
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric hydrogenation of exocyclic γ,δ-unsaturated β-ketoesters to functionalized chiral allylic alcohols via dynamic kinetic resolution†
Huai-Yu
Bin
a,
Li
Cheng
a,
Xiong
Wu
a,
Chang-Liang
Zhu
a,
Xiao-Hui
Yang
*b,
Jian-Hua
Xie
 *a and
Qi-Lin
Zhou
a
*a and
Qi-Lin
Zhou
a
aState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
bAdvanced Research Institute of Multidisciplinary Science, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: jhxie@nankai.edu.cn; xhyang@bit.edu.cn
First published on 29th April 2021
Abstract
An iridium catalyzed asymmetric hydrogenation of racemic exocyclic γ,δ-unsaturated β-ketoesters via dynamic kinetic resolution to functionalized chiral allylic alcohols was developed. With the chiral spiro iridium catalysts Ir-SpiroPAP, a series of racemic exocyclic γ,δ-unsaturated β-ketoesters bearing a five-, six-, or seven-membered ring were hydrogenated to the corresponding functionalized chiral allylic alcohols in high yields with good to excellent enantioselectivities (87 to >99% ee) and cis-selectivities (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 to >99:1). The origin of the excellent stereoselectivity was also rationalized by density functional theory calculations. Furthermore, this protocol could be performed on gram scale and at a lower catalyst loading (0.002 mol%) without the loss of reactivity and enantioselectivity, and has been successfully applied in the enantioselective synthesis of chiral carbocyclic δ-amino esters and the β-galactosidase inhibitor isogalactofagomine.
:7 to >99:1). The origin of the excellent stereoselectivity was also rationalized by density functional theory calculations. Furthermore, this protocol could be performed on gram scale and at a lower catalyst loading (0.002 mol%) without the loss of reactivity and enantioselectivity, and has been successfully applied in the enantioselective synthesis of chiral carbocyclic δ-amino esters and the β-galactosidase inhibitor isogalactofagomine.
Introduction
Chiral allylic alcohols not only are highly valuable and versatile chiral building blocks in organic synthesis but also represent common structural motifs in a variety of natural products and biologically active compounds. As a result, substantial efforts have been devoted to the development of a variety of catalytic asymmetric reactions allowing efficient and enantioselective syntheses of chiral allylic alcohols.1 Among them, catalytic asymmetric hydrogenation of enones has been demonstrated to be one of the most efficient, practical, and atom-economical approaches.2 However, despite long-standing interest, the enantioselective synthesis of highly functionalized chiral allylic alcohols such as γ,δ-unsaturated β-hydroxy esters with two contiguous stereocenters remains rare and challenging.2 The reported catalytic asymmetric methods for the synthesis of optically active chiral γ,δ-unsaturated β-hydroxy esters with two contiguous stereocenters are limited to asymmetric aldol reactions (Scheme 1a)3 and ruthenium catalyzed asymmetric transfer hydrogenation of racemic α-substituted γ,δ-unsaturated β-ketoesters via a dynamic kinetic resolution (DKR) (Scheme 1b).4 However, most of these methodologies are restricted to provide noncyclic chiral γ,δ-unsaturated β-hydroxy esters containing two contiguous stereocenters, and no reports have been published on the enantioselective synthesis of such types of highly functionalized chiral allylic alcohols through direct asymmetric hydrogenation of the corresponding racemic enone esters via DKR.5 | ||
| Scheme 1 Enantioselective synthesis of chiral allylic alcohols with two contiguous stereocenters. | ||
In recent years, we were interested in the development of highly efficient asymmetric hydrogenations for the syntheses of chiral alcohols with multiple chiral stereocenters, which could serve as valuable chiral building blocks to facilitate the concise and rapid construction of the chiral core structures of natural products.6 We noticed that chiral exocyclic allylic alcohols with contiguous stereocenters are widely found in biologically active natural products (Fig. 1).7 Inspired by these fascinating substructural features, we envisioned an asymmetric hydrogenation of racemic exocyclic γ,δ-unsaturated β-ketoesters via DKR for the synthesis of ester-functionalized chiral cyclic allylic alcohols with two contiguous stereocenters. We found that chiral spiro iridium catalysts Ir-SpiroPAP8 were highly efficient for these transformations, providing the corresponding functionalized chiral allylic alcohols with two contiguous stereocenters in high yields and excellent enantioselectivities (Scheme 1c). We successfully employed this efficient asymmetric hydrogenation as a key step for the scalable enantioselective total synthesis of the monoterpene indole alkaloid (−)-goniomitine.9 In this study, we showcase the details of the asymmetric hydrogenation of such γ,δ-unsaturated racemic cyclic β-ketoesters via DKR10 and the application of this method in the enantioselective synthesis of chiral carbocyclic δ-amino esters and the β-galactosidase inhibitor (+)-isogalactofagomine (Scheme 1c).
 | ||
| Fig. 1 Representative biologically active natural products containing chiral exocyclic allylic alcohol substructures. | ||
Results and discussion
The study commenced with the evaluation of chiral catalysts in the hydrogenation of a racemic δ-aryl substituted exocyclic enone ester bearing a six-membered ring, 7a (Scheme 2). We initially evaluated the Ru–BINAP complex Ru[(S)–BINAP]Cl2(DMF)n ((S)-1),11 a type of efficient chiral catalyst for asymmetric hydrogenation of racemic α-substituted β-ketoesters via DKR. Under the general conditions (1 mol% (S)-1, MeOH, 50 °C, 100 atm H2), 8a was obtained in 18% yield and 87% ee with 96:4 trans-selectivity. Then, another type of Ru–BINAP complex, [RuCl(p-cymene)(S)-BINAP]Cl ((S)-2), was also evaluated,12 which provided 8a in 11% yield and 84% ee with 99:1 trans-selectivity. The chiral spiro Ru–SDP complex RuCl2-(R)-Xyl-SDP/(S,S)-DPEN ((Ra,S,S)-3)13 and the iridium complex Ir-(R)-SpiroAP ((R)-4),2f developed by us previously, showed low reactivity (<5% conv.). The chiral spiro iridium complex Ir-(R)-SpiroSAP ((R)-5)14 displayed higher reactivity but a moderate ee value (41% yield, 72% ee and >99:1 cis-selectivity). However, promising results were observed by using the chiral spiro iridium complex Ir-(R)-SpiroPAP ((R)-6a),8 providing the desired product 8a in a high yield (94%) with excellent enantioselectivity (98% ee) and high cis-selectivity (99:1).
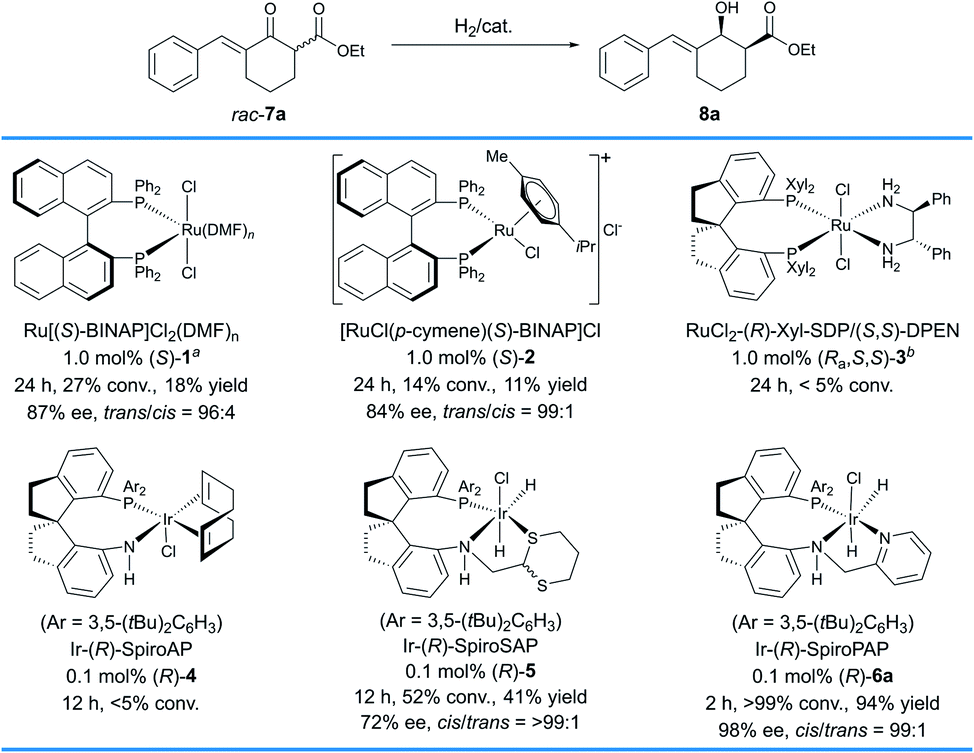 | ||
| Scheme 2 Evaluation of chiral catalysts for asymmetric hydrogenation of 7a. Reaction conditions: 1.0 mmol scale, 7a/cat. = 100:1, MeOH (4.0 mL), 50 atm H2, 50 °C, 24 h for Ru-catalysts (S)-1, (S)-2, and (Ra,S,S)-3; 1.0 mmol scale, 7a/KOtBu/cat. = 1000:10:1, EtOH (4.0 mL), 10 atm H2, 25–30 °C, 12 h for Ir-catalysts (R)-4 and 5, and 2 h for Ir-catalyst (R)-6a unless otherwise specified. The conversions of 7a were determined by 1H NMR spectroscopy. The yields are isolated yields of cis- and trans-isomers of 8a. The enantiomeric excesses (ee) of the major diastereomers and the cis/trans values were determined by HPLC analysis using a chiral column. a100 atm H2. b5 mol% KOtBu, iPrOH, 25–30 °C. | ||
Subsequently, the evaluation of other Ir-(R)-SpiroPAPs showed that the catalysts (R)-6b gave the highest enantioselectivity (99% ee) and cis-selectivity (>99:1) (Table 1, entries 1–7). In addition to KOtBu, other bases (NaOtBu, LiOtBu and K2CO3) could also be used, but the enantioselectivity and cis-selectivity were slightly lower, and a longer reaction time was needed when using K2CO3 as the base (Table 1, entries 8–10). When the H2 pressure was decreased to 1 atm, the reaction time was increased to 2 h with the enantioselectivity and cis-selectivity being maintained (Table 1, entry 11). It is worth noting that when the catalyst loading was reduced to 0.002 mol% (S/C = 50000) at 50 atm H2, the hydrogenation still occurred smoothly without change in reactivity (98% yield) and enantioselectivity (99% ee) (Table 1, entry 12).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | (R)-6 | Base | Time (h) | Yieldb (%) | cis/transc | eec (%) |
|
a Reaction conditions: 1.0 mmol scale, 7a/base/(R)-6 = 1000:10:1, EtOH (4.0 mL), 25–30 °C, >99% conversion (determined by 1H NMR spectroscopy).
b Isolated yields of cis- and trans-isomers of 8a.
c The enantiomeric excesses (ee) of the major diastereomer and the cis/trans values were determined by HPLC analysis using a chiral column.
d 1 atm H2.
e 5.2 g (20 mmol) scale, 7a/KOtBu/(R)-6b = 50000:100:1, 50 atm H2 (initial), EtOH (50 mL).
|
||||||
| 1 | (R)-6a | KOtBu | 2.0 | 94 | 99:1 |
98 |
| 2 | (R)-6b | KOtBu | 0.5 | 96 | >99:1 |
99 |
| 3 | (R)-6c | KOtBu | 0.5 | 95 | 99:1 |
98 |
| 4 | (R)-6d | KOtBu | 0.5 | 95 | 98:2 |
98 |
| 5 | (R)-6e | KOtBu | 0.5 | 96 | 99:1 |
55 |
| 6 | (R)-6f | KOtBu | 0.5 | 97 | 99:1 |
98 |
| 7 | (R)-6g | KOtBu | 0.5 | 95 | >99:1 |
98 |
| 8 | (R)-6b | NaOtBu | 0.5 | 97 | 99:1 |
98 |
| 9 | (R)-6b | LiOtBu | 0.5 | 94 | 99:1 |
98 |
| 10 | (R)-6b | K2CO3 | 10.0 | 95 | 99:1 |
98 |
| 11d | (R)-6b | KOtBu | 2.0 | 97 | >99:1 |
99 |
| 12e | (R)-6b | KOtBu | 24 | 98 | 99:1 |
99 |

With the optimized reaction conditions in hand, we first investigated the hydrogenation of racemic δ-aryl substituted exocyclic enone esters 7 bearing a six-membered ring (Scheme 3). The electronic properties and the positions of the substituents on the phenyl ring have no obvious effect on the reactivity or enantioselectivity, and the corresponding hydrogenated products 8b–q were obtained in high yields (90–98%) with excellent enantioselectivity (92–99% ee) and cis-selectivity (dr = 98:2 to >99:1). The substrates 7r–v containing a heteroaryl ring, such as 2-pyridinyl (7r) and 2-thienyl (7t), also provided the desired products 8r–v in comparable yields (92–96%) and cis-selectivities (99 to >99% ee, 96:4 to >99:1 dr). The hydrogenation of substrates 7w–y derived from 1,4-cyclohexanedione monoacetal (7w), N-Boc piperidin-4-one (7x), and tetrahydropyran-4-one (7y), respectively, also occurred smoothly and afforded chiral allylic alcohols 8w–y in high yields (93–97%), enantioselectivities (97–99% ee) and cis-selectivities (96:4 to 98:2 dr). The substrate 7z with a dienone moiety was also a suitable candidate, providing dienyl alcohol 8z in 95% yield with excellent enantioselectivity (99% ee) and cis-selectivity (>99:1 dr). The stereochemistry of the hydrogenation can be determined using the single crystals of 8v. The X-ray diffraction analysis of the crystal structure of 8v showed that the hydrogenation with the catalyst (R)-6b gave 8v with a 1S,2S-configuration.
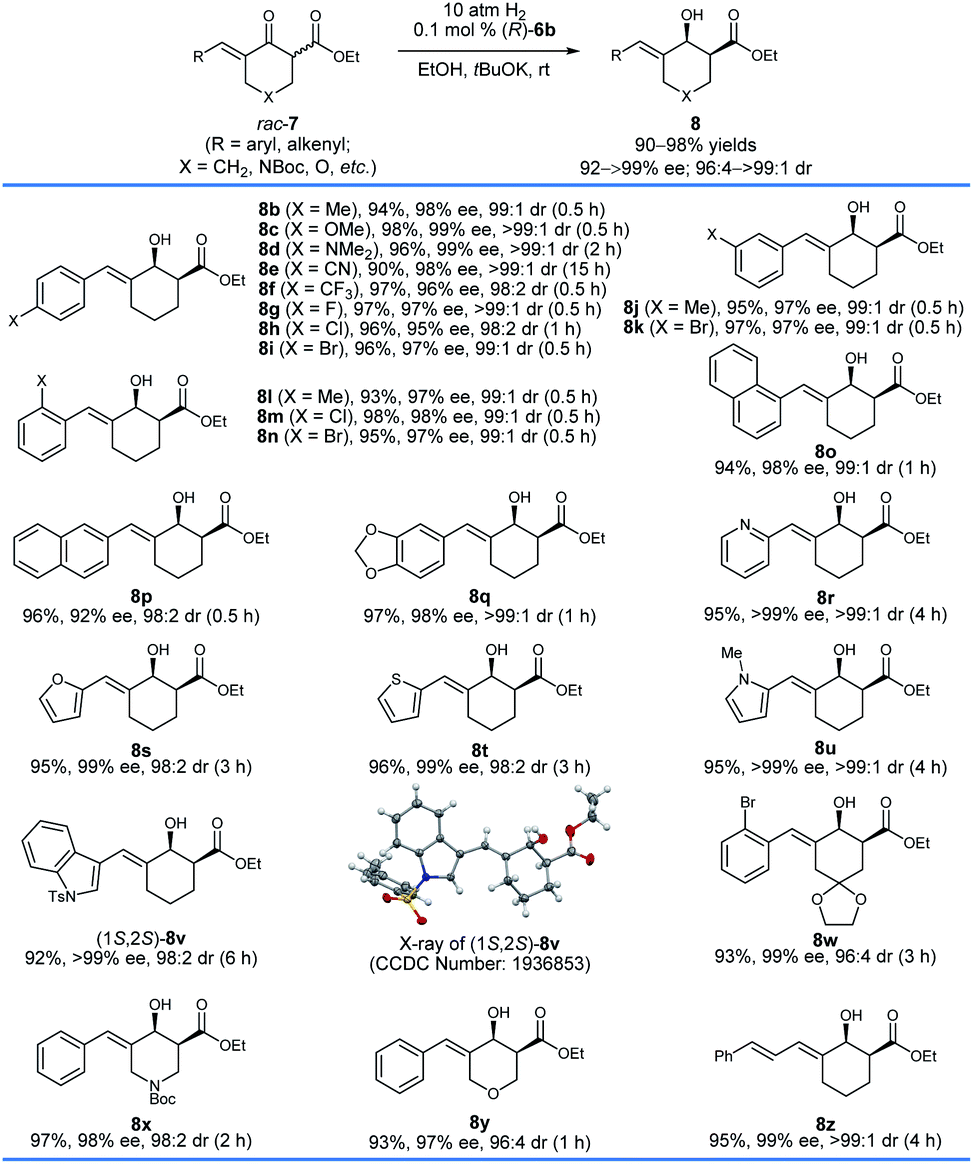 | ||
| Scheme 3 Asymmetric hydrogenation of δ-aryl substituted exocyclic enone esters bearing a six-membered ring, 7. Reaction conditions: 1.0 mmol scale, 7/tBuOK/(R)-6b = 1000:10:1, EtOH (4.0 mL), 25–30 °C, 0.5–15 h. The yields are isolated yields of cis- and trans-isomers of 8. The enantiomeric excesses (ee) of the major diastereomer and the diastereomeric ratio (dr) values were determined by HPLC analysis using a chiral column. | ||
A series of racemic δ-alkyl substituted six-membered exocyclic enone esters 7 were also evaluated with (R)-6b under the same conditions (Scheme 4). The size of the alkyl groups of the substrates has no significant effect on the enantioselectivity, and chiral allylic alcohols 8aa–aq were obtained in high yields (54–98%) with 86 to >99% ee and 93:7 to >99:1 cis-selectivity. However, a longer reaction time was required for the substrates with a relatively bulky alkyl group. The substrate 7aa with a terminal double bond gave 8aa in 54% yield with 94% ee and >99:1 cis-selectivity. The reason for the lower yield was that the catalyst (R)-6b hydrogenated the less hindered terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of 8aa, leading to the production of the corresponding saturated alcohol with both the CC and CO double bonds being hydrogenated successively.15 Notably, functional groups such as allyl ether (7aj), aldehyde acetal (7ak), ester (7al), and amide (7am) could be tolerated, and the corresponding chiral allylic alcohols 8aj–am were delivered in great reactivity with 86 to >99% ee and 93:7 to >99:1 cis-selectivity. The hydrogenation of substrates 7an–ap worked well and provided chiral allylic alcohols 8an–ap in high yields (94–97%) and enantioselectivities (94 to >99% ee). In addition, the hydrogenation of the more challenging tetrasubstituted enone ester 7aq could also be completed within 0.5 h and it provided the allylic alcohol 8aq in 98% yield with 99% ee and >99:1 cis-selectivity.
C double bond of 8aa, leading to the production of the corresponding saturated alcohol with both the CC and CO double bonds being hydrogenated successively.15 Notably, functional groups such as allyl ether (7aj), aldehyde acetal (7ak), ester (7al), and amide (7am) could be tolerated, and the corresponding chiral allylic alcohols 8aj–am were delivered in great reactivity with 86 to >99% ee and 93:7 to >99:1 cis-selectivity. The hydrogenation of substrates 7an–ap worked well and provided chiral allylic alcohols 8an–ap in high yields (94–97%) and enantioselectivities (94 to >99% ee). In addition, the hydrogenation of the more challenging tetrasubstituted enone ester 7aq could also be completed within 0.5 h and it provided the allylic alcohol 8aq in 98% yield with 99% ee and >99:1 cis-selectivity.
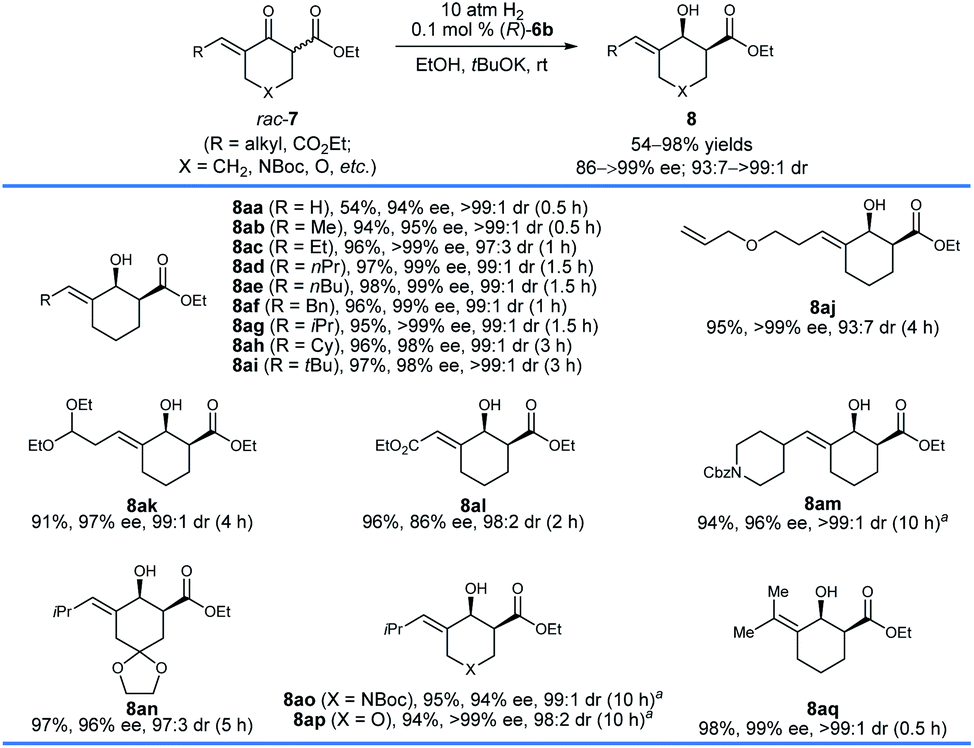 | ||
| Scheme 4 Asymmetric hydrogenation of δ-alkyl substituted exocyclic enone esters bearing a six-membered ring, 7. Reaction conditions: 1.0 mmol scale, 7/tBuOK/(R)-6b = 1000:10:1, EtOH (4.0 mL), 25–30 °C, 0.5–10 h. The yields are isolated yields of cis- and trans-isomers of 8. The enantiomeric excess (ee) of the major diastereomer and the diastereomeric ratio (dr) values were determined by HPLC analysis using a chiral column. a7/tBuOK/(R)-6b = 500:50:1. | ||
Subsequently, we found that (R)-6b was also highly efficient for the asymmetric hydrogenation of racemic exocyclic enone esters bearing a seven-membered ring, 9 (Scheme 5). The hydrogenations could be completed within 2 h which afforded chiral exocyclic allylic alcohols 10 in high yields (90–96%) with excellent enantioselectivities (98 to >99% ee) and cis-selectivities (>99:1). Likewise, the substrates bearing coordinating heteroaryl moieties such as 2-pyridinyl (9g) and 2-furanyl (9h) have no significant effect on both the reactivity and enantioselectivity of the hydrogenations.
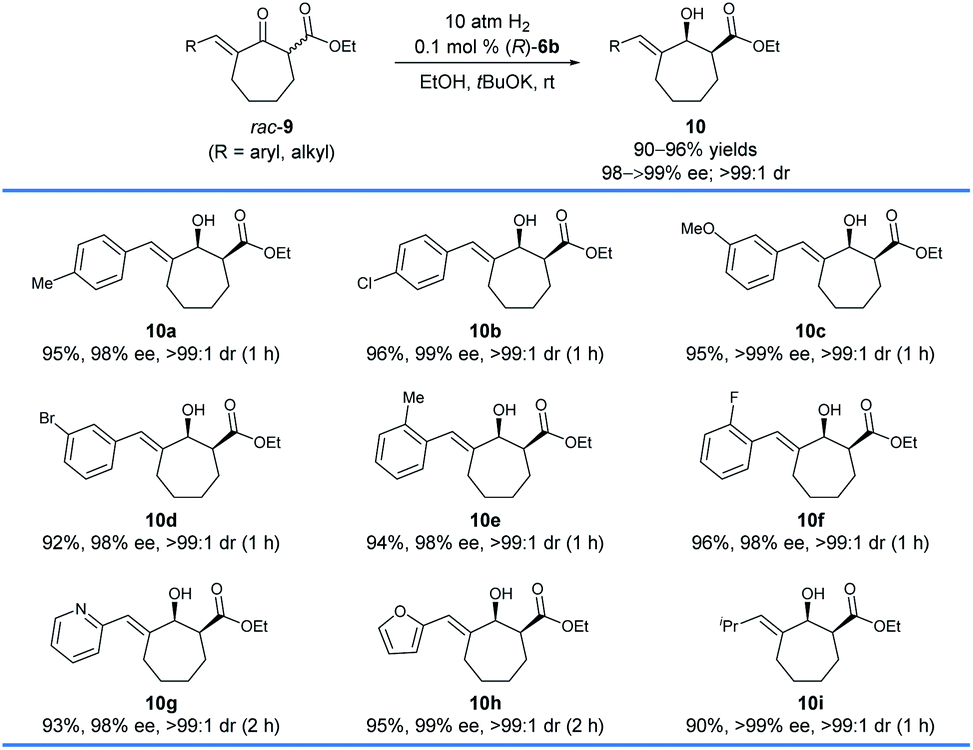 | ||
| Scheme 5 Asymmetric hydrogenation of exocyclic enone esters bearing a seven-membered ring, 9. Reaction conditions: 1.0 mmol scale, 9/tBuOK/(R)-6b = 1000:10:1, EtOH (4.0 mL), 25–30 °C, 1–2 h. The yields are isolated yields of cis- and trans-isomers of 10. cThe enantiomeric excesses (ee) of the major diastereomer and the diastereomeric ratio (dr) values were determined by HPLC analysis using a chiral column. | ||
The aforementioned asymmetric hydrogenation of racemic exocyclic enone esters bearing a five-membered ring has been successfully applied in the enantioselective synthesis of the monoterpene indole alkaloid (−)-goniomitine,9 but the substrate scope was not widely studied. Thus, we also tested the asymmetric hydrogenation of racemic exocyclic enone esters bearing a five-membered ring, 11, with the catalyst (R)-6b (Scheme 6). We found that (R)-6b provided 89–93% ee and 96:4 to >99:1 cis-selectivity for the hydrogenation of 11a–e with a para- or meta-substituent on the phenyl ring. When the five-membered substrate 11f with an ortho-F on the phenyl ring was hydrogenated, the catalyst (R)-6g with a 4-tBu on the pyridinyl ring gave the corresponding allylic alcohol product 12f in 96% yields with 92% ee and 99:1 cis-selectivity. The heteroaryl substituted allylic alcohol 12g (2-thienyl) could be obtained with 89% ee and >99:1 cis-selectivity using (R)-6b as the catalyst. Moreover, the hydrogenation of the alkyl substituted substrate 11h (R = iPr) with the catalyst (R)-6g provided allylic alcohol 12h (97%, 87% ee) with relatively low enantioselectivity and excellent cis-selectivity (99:1). The absolute configuration of 12f was determined to be 1S,2S by single-crystal X-ray crystallography. This result showed that the asymmetric hydrogenation of five-membered exocyclic enone esters 11 with (R)-6b or 6g shares a similar stereochemical control with the asymmetric hydrogenation of six-membered exocyclic enone esters 7.
 | ||
| Scheme 6 Asymmetric hydrogenation of exocyclic enone esters bearing a five-membered ring, 11. Reaction conditions: 1.0 mmol scale, 11/tBuOK/(R)-6b = 1000:10:1, EtOH (4.0 mL), 25–30 °C, 4–16 h. The yields are isolated yields of cis- and trans-isomers of 12. The enantiomeric excesses (ee) of the major diastereomer and the diastereomeric ratio (dr) values were determined by HPLC analysis using a chiral column. aWith (R)-6g as the catalyst. | ||
To understand the origins of the stereoselectivity, density functional theory (DFT) calculations were performed based on an outer-sphere mechanism.16Fig. 2 showcases the proposed models of the interaction between the substrate 7ab and catalyst (R)-6b according to the crystal structure of Ir-SpiroPAP.8b To minimize steric repulsion, the substrate 7ab approaches the catalyst (R)-6b with the ester group of the substrate away from the catalyst. Thus, the hydride from the Ir (Ir–H) and the proton from the nitrogen (N–H) of the catalyst (R)-6b prefer to transfer through six-membered-ring transition states TS-SS and TS-RR to the keto-carbonyl groups of the (S)- or (R)-isomer of the substrate 7ab. Comparing the calculated energies of TS-SS (0.0 kcal mol−1) and TS-RR (2.5 kcal mol−1), an activation Gibbs free energy difference of 2.5 kcal mol−1 was found between them. These results suggested that the catalyst (R)-6b tends to preferentially afford (1S,2S)-8ab, which is consistent with the experimental observation. The analysis of the non-covalent interactions (NCIs) with the independent gradient model (IGM) method also indicated that TS-SS was calculated to be more stable than TS-RR, owing to the attractive C–H/π interaction17 between the α-C–H bond of the ester group and one of the phenyl ring of the spiro backbone in TS-SS (for details, see the ESI†). Therefore, the 1S,2S-configured product was preferentially formed with excellent enantioselectivity.
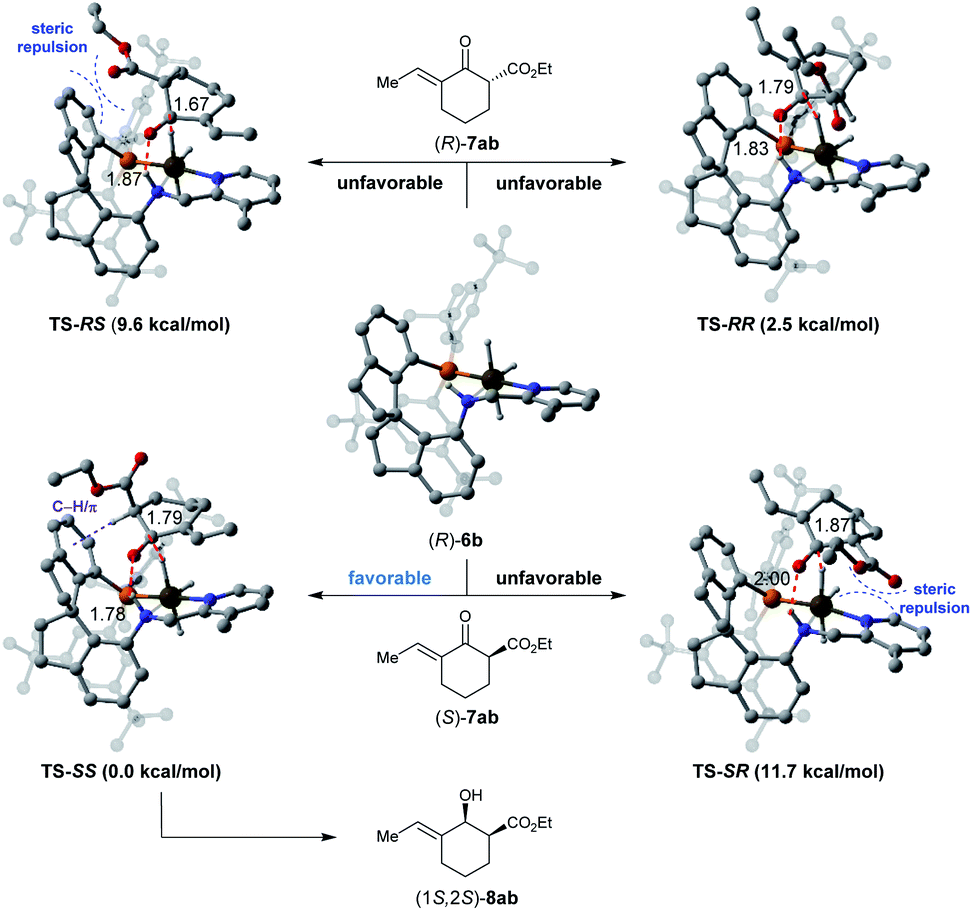 | ||
| Fig. 2 Diastereomeric hydride/proton transfer transition states. Relative Gibbs free energies are reported in parentheses. The C–H/π interactions are depicted in purple dashed lines. | ||
To exemplify the potential utility of this highly efficient asymmetric hydrogenation methodology, we initially selected δ-amino acids and their derivatives as targets. Chiral δ-amino acids are an important class of biomolecules used extensively as peptidomimetics and in the development of pharmaceuticals.18 We noticed that carbocyclic δ-amino acid ester 16 is a key chiral intermediate to peptidomimetic 17 (Scheme 7), which showed submicromolar activity on β-site amyloid precursor protein cleavage enzyme-1 (BACE-1).19 However, long synthetic sequences (at least 11 steps) were required to complete the synthesis of such a δ-amino acid and its derivatives.19b Our synthetic route started from the asymmetric hydrogenation of 7ag with (S)-6b on the multi-gram scale at 0.01 mol% catalyst loading. The hydrogenation was completed within 8 h under 30 atm H2 pressure and it afforded (−)-8ag in 97% yield with 98% ee and 97:3 cis-selectivity. Carbamoylation of (−)-8ag with trichloroacetyl isocyanate followed by dehydration with trifluoroacetic anhydride (TFAA) in the presence of NEt3 yielded an allyl isocyanate intermediate 14, which was then converted to allylic amine (+)-15 in 88% yield (2 steps) via an Ichikawa rearrangement20 and trapping with MeOH promoted by Bu3SnOMe according to Stecko's procedure.21 Hydrogenation of allylic amine (+)-15 on Pd/C afforded N-methoxycarbonyl δ-amino acid ester (−)-16 in 98% yield with 6:1 diastereoselectivity. Thus, we completed the enantioselective synthesis of the δ-amino acid derivative (−)-16 in 4 steps with 84% overall yield from exocyclic enone ester 7ag and provided an efficient and concise access to carbocyclic δ-amino acid esters.
 | ||
| Scheme 7 Enantioselective synthesis of carbocyclic δ-amino acid ester 16. | ||
The synthetic utility was further demonstrated through the enantioselective synthesis of (+)-isogalactofagomine from exocyclic enone ester 7x (Scheme 8). Isogalactofagomine (4-epi-isofagomine) is one of the most interesting members of azasugars and is an extremely potent and selective β-galactosidase inhibitor (IC50 = 12 nM; Ki = 4 nM).22 While several synthetic routes have been reported to access such azasugars, these approaches rely on chiral starting materials23 and chiral resolution by enzymes.24 The gram-scale asymmetric hydrogenation of 7x with (R)-6b provided (+)-8x in 97% yield with 98% ee and 98:2 cis-selectivity within 12 h. The reduction of (+)-8x with LiAlH4 followed by the reaction with 2,2-dimethoxypropane yielded the cyclic imino derivative (+)-18 in 76% yield. Then, ozonolysis followed by reduction with NaBH4 in a one-pot procedure converted (+)-19 to the alcohol product (+)-20 in 85% yield with >20:1 diastereoselectivity. The treatment of (+)-20 with trifluoroacetic acid (TFA) afforded the target molecule (+)-isogalactofagomine in quantitative yield. The NMR spectroscopic data and the optical rotation ([α]25D = +2.6 (c = 1.0, H2O); lit.24 [α]22D = +2.5 (c = 1.0, H2O)) of our synthetic (+)-isogalactofagomine are identical to those reported in the previous synthesis.24 In short, with the asymmetric hydrogenation of exocyclic enone ester 7x as the key step, (+)-isogalactofagomine was synthesized enantioselectively in 63% overall yield via 5 steps. This represents the first example of transition-metal catalyzed asymmetric synthesis of (+)-isogalactofagomine.
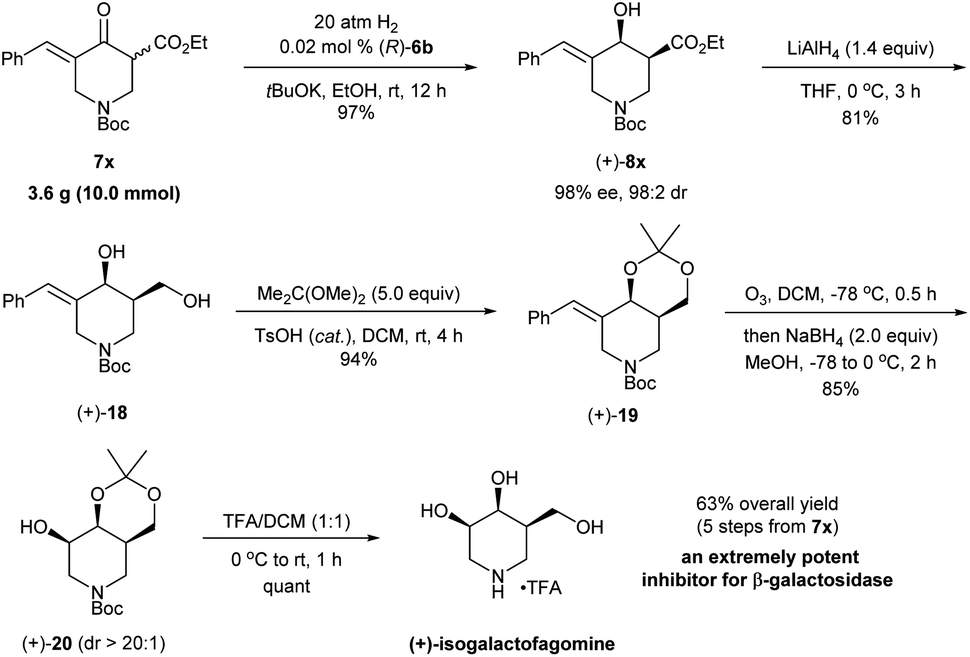 | ||
| Scheme 8 Enantioselective synthesis of (+)-isogalactofagomine. | ||
Conclusions
In conclusion, we developed an efficient asymmetric hydrogenation of racemic exocyclic enone esters to functionalized chiral allylic alcohols. With chiral spiro iridium catalysts (R)-6b and 6g, a series of racemic exocyclic enone esters bearing a five-, six-, or seven-membered ring were hydrogenated to the corresponding functionalized chiral allylic alcohols in high yields with good to excellent enantioselectivities (87 to >99% ee) and cis-selectivities (93:7 to >99:1). The asymmetric hydrogenation of six- and seven-membered exocyclic enone esters 7 and 9 afforded better enantioselectivities (95 to >99% ee) than those for five-membered exocyclic enone esters 11 (87–93% ee). The origin of excellent enantioselectivity and cis-selectivity in the catalysis was revealed by DFT calculations to be an attractive C–H/π interaction and stereo-repulsion between the substrate and the catalyst in the favored transition state. This asymmetric hydrogenation could be performed on gram scale and at a lower catalyst loading (0.002 mol%) without the loss of enantioselectivity. Based on this highly efficient asymmetric hydrogenation, the concise and efficient approaches to chiral carbocyclic δ-amino esters and the β-galactosidase inhibitor isogalactofagomine were developed.
Author contributions
H.-Y. B. performed the experiments and prepared the ESI† and paper. L. C. and C.-L. Z. prepared some substrates and repeated some experiments. X. W. carried out all computational work. X.-H. Y., J.-H. X. and Q.-L. Z. conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21871152, 92056105, 21532003, and 21790332), the “111” project (B06005) of the Ministry of Education of China, and the Fundamental Research Funds for the Central Universities (Nankai University, 020-63191746) for financial support.Notes and references
- (a) Palladium Reagents and Catalysis, ed. J. Tsuji, Wiley-VCH, Chichester, 1997, ch. 4 Search PubMed; (b) T. Katsuki, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 2, p. 621 Search PubMed; (c) R. A. Johnson and K. B. Sharpless, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Weinheim, 2000, ch. 6A Search PubMed; (d) A. Lumbroso, M. L. Cooke and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 1890–1932 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS; (b) T. Ohkuma and R. Noyori, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, p. 1106 Search PubMed; (c) Z. Zhang, M. Butt, N. Zhou, D. Liu and W. Zhang, Chin. J. Chem., 2018, 36, 443–454 CrossRef CAS . For selected papers, see:; (d) N. Arai, K. Suzuki, S. Sugizaki, H. Sorimachi and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 1770–1797 CrossRef CAS PubMed; (e) N. Arai, K. Azuma, N. Nii and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 7457–7460 CrossRef CAS PubMed; (f) J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538–4539 CrossRef CAS PubMed; (g) Q.-Q. Zhang, J.-H. Xie, X.-H. Yang, J.-B. Xie and Q.-L. Zhou, Org. Lett., 2012, 14, 6158–6161 CrossRef CAS PubMed; (h) X.-N. Chen, H. Zhou, K.-Y. Zhang, J.-W. Li and H.-M. Huang, Org. Lett., 2014, 16, 3912–3915 CrossRef CAS PubMed; (i) X.-D. Zou, S.-M. Gou, R. Yang, J.-H. Xie and Q.-L. Zhou, Chem. Sci., 2017, 8, 6202–6206 RSC; (j) Y. Wang, G. Yang, F. Xie and W. Zhang, Org. Lett., 2018, 20, 6135–6139 CrossRef CAS PubMed; (k) J. Li, Y. Lu, Y. Zhu, Y. Nie, J. Shen, Y. Liu, D. Liu and W. Zhang, Org. Lett., 2019, 21, 4331–4335 CrossRef CAS PubMed; (l) J. Li, Y. Zhu, Y. Lu, Y. Wang, Y. Liu, D. Liu and W. Zhang, Organometallics, 2019, 38, 3970–3978 CrossRef CAS.
- For selected recent reviews on catalytic asymmetric aldol reactions, see: (a) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC; (b) Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH Verlag GmbH & KGaA, 2013 Search PubMed; (c) Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC; (d) C. C. Meyer, E. Ortiz and M. J. Krische, Chem. Rev., 2020, 120, 3721–3748 CrossRef CAS PubMed . For selected recent papers, see:; (e) S. Denmark and S. K. Ghosh, Angew. Chem., Int. Ed., 2001, 40, 4759–4762 CrossRef CAS; (f) Y. Yamashita, H. Ishitani, H. Shimizu and S. Kobayashi, J. Am. Chem. Soc., 2002, 124, 3292–3302 CrossRef CAS PubMed; (g) S. E. Denmark, T. Wynn and G. L. Beutner, J. Am. Chem. Soc., 2002, 124, 13405–13407 CrossRef CAS PubMed; (h) D. A. Evans, C. Wade Downey and J. L. Hubbs, J. Am. Chem. Soc., 2003, 125, 8706–8707 CrossRef CAS PubMed; (i) H. M. L. Davies, R. E. J. Beckwith, E. G. Antoulinakis and Q. Jin, J. Org. Chem., 2003, 68, 6126–6132 CrossRef CAS PubMed; (j) A. E. Russell, N. O. Fuller, S. J. Taylor, P. Aurriset and J. P. Morken, Org. Lett., 2004, 6, 2309–2312 CrossRef CAS PubMed; (k) S. Denmark, G. Beutner, T. Wynn and M. D. Eastgate, J. Am. Chem. Soc., 2005, 127, 3774–3789 CrossRef CAS PubMed; (l) H. Nishiyama, T. Shiomi, Y. Tsuchiya and I. Matsuda, J. Am. Chem. Soc., 2005, 127, 6972–6973 CrossRef CAS PubMed; (m) Y. Mei, P. Dissanayake and M. J. Allen, J. Am. Chem. Soc., 2010, 132, 12871–12873 CrossRef CAS PubMed; (n) M. Sugiura, N. Sato, Y. Sonoda, S. Kotani and M. Nakajima, Chem.–Asian J., 2010, 5, 478–481 CrossRef CAS PubMed; (o) S. Rossi, M. Benaglia, F. Cozzi, A. Genoni and T. Benincori, Adv. Synth. Catal., 2011, 353, 848–854 CrossRef CAS; (p) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 11940–11943 CrossRef CAS PubMed; (q) T. Kitanosono, T. Ollevier and S. Kobayashi, Chem.–Asian J., 2013, 8, 3051–3062 CrossRef CAS PubMed; (r) L. Lin, K. Yamamoto, H. Mitsunuma, Y. Kanzaki, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2015, 137, 15418–15421 CrossRef CAS PubMed; (s) T. Kano, H. Maruyama, R. Sakamoto and K. Maruoka, Chem. Commun., 2015, 51, 10062–10065 RSC; (t) Y. Hayashi, K. Nagai and S. Umemiya, Chem.–Asian J., 2019, 14, 4146–4149 CrossRef CAS PubMed.
- (a) D. Cartigny, K. Püntener, T. Ayad, M. Scalone and V. Ratovelomanana-Vidal, Org. Lett., 2010, 12, 3788–3791 CrossRef CAS PubMed; (b) B. Seashore-Ludlow, F. Saint-Dizier and P. Somfai, Org. Lett., 2012, 14, 6334–6337 CrossRef CAS PubMed; (c) L. Monnereau, D. Cartigny, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Chem.–Eur. J., 2015, 21, 11799–11806 CrossRef CAS PubMed; (d) L. Yu and P. Somfai, RSC Adv., 2019, 9, 2799–2802 RSC.
- (a) E. A. Reiff, S. K. Nair, B. S. N. Reddy, J. Inagaki, J. T. Henri, J. F. Greiner and G. I. Georg, Tetrahedron Lett., 2004, 45, 5845–5847 CrossRef CAS; (b) X. Ma, W. Li, X. Li, X. Tao, W. Fan, X. Xie, T. Ayad, V. Ratovelomanana-Vidal and Z. Zhang, Chem. Commun., 2012, 48, 5352 RSC.
- For a review, see: (a) J.-H. Xie and Q.-L. Zhou, Aldrichimica Acta, 2015, 48, 33–40 CAS . For recent papers, see:; (b) H. Lin, L.-J. Xiao, M.-J. Zhou, H.-M. Yu, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2016, 18, 1434–1437 CrossRef CAS PubMed; (c) Y. Liu, L.-J. Cheng, H.-T. Yue, W. Che, J.-H. Xie and Q.-L. Zhou, Chem. Sci., 2016, 7, 4725–4729 RSC; (d) X.-D. Zuo, S.-M. Guo, R. Yang, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2017, 19, 5240–5243 CrossRef CAS PubMed; (e) Y.-T. Liu, L.-P. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2017, 56, 12708–12711 CrossRef CAS PubMed and ref. 2j.
- (a) H. Knocke, G. Ourisson, G. W. Perold, J. Foussereau and J. Maleville, Science, 1969, 166, 239–240 CrossRef PubMed; (b) G. Xu, A. Hou, Y. Zheng, Y. Zhao, X. Li, L. Peng and Q. Zhao, Org. Lett., 2007, 9, 291–293 CrossRef CAS PubMed; (c) W. Xiang, Z. Na, S.-H. Li, M.-L. Li, R.-T. Li, Q.-E. Tian and H.-D. Sun, Planta Med., 2003, 69, 1031–1305 CrossRef CAS PubMed; (d) W. Schmidt, T. M. Schulze, G. Brasse, E. Nagraodzka, M. Maczka, J. Zettel, P. G. Jones, J. Grunenberg, M. Hilker, U. Trauer-Kizilelma, U. Braun and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 7698–7702 CrossRef CAS PubMed; (e) B. Jiang, A.-J. Hou, M.-L. Li, S.-H. Li, Q.-B. Han, S.-J. Wang, Z.-W. Lin and H.-D. Sun, Planta Med., 2002, 68, 921–925 CrossRef CAS PubMed; (f) H. Watanabe and M. Nakada, J. Am. Chem. Soc., 2008, 130, 1150–1151 CrossRef CAS PubMed; (g) M. Binder and C. Tamm, Angew. Chem., Int. Ed., 1973, 12, 370–380 CrossRef CAS PubMed; (h) J. Hohmann, D. Rédei, F. Evanics, A. Kálmán, G. Argay and T. Bartók, Tetrahedron, 2000, 56, 3619–3623 CrossRef CAS.
- (a) J.-H. Xie, X.-Y. Liu, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7329–7332 CrossRef CAS PubMed; (b) J.-H. Xie, X.-Y. Liu, X.-H. Yang, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2012, 51, 201–203 CrossRef CAS PubMed; (c) X.-H. Yang, J.-H. Xie, W.-P. Liu and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 7833–7836 CrossRef CAS PubMed; (d) X.-H. Yang, K. Wang, S.-F. Zhu, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2014, 136, 17426–17429 CrossRef CAS PubMed; (e) X.-H. Yang, H.-T. Yue, N. Yu, Y.-P. Li, J.-H. Xie and Q.-L. Zhou, Chem. Sci., 2017, 8, 1811–1814 RSC; (f) Y.-T. Liu, J.-Q. Chen, L.-P. Li, X.-Y. Shao, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2017, 19, 3231–3234 CrossRef CAS PubMed and ref. 6d.
- H.-Y. Bin, K. Wang, D. Yang, X.-H. Yang, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2019, 58, 1174–1177 CrossRef CAS PubMed.
- During the preparation of this manuscript, Zhou and co-workers demonstrated that Ru-(S,S)-Ts-DPEN was efficient for the asymmetric transfer hydrogenation of ethyl (E)-3-(2-bromobenzylidene)-2-oxocyclopentane-1-carboxylate to a chiral allylic alcohol with two contiguous stereocenters, see: K. Zhang, Q. Liu, R. He, D. Chen, Z. Deng, N. Huang and H. Zhou, Green Chem., 2021, 23, 1628 RSC.
- K. Makino, T. Goto, Y. Hiroki and Y. Hamada, Angew. Chem., Int. Ed., 2004, 43, 882–884 CrossRef CAS PubMed.
- K. Mashima, K.-h. Kusano, N. Sato, Y.-i. Matsumura, K. Nozaki, H. Kumobayashi, N. Sayo, Y. Hori and T. Ishizaki, J. Org. Chem., 1994, 59, 3064–3076 CrossRef CAS.
- J.-H. Xie, L.-X. Wang, Y. Fu, S.-F. Zhu, B.-M. Fan, H.-F. Duan and Q.-L. Zhou, J. Am. Chem. Soc., 2003, 125, 4404–4405 CrossRef CAS PubMed.
- D.-H. Bao, H.-L. Wu, C.-L. Liu, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2015, 54, 8791–8794 CrossRef CAS PubMed.
- The successive hydrogenation of both the CC and CO double bonds of 8aa with (R)-6b produced cis,cis-ethyl 2-hydroxy-3-methylcyclohexane-1-carboxylate in 41% yield with 1.2% ee and >20:1 dr (determined by 1H NMR and HPLC, respectively). For examples of successive hydrogenation of both the CC and CO double bonds with Ir-SpiroPAPs, see ref. 8f..
- (a) K. Abdur-Rashid, M. Faatz, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2001, 123, 7473–7474 CrossRef CAS PubMed; (b) K. Abdur-Rashid, S. E. Clapham, A. Hadzovic, J. N. Harvey, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2002, 124, 15104–15118 CrossRef CAS PubMed; (c) C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS PubMed.
- B. Li, J. Chen, Z. Zhang, I. D. Gridnev and W. Zhang, Angew. Chem., Int. Ed., 2019, 58, 7329–7334 CrossRef CAS PubMed.
- (a) A. Trabocchi, G. Menchi and A. Guarna, in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes, Wiley-VCH, Weinheim, 2010, pp. 527–571 Search PubMed; (b) R. T. Stendall and A. J. A. Cobb, Tetrahedron, 2018, 74, 4917–4925 CrossRef CAS.
- (a) S. Hanessian, Z. Shao, C. Betschart, J.-M. Rondeau, U. Neumann and M. Tintelnot-Blomley, Bioorg. Med. Chem. Lett., 2010, 20, 1924–1927 CrossRef CAS PubMed; (b) S. Hanessian, D. K. Maji, S. Govindan, R. Matera and M. Tintelnot-Blomley, J. Org. Chem., 2010, 75, 2861–2876 CrossRef CAS PubMed.
- Y. Ichikawa, Synlett, 2007, 2927–2936 CrossRef CAS.
- P. Szcześniak, M. Pieczykolan and S. Stecko, J. Org. Chem., 2016, 81, 1057–1074 CrossRef PubMed.
- (a) Y. Ichikawa and Y. Igarashi, Tetrahedron Lett., 1995, 36, 4585–4586 CrossRef CAS; (b) Y. Ichikawa, Y. Igarashi, M. Ichikawa and Y. Suhara, J. Am. Chem. Soc., 1998, 120, 3007–3018 CrossRef CAS.
- (a) Y. Mihara, H. Ojima, T. Imahori, Y. Yoshimura, H. Ouchi and H. Takahata, Heterocycles, 2007, 72, 633–645 CrossRef CAS; (b) P. Spanu, C. de Candia and F. Ulgheri, Tetrahedron Lett., 2010, 51, 2400–2402 CrossRef CAS; (c) Y. S. Reddy, P. K. Kancharla, R. Roy and Y. D. Vankar, Org. Biomol. Chem., 2012, 10, 2760–2773 RSC; (d) A. Biela-Banaś, F. Oulaïdi, S. Front, E. Gallienne, K. Ikeda-Obatake, N. Asano, D. A. Wenger and O. R. Martin, ChemMedChem, 2014, 9, 2647–2652 CrossRef PubMed; (e) C. H. Hill, A. H. Viuff, S. J. Spratley, S. Salamone, S. H. Christensen, R. J. Read, N. W. Moriarty, H. H. Jensen and J. E. Deane, Chem. Sci., 2015, 6, 3075–3086 RSC.
- X. Liang, A. Lohse and M. Bols, J. Org. Chem., 2000, 65, 7432–7437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR, HPLC spectra, XRD diffraction analysis of compounds 8v and 12f, and computational studies. CCDC [1936853, 2054732] and Cartesian coordinates of the DFT-optimised geometries. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02044g |
| This journal is © The Royal Society of Chemistry 2021 |