence, the most suitable reaction type for DeepReac+ prefers two points: (a) several reaction components, i.e. (co-)catalysts, additives, solvents and so on, are involved, which produces a huge reaction space; (b) most of the components can be represented by the graph structure. Moreover, since the advantage of active learning here is to guide chemical synthesis by selecting informative experiments to be conducted, those who have no access to new experiments will not benefit from the “+” in DeepReac+. In other words, active learning will not reduce the number of required data points if the experiments have already been performed without the guidance of DeepReac+.
(3) By comparing the results of three datasets, we found that the performance of DeepReac+ is slightly inferior when the size of the dataset is small, i.e. Dataset C. It's a well-known limitation of the application of the deep learning technique. Combined with quantum chemical descriptors or fingerprints, the traditional machine learning models, i.e. random forest, have an advantage in the low data regime. As shown in a previous report95, it is likely that GNNs can be further enhanced by relevant descriptors. Thus, the inclusion of quantum chemical descriptors into DeepReac+ may be a future solution. Another promising direction of improvement of DeepReac+ is to pretrain the module encoding molecules on a large-scale dataset, i.e. ZINC96 or QM9.97 Recently, several unsupervised or transfer learning methods have been successfully applied to learn the universal representation of molecules.98–100 It would boost the performance of DeepReac+ in the low data regime if the Molecule GAT module is replaced by such pretrained networks.
(4) Continuous variables, such as temperature and the amount of substrates or catalysts, have not been included in our model. The main reason is the lack of large datasets where both quantitative and qualitative variables are included as well as the lack of experimental data for all combinations of these variables. Although it can't be validated so far, continuous variables can be included easily in our model. For global continuous variables, i.e. temperature and reaction time, an additional feature vector representing them can be concatenated with the output of the Capsule module. For continuous variables of the individual reaction component, i.e. the amount of substrates or catalysts, an additional feature vector representing them can be concatenated with the output of the Molecule GAT module. In summary, the architecture of DeepReac is quite flexible and more types of variables can be included in the future.
Conclusions
In summary, to accelerate the automation of chemical synthesis, a universal and generalized computational framework, DeepReac+, is proposed to predict various reaction performances, such as yield and stereoselectivity. Regardless of the reaction mechanisms and predictive targets, DeepReac, a GNN-based deep learning model, directly takes the 2D molecular structures of organic components and types of inorganic components as inputs without elaborate design or calculations based on certain hypotheses. It learns the task-related representations of the reaction conditions automatically during training and achieves state-of-the-art predictive performance on various datasets. Furthermore, we propose two active learning strategies, diversity-based and adversary-based strategies, to reduce the number of experiments necessary for model training. Based on the reaction representations learned iteratively by DeepReac, the two sampling strategies explore the reaction space selectively and train the model with only a small number of informative samples to achieve remarkable predictive performance. It should be noted that adversary strategy-based methods can be well applied to other common machine learning models in addition to DeepReac. When the adversary-based strategy is combined with the greed-based strategy, reaction conditions that achieve high yield or stereoselectivity can be identified more rapidly, and the predictive performance of DeepReac can continue to increase at the same time. Hopefully, as a universal and efficient feedback framework, DeepReac+ can facilitate the development of automated chemical synthesis platforms,86,101–106 to enable cost reduction and liberate the scientific workforce from repetitive tasks.Data availability
DeepReac+ is freely accessible at https://github.com/bm2-lab/DeepReac.Author contributions
Q. L. and Y. K. G. conceived the study. Y. K. G. D. Y. X. G. H. C. and J. Y. performed the analysis and developed the framework of DeepReac+. Q. L. and Y. K. G. wrote the manuscript with assistance from other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grant No. 2017YFC0908500, and 2016YFC1303205), National Natural Science Foundation of China (Grant No. 31970638, 61572361, and 62002264), China Postdoctoral Science Foundation (Grant No. 2019M651575), Shanghai Natural Science Foundation Program (Grant No. 17ZR1449400), Shanghai Artificial Intelligence Technology Standard Project (Grant No. 19DZ2200900) and the Fundamental Research Funds for the Central Universities.References
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- M. MacCoss and T. A. Baillie, Science, 2004, 303, 1810–1813 CrossRef CAS PubMed.
- G. M. Whitesides, Angew. Chem., Int. Ed., 2015, 54, 3196–3209 CrossRef CAS PubMed.
- G. N. Philippe, Chem, 2016, 1, 335–336 Search PubMed.
- K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC.
- M. Baker, Nature, 2016, 533, 452–454 CrossRef CAS PubMed.
- P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751–4755 CrossRef CAS PubMed.
- M. S. Lajiness, G. M. Maggiora and V. Shanmugasundaram, J. Med. Chem., 2004, 47, 4891–4896 CrossRef CAS PubMed.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, Chem. Sci., 2015, 6, 1258–1264 RSC.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 359, 429–434 CrossRef CAS PubMed.
- E. Kim, K. Huang, S. Jegelka and E. Olivetti, npj Comput. Mater., 2017, 3, 53 CrossRef.
- A. A. Lee, Q. Yang, A. Bassyouni, C. R. Butler, X. Hou, S. Jenkinson and D. A. Price, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3373–3378 CrossRef CAS PubMed.
- J. Ma, R. P. Sheridan, A. Liaw, G. E. Dahl and V. Svetnik, J. Chem. Inf. Model., 2015, 55, 263–274 CrossRef CAS PubMed.
- F. Wan, L. Hong, A. Xiao, T. Jiang and J. Zeng, Bioinformatics, 2019, 35, 104–111 CrossRef CAS PubMed.
- J. Wenzel, H. Matter and F. Schmidt, J. Chem. Inf. Model., 2019, 59, 1253–1268 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- R. Ding, R. Wang, Y. Ding, W. Yin, Y. Liu, J. Li and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 19175–19183 CrossRef CAS PubMed.
- P. Raccuglia, K. C. Elbert, P. D. Adler, C. Falk, M. B. Wenny, A. Mollo, M. Zeller, S. A. Friedler, J. Schrier and A. J. Norquist, Nature, 2016, 533, 73–76 CrossRef CAS PubMed.
- K. T. Schutt, H. E. Sauceda, P. J. Kindermans, A. Tkatchenko and K. R. Muller, J. Chem. Phys., 2018, 148, 241722 CrossRef CAS PubMed.
- T. Dimitrov, C. Kreisbeck, J. S. Becker, A. Aspuru-Guzik and S. K. Saikin, ACS Appl. Mater. Interfaces, 2019, 11, 24825–24836 CrossRef CAS PubMed.
- R. Gomez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernandez-Lobato, B. Sanchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef CAS PubMed.
- K. Kim, S. Kang, J. Yoo, Y. Kwon, Y. Nam, D. Lee, I. Kim, Y.-S. Choi, Y. Jung, S. Kim, W.-J. Son, J. Son, H. S. Lee, S. Kim, J. Shin and S. Hwang, npj Comput. Mater., 2018, 4, 67 CrossRef.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo and A. Aspuru-Guzik, Nat. Biotechnol., 2019, 37, 1038–1040 CrossRef CAS PubMed.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- W. Beker, E. P. Gajewska, T. Badowski and B. A. Grzybowski, Angew. Chem., Int. Ed., 2019, 58, 4515–4519 CrossRef CAS PubMed.
- J. A. Kammeraad, J. Goetz, E. A. Walker, A. Tewari and P. M. Zimmerman, J. Chem. Inf. Model., 2020, 60, 1290–1301 CrossRef CAS PubMed.
- X. Li, S. Q. Zhang, L. C. Xu and X. Hong, Angew. Chem., Int. Ed., 2020, 59, 13253–13259 CrossRef CAS PubMed.
- G. Pesciullesi, P. Schwaller, T. Laino and J. L. Reymond, Nat. Commun., 2020, 11, 4874 CrossRef CAS PubMed.
- P. M. Pfluger and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 18860–18865 CrossRef PubMed.
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343–348 CrossRef CAS PubMed.
- E. Walker, J. Kammeraad, J. Goetz, M. T. Robo, A. Tewari and P. M. Zimmerman, J. Chem. Inf. Model., 2019, 59, 3645–3654 CrossRef CAS PubMed.
- F. Sandfort, F. Strieth-Kalthoff, M. Kühnemund, C. Beecks and F. Glorius, Chem, 2020, 6, 1379–1390 CAS.
- F. Hase, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef CAS PubMed.
- F. Hutter, H. H. Hoos and K. Leyton-Brown, Presented in part at the Proceedings of the 5th international conference on Learning and Intelligent Optimization, Rome, Italy, 2011 Search PubMed.
- J. Snoek, H. Larochelle and R. P. Adams, Advances in Neural Information Processing Systems, 2012, vol. 25, pp. 2960–2968 Search PubMed.
- T. Desautels, A. Krause and J. Burdick, Presented in part at the Proceedings of the 29th International Conference on International Conference on Machine Learning, Edinburgh, Scotland, 2012 Search PubMed.
- J. T. Springenberg, A. Klein, S. Falkner and F. Hutter, Presented in part at the Proceedings of the 30th International Conference on Neural Information Processing Systems, Barcelona, Spain, 2016 Search PubMed.
- F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner and G. Monfardini, IEEE Trans. Neural Netw. Learn. Syst., 2009, 20, 61–80 Search PubMed.
- Z. Wu, S. Pan, F. Chen, G. Long, C. Zhang and P. S. Yu, IEEE Trans. Neural Netw. Learn. Syst., 2020, 32, 4–24 Search PubMed.
- Z. Zhang, P. Cui and W. Zhu, IEEE Trans. Knowl. Data Eng., 2020 DOI:10.1109/tkde.2020.2981333.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- D. Hwang, S. Yang, Y. Kwon, K. H. Lee, G. Lee, H. Jo, S. Yoon and S. Ryu, J. Chem. Inf. Model., 2020, 60, 5936–5945 CrossRef CAS PubMed.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, J. Comput.-Aided Mol. Des., 2016, 30, 595–608 CrossRef CAS PubMed.
- S. Y. Louis, Y. Zhao, A. Nasiri, X. Wang, Y. Song, F. Liu and J. Hu, Phys. Chem. Chem. Phys., 2020, 22, 18141–18148 RSC.
- T. Nguyen, H. Le, T. P. Quinn, T. Nguyen, T. D. Le and S. Venkatesh, Bioinformatics, 2020, 37, 1140–1147 CrossRef PubMed.
- M. Sun, S. Zhao, C. Gilvary, O. Elemento, J. Zhou and F. Wang, Briefings Bioinf., 2020, 21, 919–935 CrossRef PubMed.
- C. Chen, W. Ye, Y. Zuo, C. Zheng and S. P. Ong, Chem. Mater., 2019, 31, 3564–3572 CrossRef CAS.
- E. N. Feinberg, D. Sur, Z. Wu, B. E. Husic, H. Mai, Y. Li, S. Sun, J. Yang, B. Ramsundar and V. S. Pande, ACS Cent. Sci., 2018, 4, 1520–1530 CrossRef CAS PubMed.
- P. Hop, B. Allgood and J. Yu, Mol. Pharm., 2018, 15, 4371–4377 CrossRef CAS PubMed.
- V. Korolev, A. Mitrofanov, A. Korotcov and V. Tkachenko, J. Chem. Inf. Model., 2020, 60, 22–28 CrossRef CAS PubMed.
- Z. Xiong, D. Wang, X. Liu, F. Zhong, X. Wan, X. Li, Z. Li, X. Luo, K. Chen, H. Jiang and M. Zheng, J. Med. Chem., 2020, 63, 8749–8760 CrossRef CAS PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed.
- M.-F. Balcan, A. Beygelzimer and J. Langford, J. Comput. Syst. Sci., 2009, 75, 78–89 CrossRef.
- D. Reker and G. Schneider, Drug Discovery Today, 2015, 20, 458–465 CrossRef PubMed.
- I. Cortes-Ciriano, N. C. Firth, A. Bender and O. Watson, J. Chem. Inf. Model., 2018, 58, 2000–2014 CrossRef CAS PubMed.
- R. Garnett, T. Gartner, M. Vogt and J. Bajorath, J. Comput.-Aided Mol. Des., 2015, 29, 305–314 CrossRef CAS PubMed.
- T. Miyao and K. Funatsu, J. Chem. Inf. Model., 2019, 59, 2626–2641 CrossRef CAS PubMed.
- M. K. Warmuth, J. Liao, G. Ratsch, M. Mathieson, S. Putta and C. Lemmen, J. Chem. Inf. Comput. Sci., 2003, 43, 667–673 CrossRef CAS PubMed.
- L. Bassman, P. Rajak, R. K. Kalia, A. Nakano, F. Sha, J. Sun, D. J. Singh, M. Aykol, P. Huck, K. Persson and P. Vashishta, npj Comput. Mater., 2018, 4, 74 CrossRef.
- H. A. Doan, G. Agarwal, H. Qian, M. J. Counihan, J. Rodríguez-López, J. S. Moore and R. S. Assary, Chem. Mater., 2020, 32, 6338–6346 CrossRef CAS.
- T. Lookman, P. V. Balachandran, D. Xue and R. Yuan, npj Comput. Mater., 2019, 5, 21 CrossRef.
- M. Todorović, M. U. Gutmann, J. Corander and P. Rinke, npj Comput. Mater., 2019, 5, 35 CrossRef.
- S. J. Ang, W. Wang, D. Schwalbe-Koda, S. Axelrod and R. Gómez-Bombarelli, Chem, 2021, 7, 738–751 CAS.
- T. D. Loeffler, T. K. Patra, H. Chan, M. Cherukara and S. K. R. S. Sankaranarayanan, J. Phys. Chem. C, 2020, 124, 4907–4916 CrossRef CAS.
- O. Borkowski, M. Koch, A. Zettor, A. Pandi, A. C. Batista, P. Soudier and J. L. Faulon, Nat. Commun., 2020, 11, 1872 CrossRef CAS PubMed.
- D. Reker and J. B. Brown, Methods Mol. Biol., 2018, 1825, 369–410 CrossRef CAS PubMed.
- P. W. Battaglia, J. B. Hamrick, V. Bapst, A. Sanchez-Gonzalez, V. Zambaldi, M. Malinowski, A. Tacchetti, D. Raposo, A. Santoro, R. Faulkner, C. Gulcehre, F. Song, A. Ballard, J. Gilmer, G. Dahl, A. Vaswani, K. Allen, C. Nash, V. Langston, C. Dyer, N. Heess, D. Wierstra, P. Kohli, M. Botvinick, O. Vinyals, Y. Li and R. Pascanu, 2018, arXiv:1806.01261.
- A. J. Kirby, Stereoelectronic Effects, Oxford University Press, 1996 Search PubMed.
- G. Gonzalez, S. Gong, I. Laponogov, K. Veselkov and M. Bronstein, 2020, arXiv:2001.05724.
- N. Ravindra, A. Sehanobish, J. L. Pappalardo, D. A. Hafler and D. V. Dijk, Presented in part at the Proceedings of the ACM Conference on Health, Inference, and Learning, Toronto, Ontario, Canada, 2020 Search PubMed.
- P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò and Y. Bengio, 2017, arXiv:1710.10903.
- S. Sabour, N. Frosst and G. E Hinton, 2017, arXiv:1710.09829.
- D. Wang, Y. Liang and D. Xu, Bioinformatics, 2019, 35, 2386–2394 CrossRef CAS PubMed.
- Y. Wang, J. Hu, J. Lai, Y. Li, H. Jin, L. Zhang, L. R. Zhang and Z. M. Liu, J. Chem. Inf. Model., 2020, 60, 2754–2765 CrossRef CAS PubMed.
- B. Yang, Y. Chen, Q.-M. Shao, R. Yu, W.-B. Li, G.-Q. Guo, J.-Q. Jiang and L. Pan, IEEE Access, 2019, 7, 109956–109968 Search PubMed.
- D. Picard, P.-H. Gosselin and M.-C. Gaspard, IEEE Signal Process. Mag., 2015, 32, 95–102 Search PubMed.
- K. Wang, D. Zhang, Y. Li, R. Zhang and L. Lin, IEEE Trans. Circuits Syst. Video Technol., 2017, 27, 2591–2600 Search PubMed.
- J. P. Janet, C. Duan, T. Yang, A. Nandy and H. J. Kulik, Chem. Sci., 2019, 10, 7913–7922 RSC.
- E. D. Cubuk, B. Zoph, S. S. Schoenholz and Q. V. Le, 2017, arXiv:1711.02846.
- O. Deniz, N. Vallez and G. Bueno, Lect. Notes Comput. Sci., 2019, 11506, 569–580 Search PubMed.
- M. Ducoffe and F. Precioso, 2018, arXiv:1802.09841.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- J. M. Granda, L. Donina, V. Dragone, D. L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed.
- V. D. M. Laurens and G. Hinton, J. Mach. Learn. Res., 2008, 9, 2579–2605 Search PubMed.
- B. J. Reizman and K. F. Jensen, Acc. Chem. Res., 2016, 49, 1786–1796 CrossRef CAS PubMed.
- B. J. Reizman, Y. M. Wang, S. L. Buchwald and K. F. Jensen, React. Chem. Eng., 2016, 1, 658–666 RSC.
- L. M. Baumgartner, C. W. Coley, B. J. Reizman, K. W. Gao and K. F. Jensen, React. Chem. Eng., 2018, 3, 301–311 RSC.
- A. C. Bedard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- C. W. Coley, D. A. Thomas 3rd, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- R. W. Epps, M. S. Bowen, A. A. Volk, K. Abdel-Latif, S. Han, K. G. Reyes, A. Amassian and M. Abolhasani, Adv. Mater., 2020, 32, e2001626 CrossRef PubMed.
- S. H. M. Mehr, M. Craven, A. I. Leonov, G. Keenan and L. Cronin, Science, 2020, 370, 101–108 CrossRef CAS PubMed.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Mach. Learn. Sci. Technol., 2021, 2, 015016 CrossRef.
- Y. Guan, C. W. Coley, H. Wu, D. Ranasinghe, E. Heid, T. J. Struble, L. Pattanaik, W. H. Green and K. F. Jensen, Chem. Sci., 2020, 12, 2198–2208 RSC.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- R. Ramakrishnan, P. O. Dral, M. Rupp and O. A. von Lilienfeld, Sci. Data, 2014, 1, 140022 CrossRef CAS PubMed.
- X. C. Zhang, C. K. Wu, Z. J. Yang, Z. X. Wu, J. C. Yi, C. Y. Hsieh, T. J. Hou and D. S. Cao, Briefings Bioinf., 2021 DOI:10.1093/bib/bbab152.
- P. Li, J. Wang, Y. Qiao, H. Chen, Y. Yu, X. Yao, P. Gao, G. Xie and S. Song, Briefings Bioinf., 2021 DOI:10.1093/bib/bbab109.
- Z. Guo, C. Zhang, W. Yu, J. Herr, O. Wiest, M. Jiang and N. V. Chawla, Presented in part at the Proceedings of the Web Conference 2021, Ljubljana, Slovenia, 2021 Search PubMed.
- I. W. Davies, Nature, 2019, 570, 175–181 CrossRef CAS PubMed.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef.
- V. Dragone, V. Sans, A. B. Henson, J. M. Granda and L. Cronin, Nat. Commun., 2017, 8, 15733 CrossRef PubMed.
- F. Häse, L. M. Roch and A. Aspuru-Guzik, Trends Chem., 2019, 1, 282–291 CrossRef.
- C. Houben and A. A. Lapkin, Curr. Opin. Chem. Eng., 2015, 9, 1–7 CrossRef.
- G. Schneider, Nat. Rev. Drug Discovery, 2018, 17, 97–113 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02087k |
| This journal is © The Royal Society of Chemistry 2021 |