
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed benzylic C(sp3)–H carbonylative arylation of azaarylmethyl amines with aryl bromides†
Haoqiang
Zhao‡
ab,
Bowen
Hu‡
a,
Lijin
Xu
 *b and
Patrick J.
Walsh
*a
*b and
Patrick J.
Walsh
*a
aRoy and Diana Vagelos Laboratories, Penn/Merck Laboratory for High-Throughput Experimentation, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: pwalsh@sas.upenn.edu
bDepartment of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: 20050062@ruc.edu.cn
First published on 8th July 2021
Abstract
A highly selective palladium-catalyzed carbonylative arylation of weakly acidic benzylic C(sp3)–H bonds of azaarylmethylamines with aryl bromides under 1 atm of CO gas has been achieved. This work represents the first examples of use of such weakly acidic pronucleophiles in this class of transformations. In the presence of a NIXANTPHOS-based palladium catalyst, this one-pot cascade process allows a range of azaarylmethylamines containing pyridyl, quinolinyl and pyrimidyl moieties and acyclic and cyclic amines to undergo efficient reactions with aryl bromides and CO to provide α-amino aryl-azaarylmethyl ketones in moderate to high yields with a broad substrate scope and good tolerance of functional groups. This reaction proceeds via in situ reversible deprotonation of the benzylic C–H bonds to give the active carbanions, thereby avoiding prefunctionalized organometallic reagents and generation of additional waste. Importantly, the operational simplicity, scalability and diversity of the products highlight the potential applicability of this protocol.
Introduction
α-Amino ketones are key components of numerous biologically active natural products and synthetic compounds. They display a wide range of medicinal and biological activities, such as anti-depressant, appetite suppressant, and anti-platelet properties (Fig. 1).1 α-Amino ketones also serve as effective synthetic intermediates for the preparation of various heterocycles and 1,2-amino alcohols.1b,2 As a result of their widespread utility, there has been substantial and long-standing interest in the efficient construction of α-amino ketones in the synthetic community.3 General approaches to construct α-amino ketones from ketones or their derivatives involve nucleophilic amination,4 electrophilic amination5 or oxidative amination.6 Considerable efforts have also been made to synthesize α-amino ketones via acylation of imines with aldehydes, acylsilanes or carboxylic acids,7 Stille reaction of sulfonamides with benzoyl chlorides,8 cross-coupling of thiol esters with boronic acids or organostannanes,9 hydrogenation of α-dehydroamino ketones or α-ketoketimines,10 and rearrangement of α-hydroxyl imines or enamines.11 Moreover, recent years have witnessed the preparation of α-amino ketones starting from alkenes,12 alkynes13 and sulfonium ylides.14 Despite these promising advances, exploration of straightforward methods that enable formation of multiple C–C bonds remain appealing. | ||
| Fig. 1 Selected pharmacologically active compounds containing α-amino aryl ketones. | ||
Due to its low cost, high reactivity and abundance, CO has been extensively explored as a versatile C1 building block for the production of carbonyl-containing compounds and heterocycles.15 Impressive achievements have been recorded in transition-metal catalyzed reactions of CO in multicomponent carbonylation reactions to construct carbonyl-containing molecules from simple starting materials.15d,16 Little attention, however, has been paid to the application of this strategy for the preparation of synthetically valuable α-amino ketones. There is only one such report in the literature. In 2018, Wang, Zhang and co-workers described an elegant synthesis of α-amino ketones via a Pd(0)-catalyzed four-component carbonylation reaction of aryl iodides, N-tosylhydrazones and amines under 1 atm of CO (Scheme 1).17 Despite the synthetic potential of this 4 component coupling, this protocol is only applicable to aryl iodides and no examples with heteroaryl groups were reported.
 | ||
| Scheme 1 Carbene-based approach of Wang and co-workers. | ||
Recent progress has established the viability of palladium-catalyzed carbonylative cross-coupling reactions of acidic C(sp3)–H bonds for the concurrent formation of two new C–C bonds with the introduction of a carbonyl group.18,19 Early reports focused on the arylation of activated C(sp3)–H bonds of malonate derivatives.18 In 2012, Skrydstrup and co-workers first realized carbonylative α-arylation of ketones with aryl iodides using CO in the presence of a catalytic amount of [Pd(dba)2] and a bidentate phosphine ligand to afford 1,3-diketones (Scheme 2a).19a Subsequently, the same group accomplished carbonylative α-arylation of monoester potassium malonate,19b acetylacetones,19d ketones,19e 2-oxindoles,19g nitromethanes,19h substituted 1,3-dioxin-4-ones,19i and cyanoacetates19j with aryl iodides and aryl bromides under palladium catalysis. Meanwhile, Beller and co-workers described the Pd-catalyzed carbonylative α-arylation of ketones and nitriles with aryl iodides and pressurized CO gas (Scheme 2a).19c,f These studies take advantage of strongly activated C(sp3)–H bonds to facilitate deprotonation. The carbonylative α-arylation of weakly acidic C(sp3)–H bonds remains underdeveloped, despite the potential utility of such a method with a wide variety of pronucleophiles.
 | ||
| Scheme 2 Deprotonative carbonylative cross-coupling reactions (a) Prior art with more acidic pro-nucleophiles, (b) Our prior work on the direct coupling reaction in the absence of CO, (c) The current study using weakly acidic Azaarylmethylamine pro-nucleophiles. | ||
In recent years, our team has built a program around direct arylation of weakly acidic C(sp3)–H bonds (pKa 25–43 in DMSO)20 with aryl halides by employing palladium catalysts and suitable bases (Scheme 2b). We called these reactions deprotonative cross-coupling processes (DCCP).21 Based on these studies from our lab, we envisioned that merging DCCP with carbonylation reactions would enable preparation of a host of new ketones. Herein we describe such a new and efficient process that allows highly selective carbonylative arylation of weakly acidic benzylic C(sp3)–H bonds of azaarylmethylamines with aryl bromides. These reactions are conveniently conducted with 1 atm of CO and a palladium catalyst to deliver α-amino aryl-azaarylmethyl-ketone products in good to excellent yields. The reaction has a broad substrate scope and good tolerance of functional groups (Scheme 2c).
Results and discussion
We started our studies by exploring the reaction conditions of benzylic C–H carbonylative arylation of 4-(pyridin-2-ylmethyl)morpholine (1a) with 1-bromo-4-(tert-butyl)benzene (2a) under 1 atm of CO gas (Table 1). Considering that we have recently achieved the coupling of azaarylmethyl amines with aryl halides to generate aryl(azaaryl)methyl amines in 1,4-dioxane using a Pd(OAc)2/NIXANTPHOS-based catalyst together with LiN(SiMe3)2 as the base,21c we first assessed the feasibility of the proposed reaction of 1a, 2a and CO (1 atm) at 65 °C for 16 h by employing the same catalytic system. To our delight, the carbonylative arylation reaction indeed occurred, delivering the expected product 3aa in 27% AY (AY = assay yield, determined by 1H NMR spectroscopy) with the formation of the non-carbonylative coupling product (also called the direct coupling product) 3aa′ in 8% AY (Table 1, entry 1 and ESI, Table S2†).|
|
||||
|---|---|---|---|---|
| Entry | Pd source | Solvent | Temp (°C) | 3aa Assay yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol), LiN(SiMe3)2 (2 equiv.), [Pd] (5 mol%), NIXANTPHOS (6 mol%), solvent (1 mL). b Yields were determined by 1H NMR analysis of unpurified reaction mixtures with internal standard CH2Br2. c LiN(SiMe3)2 (3 equiv.) was employed. d 2a (1.5 equiv.) was employed. e Isolated yield. f In the absence of NIXANTPHOS. g CO (8.6 atm) was applied. Pd G4 dimer: Buchwald G4 precatalysts; dba: dibenzylideneacetone. | ||||
| 1 | Pd(OAc)2 | 1,4-Dioxane | 65 | 27 |
| 2 | Pd(OAc)2 | THF | 65 | 22 |
| 3 | Pd(OAc)2 | CPME | 65 | 21 |
| 4 | Pd(OAc)2 | DME | 65 | 10 |
| 5 | Pd2(dba)3 | 1,4-Dioxane | 65 | 31 |
| 6 | Pd G4 dimer | 1,4-Dioxane | 65 | 37 |
| 7 | [PdCl(allyl)]2 | 1,4-Dioxane | 65 | 7 |
| 8 | Pd(dba)2 | 1,4-Dioxane | 65 | 43 |
| 9c | Pd(dba)2 | 1,4-Dioxane | 65 | 89 |
| 10c | Pd(dba)2 | 1,4-Dioxane | 80 | 93 |
| 11c | Pd(dba)2 | 1,4-Dioxane | 100 | 89 |
| 12 , | Pd(dba) 2 | 1,4-Dioxane | 80 | 97 (92) |
| 13c,d,f | Pd(dba)2 | 1,4-Dioxane | 80 | 0 |
| 14c,d | — | 1,4-Dioxane | 80 | 0 |
| 15c,d,g | Pd(dba)2 | 1,4-Dioxane | 80 | 4 |
With an aim to improve the reaction efficiency, the performance of the coupling in other solvents, such as THF, CPME (cyclopentyl methyl ether) and DME were examined (Table 1, entries 2–4), but none outperform 1,4-dioxane. Replacement of LiN(SiMe3)2 with NaN(SiMe3)2, or KN(SiMe3)2 failed to give better results, and the reaction was not promoted with LiOtBu, NaOtBu, or KOtBu as the base (ESI, Table S2†). The subsequent screening of palladium salts revealed the superiority of Pd(dba)2 in this reaction, allowing the generation of product 3aa in 43% yield (Table 1, entry 8). Unexpectedly, increasing the loading of LiN(SiMe3)2 to 3 equiv. improved the yield of 3aa to 89% with only a small amount of direct coupling byproduct 3aa′ (7%) (Table 1, entry 9 and ESI, Table S2†). The yield of 3aa could be further enhanced to 93% with an increase of reaction temperature to 80 °C (Table 1, entry 10), but a higher reaction temperature of 100 °C was found to be detrimental (Table 1, entry 11). Further examination of the stoichiometry indicated that increasing the amount of 2a from 1.2 to 1.5 equiv. resulted in almost exclusive formation of 3aa in an excellent assay yield of 97% with 92% isolated yield (Table 1, entry 12). The phosphine ligand bound to palladium also proved to be critical. Variation of the bidentate phosphine ligand to dppe, dppb, dppp, dppf and xantphos all led to substantial decreases in the reaction conversion (ESI, Table S2†). Control experiments confirmed the dependency on both the phosphine ligand and the palladium source in this transformation (Table 1, entries 13 and 14). Increasing the CO pressure to 8.6 atm resulted in only 4% AY (Table 1, entry 15), which suggested a higher CO pressure could potentially saturate the metal catalyst and deactivate it.
With the optimized reaction conditions in hand, we then evaluated the substrate scope of azaarylmethylamines and the results are summarized in Table 2. 2-Pyridylmethylamines (1b–1f) bearing thiomorpholine, methylpiperazine, dimethylamine, pyrrolidine and piperidine underwent smooth C–H carbonylative arylation to afford the expected α-amino ketone products (3ba–3fa) in 64–87% yield. Substrate 1g bearing a sterically hindered ortho-substituent on the pyridine ring was also reactive, delivering the desired product 3ga in 52% yield. The more acidic 4-pyridylmethylamines (1h–1k) containing different amino groups reacted efficiently to give the desired products (3ah–3ak) in 61–88% yield. In the case of 3-pyridylmethylamine, however, the reaction failed to yield any desired product, with recovery of most of the starting materials. It is interesting to note that in the that direct coupling with the 3-pyridylmethylamine (in the absence of CO), the reaction was successful with KN(SiMe3)2 as base.21c Use of KN(SiMe3)2 instead of LiN(SiMe3)2 under CO at 110 °C, however, did not lead to ketone product.21c It is likely that the higher pKa of this pronucleophile, in combination with the less reactive base LiN(SiMe3)2 disfavors formation of sufficient quantities of the nucleophile and prevents the reaction from proceeding.20 This result shows the crucial dependency on the main group metal in such multistep reactions. To further extend the scope of this carbonylative arylation, other azaarylmethylamines were tested. Notably, when 4-(benzo[d]thiazol-2-ylmethyl)morpholine 1l was employed, the product (3la) was obtained in 50% yield as a mixture of the ketone and enol isomers (the ratio was ketone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enol = 3:1). However, for other kinds of azaarylmethylamines like 2-(morpholinomethyl)benzo[d]oxazole, 4-((1-methyl-1H-imidazol-2-yl)methyl)morpholine, 4-benzylmorpholine or even 2-(ethoxymethyl)pyridine, the reaction failed to yield the desired products (See ESI, Table S3† for substrates that were unsuccessful with this catalyst system). When 2-quinolinylmethyl amines 1m and 1n were employed, the corresponding products 3ma and 3na were obtained in 74% and 72% yields, respectively. Moreover, 2-pyrimidylmethylamine 1o also reacted smoothly, affording product 3oa in 93% yield.
:enol = 3:1). However, for other kinds of azaarylmethylamines like 2-(morpholinomethyl)benzo[d]oxazole, 4-((1-methyl-1H-imidazol-2-yl)methyl)morpholine, 4-benzylmorpholine or even 2-(ethoxymethyl)pyridine, the reaction failed to yield the desired products (See ESI, Table S3† for substrates that were unsuccessful with this catalyst system). When 2-quinolinylmethyl amines 1m and 1n were employed, the corresponding products 3ma and 3na were obtained in 74% and 72% yields, respectively. Moreover, 2-pyrimidylmethylamine 1o also reacted smoothly, affording product 3oa in 93% yield.

We next investigated the reaction of various aryl bromides with 1a under 1 atm CO (Table 3). A variety of para-substituted aryl bromides bearing electron-donating groups (2b–2f, 2j and 2k) and electron-withdrawing groups (2g–2i) were all effective reaction partners, providing the corresponding products (3ab–3ak) in moderate to high yields (57–90%). Moreover, the reaction could be successfully extended to meta-substituted aryl bromides (2l–2n), delivering the desired products (3al–3an) in 61–84% yields. Lower yields were generally observed with electron-poor aryl bromides, possibly due to the partial decomposition of these aryl bromides in the presence of the base. Notably, 1-bromo-4-chlorobenzene (2h) and 1-bromo-3-chlorobenzene (2m) led to the formation of products 3ah and 3am in 77% and 71% yields, respectively, with the chloro group remaining intact during the reaction process. The Pd(NIXANTPHOS)-based catalysts is known to oxidatively add aryl chlorides at room temperature (see ESI, Table S2†).21 In the presence of CO, however, aryl chlorides were not reactive. These observations suggest to us that the palladium catalyst bears a CO ligand that tempers its ability to oxidatively add the stronger C–Cl bond of aryl chlorides.

Multi-substituted aryl bromides (2o–2r) also readily engaged in the transformation to afford the corresponding products (3ao–3ar) in 65–87% yields. The generality of the current catalytic system was further demonstrated by the success of heteroaryl bromides 2s and 2t to generate 3as and 3at in 50% and 55% yields, respectively. It is noteworthy that the products of these reactions are rich in heterocycles.
Preliminary studies were conducted to gain insight into the reaction product. We hypothesized that the product generated under the reaction conditions before workup was not the ketone, but the enolate that is protonated upon aqueous workup. Thus, we first prepared the enolate 3aA by deprotonation of ketone 3aa with LiN(SiMe3)2 (Scheme 3a). Next, the carbonylative α-arylation reaction of 1a, 2a and CO was conducted and the product characterized by NMR spectroscopy before quenching with water (Scheme 3b). This product was found to be identical to the independently synthesized enolate 3aA, confirming our hypothesis that the enolate is the product of the reaction (Scheme 3). This result also helps to explain why 3 equiv. of LiN(SiMe3)2 are optimal. An equivalent is needed to consume the starting material 1 and a second to deprotonate the ketone. The role of the third is to deprotonate the catalyst backbone N–H and to react with any advantageous water.
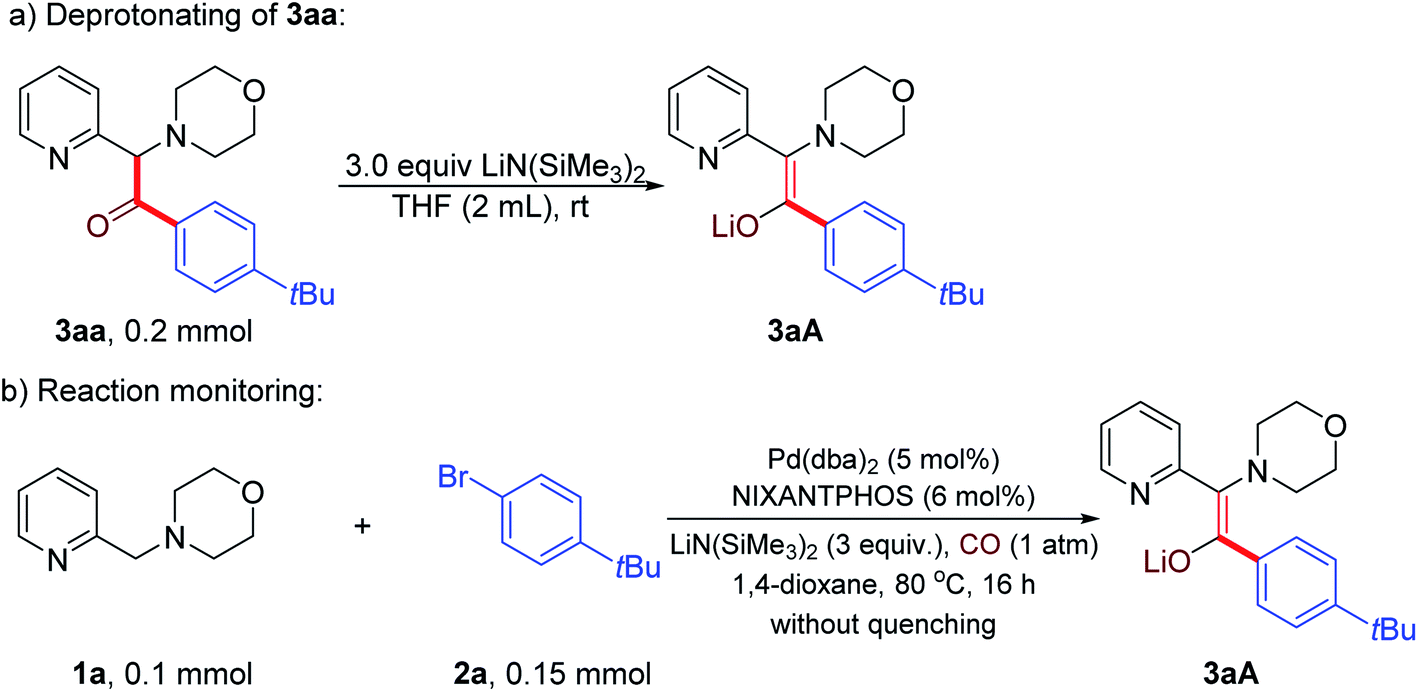 | ||
| Scheme 3 Detection of product precursor (a) Independent synthesis of enolate 3aA, (b) Determination of the reaction product before workup. | ||
To showcase the synthetic utility of our method, we first conducted a gram-scale reaction of 1a, 2a and CO under the standard reaction conditions. The desired product 3aa was obtained in 71% yield (1.2 g) (Scheme 4a). Next, we attempted the transformation of the product ketones into other useful derivatives. Although Baeyer–Villiger oxidation of 3aa with m-CPBA in CH2Cl2 failed to give the desired ester, an unexpected oxidative cleavage reaction occurred to give product 4 in 91% yield (Scheme 4b). Notably, further study revealed that this oxidative cleavage reaction could take place under air oxidation to give a high yield of 4 (83%). Moreover, reduction of 3aa with NaBH4 in MeOH at room temperature for 16 h resulted in the formation of amino alcohol product 5 in 68% yield (Scheme 4c). Finally, treatment of 3aa with LiN(SiMe3)2 and Me2NEt followed by addition of allyl chloroformate gave the allyl enol carbonate product 6 in 87% isolated yield as a single diastereomer (Scheme 4d). As reported by the Stoltz group,22 this enol carbonate product can undergo palladium-catalyzed enantioselective decarboxylative allylic alkylation to afford a chiral α-quaternary ketone.
 | ||
| Scheme 4 Synthetic applications (a) scale-up of reaction, (b) product oxidation, (c and d) further transformations of the product. | ||
Based on the aforementioned results in DCCP chemistry (no CO)21 and previous reports on carbonylative arylation,19,23 a plausible mechanism is proposed in Scheme 5. The catalytic cycle starts with complexation of NIXANTPHOS (see Scheme 2b for structure) and Pd(dba)2 to yield the (NIXANTPHOS)Pd(0) species A.21a We previously demonstrated that the NIXANTPHOS N–H (pKa 21 in DMSO) will be deprotonated by silyl amide bases to afford bimetallic adducts that have exhibited cooperativity between the Pd and main group cation.21m In the absence of CO, the resulting heterobimetallic complex will oxidatively add aryl chlorides at room temperature through a mechanism involving cooperativity between the main group metal and the palladium.21m In the presence of CO we also expect the ligand N–H to be deprotonated by LiN(SiMe3)2. The oxidative addition does not appear to be accelerated and the carbonylation reaction does not work with aryl chlorides. Under CO atmosphere, the Pd(0) species is proposed to undergo CO coordination to generate Pd(0) carbonyl (B) and dicarbonyl C complexes.23b This proposal is consistent with inhibition at high CO pressures. Upon dissociation of CO from C to generate the 16 electron mono-carbonyl adduct B, oxidative addition of the aryl bromide 2 takes place to produce (NIXANTPHOS)Pd(CO)(Ar)Br-complex D. Intermediate D likely undergoes CO insertion into the Pd–Ar bond to furnish the acyl–Pd(II) complex E. Intermediate E reacts with the deprotonated pronucleophile 1′ in a transmetallation step to deliver the reductive elimination precursor F. Reductive elimination of F gives the ketone product 3 and Pd(0) species A to close the catalytic cycle. In the presence of LiN(SiMe3)2 the ketone 3 is rapidly deprotonated, furnishing the enolate 3′. Quenching the reaction with H2O results in the formation of the observed ketone product 3. The direct coupling product likely forms when transmetallation takes place before CO insertion or if CO insertion is reversible. At this stage, we cannot rule out the possibility of an adduct between Pd(0) and the enolate, as described by Skrydstrup and coworkers.19e Further investigations into the reaction mechanism will be presented in due course.
 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusion
In conclusion, we have developed the carbonylative arylation of weakly acidic benzylic C(sp3)–H bonds of azaarylmethylamines with aryl bromides and CO using a Pd catalyst. This work is unique in that it employs pronucleophiles with high pKa values, suggesting a wide variety of previously overlooked substrates may be viable coupling partners in carbonylation reactions. The reaction is operative under 1 atm of CO and does not require high pressure equipment. This one-pot cascade process is applicable to the coupling of a wide range of azaarylmethylamines and aryl bromides, enabling facile access to useful α-amino aryl-azaarylmethyl-ketones in moderate to high yields with good functional group tolerance. This work provides an attractive and complementary approach to prepare heteroatom-rich α-amino ketones.Data availability
The datasets supporting this article have been uploaded as part of the ESI.Author contributions
HZ performed the optimization of the reaction with help from BH. The substrate scope and product characterization was performed by HZ and BH. The first draft was written by HZ and all authors contributed to revising the draft. The project was conceived by PJW and the research directed by PJW and LX.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
P. J. W. thanks the U.S. National Science Foundation (CHE-1902509). We acknowledge the Program for China Scholarship Council (No. 201806360122), National Natural Science Foundation of China (21372258) for financial support. Yue Fu is acknowledged for preliminary experiments.References
- (a) M. C. Myers, J. Wang, J. A. Iera, J.-k. Bang, T. Hara, S. i. Saito, G. P. Zambetti and D. H. Appella, J. Am. Chem. Soc., 2005, 127, 6152–6153 CrossRef CAS PubMed; (b) F. I. Carroll, B. E. Blough, P. Abraham, A. C. Mills, J. A. Holleman, S. A. Wolckenhauer, A. M. Decker, A. Landavazo, K. T. McElroy and H. A. Navarro, J. Med. Chem., 2009, 52, 6768–6781 CrossRef CAS PubMed; (c) P. C. Meltzer, D. Butler, J. R. Deschamps and B. K. Madras, J. Med. Chem., 2006, 49, 1420–1432 CrossRef CAS PubMed; (d) T. O. Kolesnikova, S. L. Khatsko, K. A. Demin, V. A. Shevyrin and A. V. Kalueff, ACS Chem. Neurosci., 2019, 10, 168–174 CrossRef CAS PubMed; (e) A. T. Nchinda, K. Chibale, P. Redelinghuys and E. D. Sturrock, Bioorg. Med. Chem. Lett., 2006, 16, 4612–4615 CrossRef CAS PubMed; (f) A. Váradi, T. C. Palmer, N. Haselton, D. Afonin, J. J. Subrath, V. Le Rouzic, A. Hunkele, G. W. Pasternak, G. F. Marrone and A. Borics, ACS Chem. Neurosci., 2015, 6, 1570–1577 CrossRef PubMed; (g) G. Gevorgyan, N. Gasparyan, O. Papoyan, D. Avakimyan, A. Tatevosyan and H. Panosyan, Pharm. Chem. J., 2017, 51, 107–110 CrossRef CAS.
- (a) R. K. Hill, P. N. Nugara, E. M. Holt and K. P. Holland, J. Org. Chem., 1992, 57, 1045–1047 CrossRef CAS; (b) R. G. Franzén, J. Comb. Chem., 2000, 2, 195–214 CrossRef PubMed; (c) C. Bouteiller, J. Becerril-Ortega, P. Marchand, O. Nicole, L. Barré, A. Buisson and C. Perrio, Org. Biomol. Chem., 2010, 8, 1111–1120 RSC; (d) S. K. Gediya, G. J. Clarkson and M. Wills, J. Org. Chem., 2020, 85, 11309–11330 CrossRef CAS PubMed.
- (a) C. Greck, B. Drouillat and C. Thomassigny, Eur. J. Org. Chem., 2004, 1377–1385 CrossRef CAS; (b) A. M. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed; (c) A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441–4557 CrossRef CAS PubMed; (d) F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS; (e) T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917–958 CrossRef CAS PubMed; (f) M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001–2011 RSC; (g) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS PubMed; (h) A. de la Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef CAS PubMed.
- (a) L. E. Fisher and J. M. Muchowski, Org. Prep. Proced. Int., 1990, 22, 399–484 CrossRef CAS; (b) G. Cecere, C. M. König, J. L. Alleva and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed; (c) D. J. Fisher, G. L. Burnett, R. Velasco and J. Read de Alaniz, J. Am. Chem. Soc., 2015, 137, 11614–11617 CrossRef CAS PubMed; (d) I. Ramakrishna, H. Sahoo and M. Baidya, Chem. Commun., 2016, 52, 3215–3218 RSC; (e) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed; (f) I. Ramakrishna, V. Bhajammanavar, S. Mallik and M. Baidya, Org. Lett., 2017, 19, 516–519 CrossRef CAS PubMed; (g) Z. Zhou, Q.-Q. Cheng and L. s. Kürti, J. Am. Chem. Soc., 2019, 141, 2242–2246 CrossRef CAS PubMed.
- (a) E. Erdik, Tetrahedron, 2004, 40, 8747–8782 CrossRef; (b) Y. Wei, S. Lin and F. Liang, Org. Lett., 2012, 14, 4202–4205 CrossRef CAS PubMed; (c) M. N. Vander Wal, A. K. Dilger and D. W. MacMillan, Chem. Sci., 2013, 4, 3075–3079 RSC; (d) B. Xu, S. F. Zhu, X. D. Zuo, Z. C. Zhang and Q. L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913–3916 CrossRef CAS PubMed; (e) D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632–7635 CrossRef CAS PubMed; (f) S. Guha, V. Rajeshkumar, S. S. Kotha and G. Sekar, Org. Lett., 2015, 17, 406–409 CrossRef CAS PubMed.
- (a) M. Lamani and K. R. Prabhu, Chem.–Eur. J., 2012, 18, 14638–14642 CrossRef CAS PubMed; (b) R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed; (c) V. Rajeshkumar, S. Chandrasekar and G. Sekar, Org. Biomol. Chem., 2014, 12, 8512–8518 RSC; (d) Y. Lv, Y. Li, T. Xiong, Y. Lu, Q. Liu and Q. Zhang, Chem. Commun., 2014, 50, 2367–2369 RSC; (e) C. Xu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2014, 126, 4233–4237 CrossRef; (f) Q. Jiang, B. Xu, A. Zhao, J. Jia, T. Liu and C. Guo, J. Org. Chem., 2014, 79, 8750–8756 CrossRef CAS PubMed; (g) S. Liang, C.-C. Zeng, H.-Y. Tian, B.-G. Sun, X.-G. Luo and F.-z. Ren, J. Org. Chem., 2016, 81, 11565–11573 CrossRef CAS PubMed; (h) J. Strehl and G. Hilt, Org. Lett., 2020, 22, 5968–5972 CrossRef CAS PubMed.
- (a) J. A. Murry, D. E. Frantz, A. Soheili, R. Tillyer, E. J. Grabowski and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 9696–9697 CrossRef CAS PubMed; (b) A. E. Mattson and K. A. Scheidt, Org. Lett., 2004, 6, 4363–4366 CrossRef CAS PubMed; (c) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654–1655 CrossRef CAS PubMed; (d) G.-Q. Li, L.-X. Dai and S.-L. You, Chem. Commun., 2007, 852–854 RSC; (e) M. R. Garrett, J. C. Tarr and J. S. Johnson, J. Am. Chem. Soc., 2007, 129, 12944–12945 CrossRef CAS PubMed; (f) L. H. Sun, Z. Q. Liang, W. Q. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 125, 5915–5918 CrossRef; (g) H.-H. Zhang and S. Yu, Org. Lett., 2019, 21, 3711–3715 CrossRef CAS PubMed; (h) X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed.
- K. W. Kells and J. M. Chong, J. Am. Chem. Soc., 2004, 126, 15666–15667 CrossRef CAS PubMed.
- (a) H. Yang, H. Li, R. Wittenberg, M. Egi, W. Huang and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 1132–1140 CrossRef CAS PubMed; (b) H. Yang and L. S. Liebeskind, Org. Lett., 2007, 9, 2993–2995 CrossRef CAS PubMed; (c) H. Li, H. Yang and L. S. Liebeskind, Org. Lett., 2008, 10, 4375–4378 CrossRef CAS PubMed; (d) L. S. Liebeskind, H. Yang and H. Li, Angew. Chem., Int. Ed., 2009, 121, 1445–1449 CrossRef.
- (a) T. Sun, G. Hou, M. Ma and X. Zhang, Adv. Synth. Catal., 2011, 353, 253–256 CrossRef CAS; (b) W. Wen, Y. Zeng, L.-Y. Peng, L.-N. Fu and Q.-X. Guo, Org. Lett., 2015, 17, 3922–3925 CrossRef CAS PubMed.
- (a) T. Ooi, M. Takahashi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2002, 124, 7640–7641 CrossRef CAS PubMed; (b) A. Frongia, F. Secci, F. Capitta, P. P. Piras and M. L. Sanna, Chem. Commun., 2013, 49, 8812–8814 RSC; (c) X. Zhang, R. J. Staples, A. L. Rheingold and W. D. Wulff, J. Am. Chem. Soc., 2014, 136, 13971–13974 CrossRef CAS PubMed; (d) D. Yadagiri and P. Anbarasan, Chem. Commun., 2015, 51, 14203–14206 RSC.
- (a) A. Villar, C. H. Hövelmann, M. Nieger and K. Muñiz, Chem. Commun., 2005, 3304–3306 RSC; (b) P. K. Prasad, R. N. Reddi and A. Sudalai, Org. Lett., 2016, 18, 500–503 CrossRef CAS PubMed; (c) M. H. Shinde and U. A. Kshirsagar, Org. Biomol. Chem., 2016, 14, 858–861 RSC; (d) S. Xu, P. Wu and W. Zhang, Org. Biomol. Chem., 2016, 14, 11389–11395 RSC.
- (a) I. Tellitu, S. Serna, M. T. Herrero, I. Moreno, E. Domínguez and R. SanMartin, J. Org. Chem., 2007, 72, 1526–1529 CrossRef CAS PubMed; (b) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi, E. Filisti, A. Goggiamani, A. Iazzetti and L. Maurone, Org. Biomol. Chem., 2012, 10, 4699–4703 RSC; (d) T. Sueda, A. Kawada, Y. Urashi and N. Teno, Org. Lett., 2013, 15, 1560–1563 CrossRef CAS PubMed; (e) N. Chalotra, M. A. Rizvi and B. A. Shah, Org. Lett., 2019, 21, 4793–4797 CrossRef CAS PubMed; (f) Z. Zhang, Y. Luo, H. Du, J. Xu and P. Li, Chem. Sci., 2019, 10, 5156–5161 RSC.
- W. Guo, Y. Luo, H. H.-Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed.
- (a) I. Ojima, Chem. Rev., 1988, 88, 1011–1030 CrossRef CAS; (b) A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef; (c) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS; (d) J. S. Quesnel and B. A. Arndtsen, Pure Appl. Chem., 2013, 85, 377–384 CAS; (e) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed; (f) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS; (g) P. Gautam and B. M. Bhanage, Catal. Sci. Technol., 2015, 5, 4663–4702 RSC; (h) Y. Bai, D. C. Davis and M. Dai, J. Org. Chem., 2017, 82, 2319–2328 CrossRef CAS PubMed; (i) Y. Li, Y. Hu and X.-F. Wu, Chem. Soc. Rev., 2018, 47, 172–194 RSC; (j) J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS; (k) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2018, 119, 2090–2127 CrossRef PubMed.
- (a) J. Zhu and H. Bienaymé, Multicomponent reactions, John Wiley & Sons, 2006 Search PubMed; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed.
- Y. Liu, Z. Zhang, S. Zhang, Y. Zhang, J. Wang and Z. Zhang, Chem.–Asian J., 2018, 13, 3658–3663 CrossRef CAS PubMed.
- (a) T.-a. Kobayashi and M. Tanaka, Tetrahedron Lett., 1986, 27, 4745–4748 CrossRef CAS; (b) E.-i. Negishi, C. Copéret, T. Sugihara, I. Shimoyama, Y. Zhang, G. Wu and J. M. Tour, Tetrahedron, 1994, 50, 425–436 CrossRef CAS; (c) E.-i. Negishi, H. Makabe, I. Shimoyama, G. Wu and Y. Zhang, Tetrahedron, 1998, 54, 1095–1106 CrossRef CAS; (d) Z. Zheng and H. Alper, Org. Lett., 2009, 11, 3278–3281 CrossRef CAS PubMed.
- (a) T. M. Gøgsig, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 124, 822–825 CrossRef; (b) S. Korsager, D. U. Nielsen, R. H. Taaning and T. Skrydstrup, Angew. Chem., Int. Ed., 2013, 52, 9763–9766 CrossRef CAS PubMed; (c) J. Schranck, A. Tlili, P. G. Alsabeh, H. Neumann, M. Stradiotto and M. Beller, Chem.–Eur. J., 2013, 19, 12624–12628 CrossRef CAS PubMed; (d) S. Korsager, D. U. Nielsen, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, Chem.–Eur. J., 2013, 19, 17687–17691 CrossRef CAS PubMed; (e) D. U. Nielsen, C. Lescot, T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Chem.–Eur. J., 2013, 19, 17926–17938 CrossRef CAS PubMed; (f) J. Schranck, M. Burhardt, C. Bornschein, H. Neumann, T. Skrydstrup and M. Beller, Chem.–Eur. J., 2014, 20, 9534–9538 CrossRef CAS PubMed; (g) Z. Lian, S. D. Friis and T. Skrydstrup, Angew. Chem., Int. Ed., 2014, 53, 9582–9586 Search PubMed; (h) Z. Lian, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 3600–3603 RSC; (i) I. S. Makarov, T. Kuwahara, X. Jusseau, I. Ryu, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2015, 137, 14043–14046 CrossRef CAS PubMed; (j) M. T. Jensen, M. Juhl, D. U. Nielsen, M. F. Jacobsen, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2016, 81, 1358–1366 CrossRef CAS PubMed.
- (a) R. R. Fraser, T. S. Mansour and S. Savard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS; (b) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- (a) J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765–13772 CrossRef CAS PubMed; (b) A. Bellomo, J. Zhang, N. Trongsiriwat and P. J. Walsh, Chem. Sci., 2013, 4, 849–857 RSC; (c) B.-S. Kim, J. Jimenez, F. Gao and P. J. Walsh, Org. Lett., 2015, 17, 5788–5791 CrossRef CAS PubMed; (d) M. Li, M. González-Esguevillas, S. Berritt, X. Yang, A. Bellomo and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 128, 2875–2879 CrossRef; (e) A. R. Rivero, B.-S. Kim and P. J. Walsh, Org. Lett., 2016, 18, 1590–1593 CrossRef CAS PubMed; (f) X. Yang, B.-S. Kim, M. Li and P. J. Walsh, Org. Lett., 2016, 18, 2371–2374 CrossRef CAS PubMed; (g) J. Jiménez, B. S. Kim and P. J. Walsh, Adv. Synth. Catal., 2016, 358, 2829–2837 CrossRef PubMed; (h) J. Zhang, S.-C. Sha, A. Bellomo, N. Trongsiriwat, F. Gao, N. C. Tomson and P. J. Walsh, J. Am. Chem. Soc., 2016, 138, 4260–4266 CrossRef CAS PubMed; (i) M. Li, B. Yucel, J. Jiménez, M. Rotella, Y. Fu and P. J. Walsh, Adv. Synth. Catal., 2016, 358, 1910–1915 CrossRef CAS PubMed; (j) K. Ablajan, G. B. Panetti, X. Yang, B. S. Kim and P. J. Walsh, Adv. Synth. Catal., 2017, 359, 1927–1932 CrossRef CAS PubMed; (k) G. Gao, Y. Fu, M. Li, B. Wang, B. Zheng, S. Hou and P. J. Walsh, Adv. Synth. Catal., 2017, 359, 2890–2894 CrossRef CAS PubMed; (l) S.-C. Sha, S. Tcyrulnikov, M. Li, B. Hu, Y. Fu, M. C. Kozlowski and P. J. Walsh, J. Am. Chem. Soc., 2018, 140, 12415–12423 CrossRef CAS PubMed; (m) J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276–6287 CrossRef CAS PubMed.
- R. Lavernhe, E. J. Alexy, H. Zhang and B. M. Stoltz, Org. Lett., 2020, 11, 4272–4275 CrossRef PubMed.
- (a) F. M. Miloserdov, C. L. McMullin, M. M. Belmonte, J. B. Buchholz, V. I. Bakhmutov, S. A. Macgregor and V. V. Grushin, Organometallics, 2014, 33, 736–752 CrossRef CAS; (b) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979–7993 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02078a |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2021 |