
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the synthesis of value-added aromatics directly from syngas via a Cr2O3 and Ga doped zeolite capsule catalyst†
Yang
Wang
ab,
Weizhe
Gao
b,
Kangzhou
Wang
b,
Xinhua
Gao
 c,
Baizhang
Zhang
b,
Heng
Zhao
b,
Qingxiang
Ma
c,
Peipei
Zhang
b,
Guohui
Yang
bd,
Mingbo
Wu
*a and
Noritatsu
Tsubaki
*b
c,
Baizhang
Zhang
b,
Heng
Zhao
b,
Qingxiang
Ma
c,
Peipei
Zhang
b,
Guohui
Yang
bd,
Mingbo
Wu
*a and
Noritatsu
Tsubaki
*b
aCollege of New Energy, State Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao 266580, China. E-mail: wumb@upc.edu.cn
bDepartment of Applied Chemistry, Graduate School of Engineering, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan. E-mail: tsubaki@eng.u-toyama.ac.jp
cState Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, College of Chemistry & Chemical Engineering, Ningxia University, Yinchuan 750021, China
dState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
First published on 26th April 2021
Abstract
Even though the transformation of syngas into aromatics has been realized via a methanol-mediated tandem process, the low product yield is still the bottleneck, limiting the industrial application of this technology. Herein, a tailor-made zeolite capsule catalyst with Ga doping and SiO2 coating was combined with the methanol synthesis catalyst Cr2O3 to boost the synthesis of value-added aromatics, especially para-xylene, from syngas. Multiple characterization studies, control experiments, and density functional theory (DFT) calculation results clarified that Ga doped zeolites with strong CO adsorption capability facilitated the transformation of the reaction intermediate methanol by optimizing the first C–C coupling step under a high-pressure CO atmosphere, thereby driving the reaction forward for aromatics synthesis. This work not only reveals the synergistic catalytic network in the tandem process but also sheds new light on principles for the rational design of a catalyst in terms of oriented conversion of syngas.
Introduction
Syngas (CO + H2) conversion, as an alternative strategy for value-added chemicals synthesis in the petroleum industry, has attracted more and more attention due to the wide range of syngas sources such as biomass, combustible ice, shale gas, etc.1–3 Aromatics, as the key building blocks in the polymer industry, are mainly produced from the petroleum refinement process.4 With the diminishing of crude oil and the fluctuations in the oil market, the exploration of a novel pathway for aromatics synthesis, especially for light aromatics (BTX, B = benzene, T = toluene, and X = xylene) production, is highly needed. The direct conversion of syngas into aromatics is a hot topic in the field of C1 chemistry, but still presents a challenge due to the low selectivity or yield of targeted chemicals.5–8The Fischer–Tropsch synthesis (FTs) technology is well known for the transformation of syngas into valuable commodity chemicals, but the wide product distribution obeying the Anderson–Schulz–Flory (ASF) law restricts the oriented conversion of syngas. Recently, Ma et al. upgraded the traditional FTs method by combining of an Fe-based FTs catalyst with the acidic zeolite H-ZSM-5 for highly selective aromatics synthesis, during which the alkenes produced from the alkali-modified Fe-based catalyst can be converted to aromatics with the help of shape-selective H-ZSM-5.9 Even though a high yield of aromatics (∼30%) was achieved through a single-pass process, the relatively low aromatics selectivity (∼50%) still needs to be improved. Compared with the upgraded FTs technology, the methanol-mediated pathway for synthesis of aromatics from syngas exhibited high aromatics selectivity (60–80%), which is a promising factor for industrial application.10–12 During the methanol-mediated syngas conversion process, oxygen vacancy-rich metal oxides are responsible for the activation of the CO molecule to produce methanol, then methanol can be converted to aromatics by the acidic H-ZSM-5 with alkenes as the intermediates. Wang et al. and Bao et al. designed Zn–ZrO2/H-ZSM-5 and Zn–CrOx/H-ZSM-5 bifunctional catalysts for the direct conversion of syngas into aromatics, respectively.13,14 However, the drawbacks of the methanol-mediated pathway lie in the low CO conversion and high CO2 selectivity due to the water gas shift reaction (WGS, CO + H2O → CO2 + H2), which lead to the low yield of targeted aromatics. To overcome these shortcomings, Wang et al. co-fed CO2 to suppress the formation of net CO2 by shifting the equilibrium of the WGS reaction that occurred during the tandem process.15 More attention has also been paid to the rational design of a metal oxide catalyst to boost the aromatics synthesis performance by increasing the methanol synthesis activity of the bifunctional catalyst.16,17 A closer look at the tandem reaction network reveals that the physicochemical properties of the zeolite component in the bifunctional catalyst also play a vital role in determining the aromatics synthesis performance, but they have been rarely studied.
In this work, the physicochemical properties of the parent H-ZSM-5 were tailored by metal ion doping (M-ZSM-5, M = Zn, Ga, La, and Mg) and subsequent SiO2 coating (M-ZSM-5@SiO2) to study the effect of the zeolite component on the aromatics synthesis performance. The highest aromatics yield, especially for para-xylene, was achieved via the bifunctional catalyst composed of Cr2O3 and Ga-ZSM-5@SiO2. Understanding of the in-depth mechanism indicated that the strong CO adsorption capacity and the lower C–C coupling barrier derived from Ga doping and the high-pressure CO reaction atmosphere guaranteed the superior aromatics yield. The blocking of the external acid sites of H-ZSM-5 by SiO2 deposition is beneficial to suppress undesirable side reactions, such as alkyl-transfer and isomerization of BTX, therefore boosting light aromatics synthesis.
Results and discussion
Cr2O3, a typical oxygen vacancy-rich metal oxide, was employed as a methanol synthesis component for the bifunctional catalyst. As shown in the transmission electron microscopy (TEM) image (Fig. S1a†), Cr2O3 obtained via the precipitation method was composed of uniform nanoparticles with a diameter of ∼200 nm. The X-ray diffraction (XRD) pattern (Fig. S1b†) of Cr2O3 corresponded well with the eskolaite (PDF #38-1479) that has a hexagonal structure. Oxygen vacancies in the reducible metal oxide have been recognized as active sites to activate the CO molecule during the methanol synthesis process,18,19 which can be characterized by CO2 temperature-programmed desorption (CO2-TPD) and X-ray photoelectron spectroscopy (XPS) techniques. In the high-resolution O 1s XPS spectra (Fig. S1c†), the peak at 531.0 eV was attributed to oxygen atoms adjacent to the oxygen vacancies. The desorption peak at high temperature in the CO2-TPD profile (Fig. S1d†) can be attributed to the desorption of CO2 molecules adsorbed on the oxygen vacancies of Cr2O3. The oxygen vacancy-dominated CO activation pathway on the reducible metal oxide has been clarified by multiple characterization studies and theoretical analysis,20,21 during which CO molecules adsorbed and activated by the oxygen vacancies first combined with the H* species to form formaldehyde (*HCO and *H2CO) and formate (*HCOO) intermediates, and the successive hydrogenation step guaranteed the formation of methoxyl (*CH3O) species and the final product, methanol. It should be noted that the formaldehyde or formate intermediates may directly diffuse into the channels of the acidic zeolite for the C–C coupling and cyclization reactions without undergoing the following hydrogenation step mentioned above when two catalytic components, reducible metal oxide and zeolite, are present in the tandem process.The bifunctional catalyst was fabricated by physically mixing Cr2O3 with the zeolite capsule catalyst H-ZSM-5@SiO2, and its catalytic performance for tandem catalysis of syngas to value-added aromatics was tested at 380 °C and 3 MPa. The topology of H-ZSM-5 (1D straight channels: 0.53 × 0.56 nm, 2D sinusoidal channels: 0.51 × 0.55 nm) indicates that it is difficult for C9+ aromatics with larger molecule diameters to diffuse out of the channels of H-ZSM-5.22,23 It has been widely accepted that heavier aromatics are mainly produced on the external acid sites of H-ZSM-5, where there is a lack of confinement effect for shape-selective synthesis. The external acid sites where undesirable side reactions such as isomerization and alkyl-transfer of BTX usually occurred were blocked by the SiO2 shell. Furthermore, the pore mouth size of H-ZSM-5 optimized by SiO2 permeation is beneficial to sieve para-xylene from xylene isomers (ortho-xylene and meta-xylene) due to its smaller dynamic diameter and diffusion resistance, therefore enhancing its selectivity in the aromatics synthesis reaction catalysed by the H-ZSM-5 zeolite.24 The effect of the thickness of the SiO2 shell on the distribution of aromatics was investigated for the first time. The thickness of the SiO2 shell on H-ZSM-5 can be controlled by varying the dosage of the Si source and the number of deposition times. The zeolite capsule catalyst H-ZSM-5 with a SiO2 deposition of 13.8 wt% (H-ZSM-5@SiO2-13.8%) on its external surface exhibited lower BTX selectivity (34.5%, Table S1†) due to the incomplete suppression of the external acid sites. High selectivity of BTX (44.6%) was achieved with the bifunctional catalyst Cr2O3/H-ZSM-5@SiO2-39.0%, in which the selectivity of para-xylene accounted for 33.0%. However, further increasing the SiO2 thickness had no obvious effect on the selectivity of BTX, but the CO conversion (18.7%) was decreased due to the weakened driving force caused by the decrease of acid site density per unit mass of the zeolite capsule catalyst. Therefore, a SiO2 deposition of 39.0 wt% is appropriate to suppress the undesirable isomerization and alkyl-transfer reaction of BTX. The texture properties of the zeolite capsule catalyst H-ZSM-5@SiO2 evolved obviously with the increasing SiO2 coating amount. As shown in Fig. S2,† the average particle sizes measured along the b-axis and c-axis respectively were slightly increased from ∼0.35 and ∼0.89 μm of the parent H-ZSM-5 zeolite to ∼0.38 and ∼0.92 μm of the zeolite capsule catalyst H-ZSM-5@SiO2-39.0% due to the deposition of SiO2 on its external surface. The decrease of the pore size and volume verified the permeation of the SiO2 species into the pore mouths of H-ZSM-5, which is beneficial to boost the synthesis of para-xylene with less diffusion resistance as mentioned above (Table S2†). Furthermore, increased SiO2 deposition led to the decrease of the specific surface area of the zeolite capsule catalyst due to the blockage of the zeolite channels by SiO2 and the aggregation of the zeolite particles as confirmed from the SEM images (Fig. S3†). Even though high BTX selectivity was achieved via Cr2O3 and zeolite capsule catalyst H-ZSM-5@SiO2, the yield of BTX remains to be improved.
During the tandem reaction, the consumption rate of methanol on the zeolite component is a vital factor affecting the yield of the product. Therefore, our attention has been turned to the modification of the zeolite by metal doping to accelerate the methanol conversion process, which has been rarely investigated in the tandem syngas conversion process. Zn, Ga, La, or Mg was doped into the parent H-ZSM-5 zeolite via an ion-exchange method, and then SiO2 of 39.0 wt% was coated onto the surface of the M-ZSM-5 crystals (M = Zn, Ga, La, and Mg) to boost the synthesis of more valuable aromatics such as BTX by blocking undesirable external acid sites. After combining with Cr2O3, Ga and Zn doped capsule catalysts exhibited enhanced aromatics yield, but La and Mg doping lowered the yield of aromatics (Fig. 1a, b and Table S3†). Furthermore, the enhanced aromatics yield from the Zn or Ga doped catalyst was mainly derived from the significant increase of CO conversion, especially for the Ga doped catalyst, with CO conversion and aromatics space time yield (STY) increasing from 20.8% and 26.3 gC h−1 kgcat−1 to 28.4% and 34.6 gC h−1 kgcat−1, respectively. Therefore, doping of zeolite crystals with Ga provides an efficient strategy to enhance the aromatics yield of the single-pass syngas conversion process, which is higher than most of the results reported in other literature (Fig. 1c and Table S4†).13–15,25–29 The following work will focus on discovering the mechanism of the enhanced aromatics yield by Ga doping, which is significant to guide the rational design of efficient catalysts for oriented conversion of syngas to value-added aromatics.
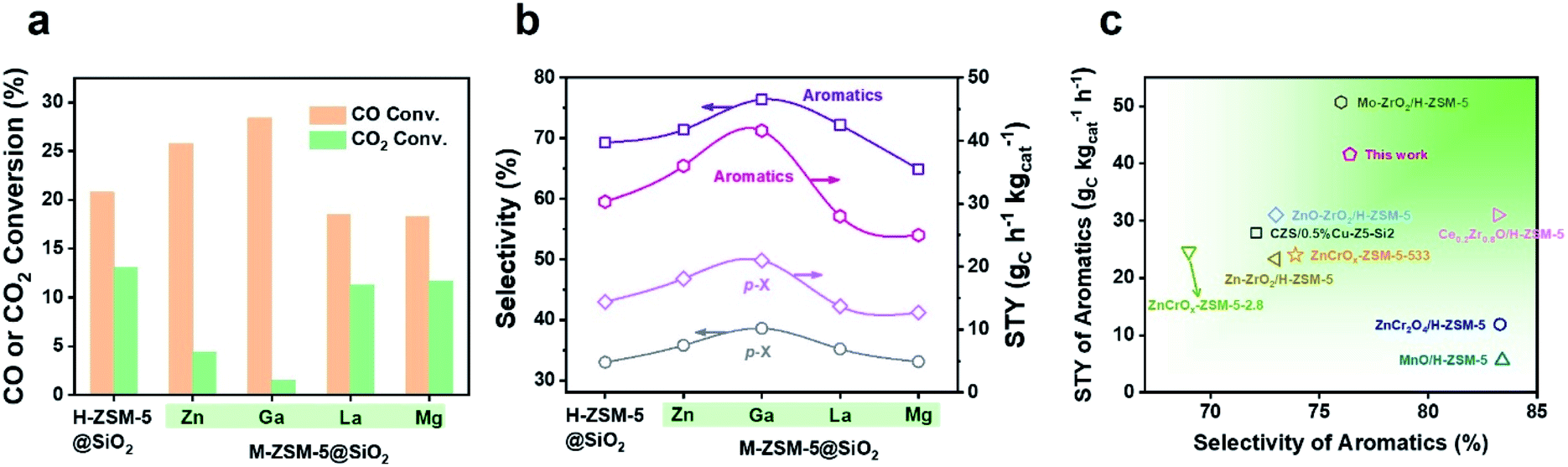 | ||
| Fig. 1 Catalytic performances of bifunctional catalysts composed of Cr2O3 and H-ZSM-5@SiO2 or M-ZSM-5@SiO2: CO or CO2 conversion (a), STY and selectivity of para-xylene and aromatics (b). Reaction conditions: 380 °C, 3 MPa, (Ar 3%, CO 29.6%, CO2 4.87%, H2 balance), GHSV = 1200 mL gcat−1 h−1, time on stream (TOS) = 8 h, metal oxide/zeolite mass ratio = 1, 0.5 g catalyst. (c) Comparisons of the STY and selectivity of aromatics obtained from Cr2O3/Ga-ZSM-5@SiO2 with those from bifunctional catalysts reported in other literature. | ||
The physicochemical properties of the Ga doped zeolite were characterized by multiple techniques. Some island-like defects were detected in the bare Ga-ZSM-5 zeolite as shown in the transmission/scanning electron microscopy images (TEM/SEM/STEM, Fig. 2 and S4†), which can be repaired by SiO2 deposition, providing a smooth external surface for Ga-ZSM-5@SiO2. Many Ga-based particles were detected in the TEM images of Ga-ZSM-5 and Ga-ZSM-5@SiO2 (Fig. S5 and S6†). This phenomenon can be attributed to the fact that Ga was predominantly present in the hydrated form and only a small fraction of Ga3+ species were contained in the aqueous solution during the ion-exchange process. The larger Ga hydrated species cannot diffuse into the channels of H-ZSM-5, which resulted in the deposition of Ga2O3 on the external surface of the zeolite after calcination.30,31 Even though Ga-based particles were formed on the external surface of zeolitic crystals, the homogeneous distribution of Ga as shown in the elemental mapping image (Fig. 2) confirmed that some nanosized Ga species have been anchored onto the surface of the zeolite or even exchanged into the framework of the zeolite. No typical metal crystal phases were detected in the XRD patterns (Fig. S7a†), which is mainly due to the presence of Ga species in the form of exchanged Ga cations (GaO+), amorphous oxides, or small particles in Ga-ZSM-5 and Ga-ZSM-5@SiO2. Furthermore, SiO2 deposition has no obvious effect on the crystal structure of Ga-ZSM-5@SiO2 as confirmed by the strong typical diffraction peak of the XRD pattern (Fig. S7b†). The binding energy at 19.4 eV as characterized by X-ray photoelectron spectroscopy (XPS) can be attributed to the Ga–O species in the Ga doped zeolites (Fig. S8†).32 The intensity of the Ga 3d XPS signal was decreased after SiO2 deposition, with the surface Ga content decreasing from 6.6 to 5.3 atomic%. The Ga content in Ga-ZSM-5 and Ga-ZSM-5@SiO2 as determined by inductive coupled plasma-atomic emission spectroscopy (ICP-AES) was 5.5 and 4.6 wt%, respectively.
 | ||
| Fig. 2 TEM images of Ga-ZSM-5 (a and b) and Ga-ZSM-5@SiO2 (c and d), STEM images and the corresponding Si, Al, and Ga elemental mapping images of Ga-ZSM-5 (e–h) and Ga-ZSM-5@SiO2 (i–l). | ||
The variations of the acidic property after Ga doping into the H-ZSM-5 were characterized by pyridine-adsorbed Fourier transform infrared (pyridine-FTIR) spectroscopy. The number of Lewis acid sites (L, 1455 cm−1) increased at the expense of Brønsted acid sites (B, 1545 cm−1) with the amount ratio of L/B increasing from 1.02 to 1.26 after Ga doping (Fig. S9†). This phenomenon can be attributed to the substitution of hydroxyl protons in Si–(OH)–Al groups by = cationic Ga species.33,34 Therefore, in addition to the extra-zeolitic Ga-based oxide, cationic Ga species GaO+ could also be formed in the framework of H-ZSM-5 by the ion-exchange strategy, which has been proven to be catalytically active.32,35 The number of B and L acid sites in Ga-ZSM-5@SiO2 decreases simultaneously due to the suppression of the external acid sites by SiO2 deposition. In addition to the variation in the type of acid site, Ga doping also alters the acidity strength of H-ZSM-5 with the NH3 desorption peak derived from the strong acid site shifting to a higher temperature (Fig. S10†). The synergistic effect generated by the spatial interaction/proximity between Brønsted acid sites and Ga species has been proven to be responsible for the enhanced Brønsted acidity.36 Di-tert-butyl-pyridine (DTBPy), a larger molecule that cannot diffuse into the internal acid sites of the zeolites was employed as a probe molecule to monitor the variations of the external acid sites in Ga-ZSM-5 and Ga-ZSM-5@SiO2 (Fig. S11†). The signal intensity at ∼1630 cm−1 derived from the interactions between H+ protons and DTBPy molecules was decreased after SiO2 deposition due to the blocking of the external acid sites, which is beneficial to suppress the undesirable alkyl-transfer and isomerization reactions of BTX. As shown in the N2 adsorption–desorption isotherm (Fig. S12†), the parent H-ZSM-5 exhibited microporous properties with sharp N2 uptake at a low relative pressure (P/P0), as well as no obvious hysteresis loop. The specific surface area of Ga-ZSM-5 was decreased compared with that of H-ZSM-5 due to the occupation of the cavities by the metal species (Table S2†). The specific surface area of the SiO2 coated catalysts (H/Ga-ZSM-5@SiO2) was further decreased with the decreasing pore size and volume, this phenomenon can be attributed to the penetration of SiO2 into the pore mouths of H/Ga-ZSM-5.
As mentioned above, Ga species in the zeolite exist in the form of Ga-based oxide and framework cationic Ga. In order to figure out the exact factors that promote the CO conversion and the synthesis of valuable aromatics, we performed the tandem syngas conversion on the bifunctional catalyst Cr2O3/Ga2O3/H-ZSM-5@SiO2 prepared by physically mixing the methanol synthesis component Cr2O3 with Ga2O3, and H-ZSM-5@SiO2 (Table S5†). However, the reaction over Cr2O3/Ga2O3/H-ZSM-5@SiO2 exhibited no obvious enhancement in CO conversion (16.2%) and aromatics yield (23.7 gC kgcat−1 h−1) compared with that over the Cr2O3/H-ZSM-5@SiO2 catalyst. Therefore, the framework cationic Ga species seemed to be more responsible for the enhancement of the catalytic performance of the bifunctional catalyst.
Recently, Chen et al. reported that high-pressure CO participates in the methanol conversion process via the carbonylation reaction on the H-ZSM-5 catalyst.37 Combined with the variation of CO conversion after Ga doping, we speculated that the doping Ga species, especially the framework cationic Ga species, will affect the reaction network by changing the capability of CO to be inserted into the intermediates during the methanol to aromatics (MTA) conversion process. In order to verify this hypothesis, MTA conversion catalyzed by H/Ga-ZSM-5@SiO2 (Fig. 3a and Table S6†) was carried out under a CO atmosphere at 2 MPa and 380 °C. For the H-ZSM-5@SiO2 catalyst, CO was produced from the MTA reaction with negative CO conversion (−1.8%) under such a CO-rich atmosphere. Interestingly, higher aromatics selectivity (81.3%) with positive CO conversion (2.1%) was achieved over the Ga-ZSM-5@SiO2 catalyst, which verified that much more CO was inserted into the reaction intermediates for aromatics synthesis during the MTA process. The variation of CO conversion after Ga doping is consistent with the CO-TPD profiles (Fig. 3b). The stronger CO adsorption ability of Ga-ZSM-5@SiO2, as confirmed by the higher CO desorption temperature (∼400 °C), is beneficial to increase the coverage of CO on the active sites, thus increasing the probability of CO being inserted into the intermediates of the methanol conversion process. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed to determine the reaction intermediates formed during the CO-assisted MTA process (Fig. 3c). The infrared (IR) peak at 2830 cm−1 corresponding to the CH3O* species was detected due to the continuous introduction of methanol via CO flow.38,39 In addition to the IR peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration (1640 cm−1) and aromatics skeleton (1510 and 1550 cm−1),40,41 the appearance of an IR peak for unsaturated carbonyl (1690 cm−1) as the reaction progressed confirmed that carbonyl group-containing species are the key intermediates in the methanol conversion process, which has been proven by spectroscopic and theoretical evidence.42–46 The catalytic performances of the CO-assisted MTA process catalyzed by M-ZSM-5@SiO2 catalysts (M = Zn, La, and Mg) were also tested (Fig. 3a and Table S6†). Both La-ZSM-5@SiO2 and Mg-ZSM-5@SiO2 exhibited high aromatics selectivity (>70%) with negative CO conversion under high CO pressure. Even though positive CO conversion (0.2%) was achieved over the Zn-ZSM-5@SiO2 catalyst, the CO conversion is still lower than that of Ga-ZSM-5@SiO2, which confirmed that Ga doping is more favorable than Zn for the insertion of CO into the reaction intermediates. The similar CO conversion variation trend between the tandem syngas conversion process and the CO-assisted MTA process (Fig. 1a and 3a) verified that the CO conversion achieved in the tandem syngas conversion process can be affected by the CO insertion ability of the metal doped zeolite.
C stretching vibration (1640 cm−1) and aromatics skeleton (1510 and 1550 cm−1),40,41 the appearance of an IR peak for unsaturated carbonyl (1690 cm−1) as the reaction progressed confirmed that carbonyl group-containing species are the key intermediates in the methanol conversion process, which has been proven by spectroscopic and theoretical evidence.42–46 The catalytic performances of the CO-assisted MTA process catalyzed by M-ZSM-5@SiO2 catalysts (M = Zn, La, and Mg) were also tested (Fig. 3a and Table S6†). Both La-ZSM-5@SiO2 and Mg-ZSM-5@SiO2 exhibited high aromatics selectivity (>70%) with negative CO conversion under high CO pressure. Even though positive CO conversion (0.2%) was achieved over the Zn-ZSM-5@SiO2 catalyst, the CO conversion is still lower than that of Ga-ZSM-5@SiO2, which confirmed that Ga doping is more favorable than Zn for the insertion of CO into the reaction intermediates. The similar CO conversion variation trend between the tandem syngas conversion process and the CO-assisted MTA process (Fig. 1a and 3a) verified that the CO conversion achieved in the tandem syngas conversion process can be affected by the CO insertion ability of the metal doped zeolite.
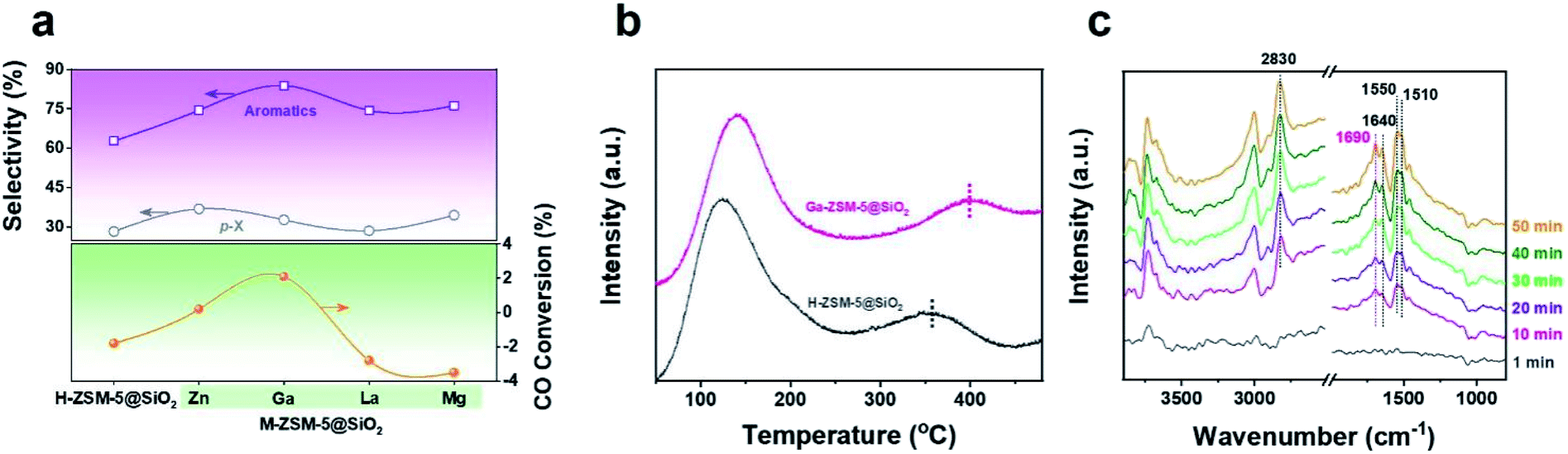 | ||
| Fig. 3 (a) CO conversion and selectivity of para-xylene and aromatics of the CO-assisted MTA process catalyzed by H-ZSM-5@SiO2 or M-ZSM-5@SiO2 (M = Zn, Ga, La, and Mg). Reaction conditions: 380 °C, 2 MPa, liquid methanol flow rate 0.005 mL min−1, gas flow (CO 90%, Ar balance) rate 15 mL min−1, TOS = 8 h, 1.0 g zeolite catalyst. (b) CO-TPD profiles of H-ZSM-5@SiO2 and Ga-ZSM-5@SiO2. (c) In situ DRIFTS of the CO-assisted MTA process on the Ga-ZSM-5@SiO2 catalyst. | ||
The conversion of methanol on the acidic sites of H-ZSM-5 was initiated by the first C–C bond formation through coupling between nucleophilic and electrophilic carbon atoms. Even though numerous direct C–C bond formation mechanisms, such as carbene-insertion,47 the methoxymethyl mechanism,48 and the methyleneoxy mechanism,49 have been proposed with the help of advanced characterization techniques, the Koch carbonylation mechanism seemed to be more feasible for the C–C bond formation during the CO-assisted MTA and the tandem syngas conversion processes due to the CO-rich atmosphere. As shown in Fig. 4a, methanol is first dehydrated to methoxy on the Brønsted acid sites.50,51 Then the carbonylation of the methoxy group guarantees the formation of acetyl group C–C bonds, which are subsequently converted into methyl acetate, acetic acid, or even acetaldehyde. Finally, the C–C bond containing intermediates undergo a series of acid-catalyzed reactions including decarboxylation, aldol condensation, and cracking for hydrocarbons synthesis.52 The optimized carbonylation process for the C–C bond formation triggers the conversion of methanol into hydrocarbons.46 Herein, the energy barrier of the carbonylation reaction for the first C–C bond formation was analyzed by density functional theory (DFT) calculations (Fig. 4b). The active site structures of H-ZSM-5 and Ga-ZSM-5 employed in the DFT calculations are illustrated in Fig. S13† and the reaction schemes of the first C–C bond formation on H-ZSM-5 and Ga-ZSM-5 under a CO-free or CO-rich atmosphere are compared in Fig. 4c and S14.† An energy barrier of 136 kJ mol−1 was obtained over the H-ZSM-5 catalyst under a CO-free atmosphere. With the CO coverage increasing, the energy barrier was decreased to 113 kJ mol−1. Therefore, high CO pressure during the tandem syngas conversion process was favorable for the methanol conversion process by facilitating the formation of the first C–C bond. Interestingly, the cationic Ga species in the zeolite framework with strong CO adsorption capacity as confirmed by the CO-TPD and control experiments further lowered the carbonylation energy barrier to 49 kJ mol−1, which implied that the enhanced CO coverage on the active sites induced by Ga doping further facilitated the methanol conversion process by optimizing the carbonylation step for C–C bond formation. Positive CO conversion in the CO-assisted MTA process, the enhanced CO conversion and the boosted aromatics yield in the tandem syngas conversion are consistent with the DFT calculation results.
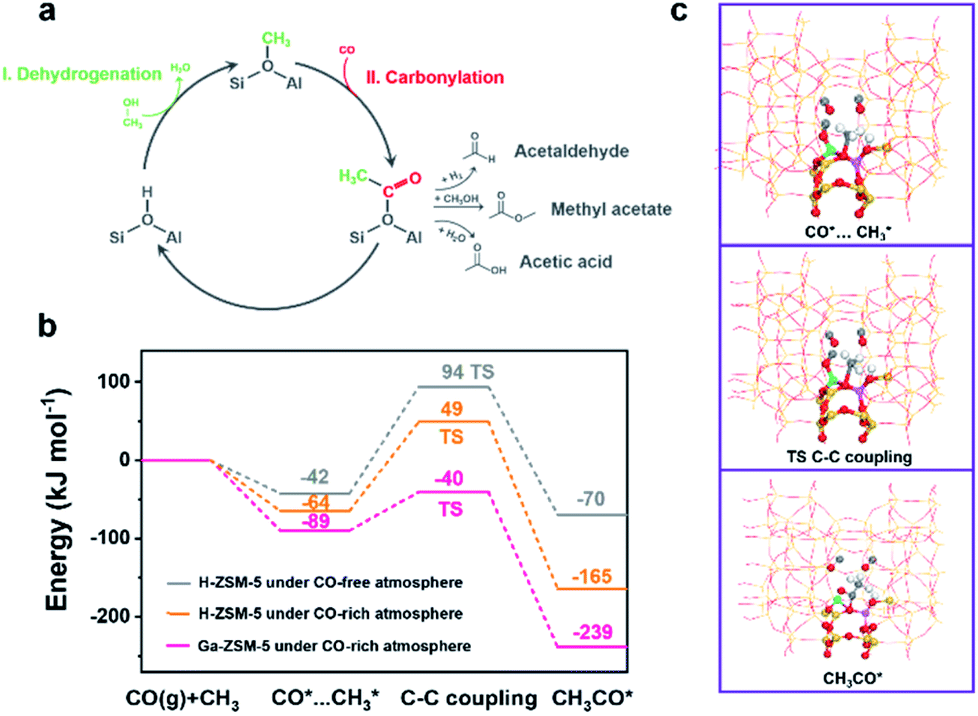 | ||
| Fig. 4 (a) Koch carbonylation mechanism for the first C–C bond formation in the MTA reaction. (b) Free energy diagrams of the first C–C bond formation on H-ZSM-5 under a CO-free or CO-rich atmosphere and Ga-ZSM-5 under a CO-rich atmosphere. (c) The reaction scheme of the first C–C bond formation on Ga-ZSM-5 under a CO atmosphere (yellow: Si, red: O, pink: Al, white: H, grey: C). | ||
Based on the characterization data, control experiments, and DFT calculation results, the mechanism of the bifunctional catalyst Cr2O3/Ga-ZSM-5@SiO2 to enhance the aromatics yield was established. The oxygen vacancies in Cr2O3 is responsible for the activation of syngas into methanol, which can be adsorbed and transformed into aromatics on the acidic sites of the H-ZSM-5 zeolite. The formation of the first C–C bond, as the key step for aromatics synthesis from methanol, can be optimized under a CO-rich atmosphere. On the other hand, the cationic Ga species in the framework of H-ZSM-5 with strong CO adsorption capability facilitates the process of aromatics synthesis from methanol. The accelerated methanol consumption rate driven by the Ga cationic species in the Ga-ZSM-5@SiO2 component and the CO-rich atmosphere pushes the tandem reaction forward, therefore enhancing the aromatics yield of the tandem syngas conversion process. Furthermore, the enhanced aromatics selectivity derived from the strong dehydrogenation capability of the Ga species and the synergistic effect of the Brønsted and Lewis acid sites is also an important factor to increase the yield of aromatics in the tandem process. In earlier studies, Ga doping has been widely employed to increase the aromatics selectivity of the conversion of methanol, alkanes, biomass, etc.31,33,53 due to the strong dehydrogenation capability of Ga. Recently, Deng et al. discovered that a dehydrogenation–aromatization route mediated by the cooperation of cationic Ga species and Brønsted acid sites could enhance the aromatics synthesis performance of the Ga/ZSM-5 zeolite.54 There are two H-subtraction pathways for the aromatics synthesis process; H-transfer from H-donors to H-acceptors and the dehydrogenation process with H2 release. The dominating H-transfer process on Brønsted acid sites results in the consumption of the aromatics synthesis intermediates of alkenes or cycloalkenes, therefore lowering the aromatics selectivity. However, the cooperative effect between the Ga species and the Brønsted acid sites alters the H species subtraction pathway to a dehydrogenation-dominant process that is favorable for aromatics synthesis. In the dehydrogenation-dominant pathway, the Lewis acid sites (GaO+) first subtract H− species from the intermediates. Then the H− species combine with the protons in the Brønsted acid sites for H2 production. Finally, the deprotonation of carbenium ions guarantee the recovery of the Brønsted acid sites and the formation of unsaturated intermediates or aromatics. Based on the above discussion, the enhanced CO adsorption and dehydrogenation capabilities induced by Ga doping are both beneficial to drive the conversion of syngas to aromatics forward.
Conclusions
A highly efficient bifunctional catalyst composed of a Cr2O3 and Ga doped H-ZSM-5 zeolite capsule catalyst has been developed for the direct conversion of syngas into valuable aromatics, especially para-xylene. With the help of multiple characterization studies, control experiments, and DFT calculations, the driving force during the tandem process for enhanced aromatics synthesis from syngas has been successfully clarified. The optimized C–C coupling step induced by the cationic Ga species under a high CO pressure atmosphere could accelerate the consumption of the reaction intermediate, methanol, thereby triggering the tandem reaction forward for aromatics synthesis. The results presented herein can provide insights into the driving force for the tandem conversion of syngas into aromatics, which will guide the rational design of bifunctional catalysts for targeted syngas conversion.Author contributions
Y. Wang, W. Gao, K. Wang and B. Zhang performed most of the experiments and analyzed the experimental data. X. Gao, H. Zhao and Q. Ma performed the DRIFTS characterization studies. P. Zhang and M. Wu performed the DFT calculations. Y. Wang wrote the original manuscript. G. Yang and N. Tsubaki designed the study, analyzed the data, and polished the manuscript. N. Tsubaki and M. Wu supervised the whole project. All the authors discussed the results and commented on the manuscript at all stages.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the JST-MIRAI project (JPMJMI17E2) of the Japan Science and Technology Agency, China Postdoctoral Science Foundation (No. 2020M672169), CNPC Innovation Foundation (2020D-5007-0407), and the Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (2020-KF-34).Notes and references
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- K. H. Choi, J. H. Lee, E. N. Shafei and M. Al-Faqeer, US Pat., 9505678B2, 2016 Search PubMed.
- X. Yang, X. Su, D. Chen, T. Zhang and Y. Huang, Chin. J. Catal., 2020, 41, 561–573 CrossRef CAS.
- S. Kasipandi and J. W. Bae, Adv. Mater., 2019, 31, 1803390 CrossRef PubMed.
- Y. Xu, J. Liu, J. Wang, G. Ma, J. Lin, Y. Yang, Y. Li, C. Zhang and M. Ding, ACS Catal., 2019, 9, 5147–5156 CrossRef CAS.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- B. Zhao, P. Zhai, P. Wang, J. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang, Y. W. Li, Q. Zhang, W. Fan and D. Ma, Chem, 2017, 3, 323–333 CAS.
- D. Miao, Y. Ding, T. Yu, J. Li, X. Pan and X. Bao, ACS Catal., 2020, 10, 7389–7397 CrossRef CAS.
- Y. Fu, Y. Ni, W. Zhu and Z. Liu, J. Catal., 2020, 383, 97–102 CrossRef CAS.
- P. Zhang, L. Tan, G. Yang and N. Tsubaki, Chem. Sci., 2017, 8, 7941–7946 RSC.
- J. Yang, X. Pan, F. Jiao, J. Li and X. Bao, Chem. Commun., 2017, 53, 11146–11149 RSC.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- W. Zhou, C. Zhou, H. Yin, J. Shi, G. Zhang, X. Zheng, X. Min, Z. Zhang, K. Cheng, J. Kang, Q. Zhang and Y. Wang, Chem. Commun., 2020, 56, 5239–5242 RSC.
- W. Zhou, S. Shi, Y. Wang, L. Zhang, Y. Wang, G. Zhang, X. Min, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2019, 11, 1681–1688 CrossRef CAS.
- Y. Wang, W. Zhan, Z. Chen, J. Chen, X. Li and Y. Li, ACS Catal., 2020, 10, 7177–7187 CrossRef CAS.
- A. M. Abdel-Mageed, A. Klyushin, A. Knop-Gericke, R. Schlögl and R. J. Behm, J. Phys. Chem. Lett., 2019, 10, 3645–3653 CrossRef CAS PubMed.
- J. Strunk, K. Kähler, X. Xia, M. Comotti, F. Schüth, T. Reinecke and M. Muhler, Appl. Catal., A, 2009, 359, 121–128 CrossRef CAS.
- S. Wang, P. Wang, D. Shi, S. He, L. Zhang, W. Yan, Z. Qin, J. Li, M. Dong, J. Wang, U. Olsbye and W. Fan, ACS Catal., 2020, 10, 2046–2059 CrossRef CAS.
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Chem. Sci., 2018, 9, 4708–4718 RSC.
- K. Miyake, Y. Hirota, K. Ono, Y. Uchida, S. Tanaka and N. Nishiyama, J. Catal., 2016, 342, 63–66 CrossRef CAS.
- J. Zhang, W. Qian, C. Kong and F. Wei, ACS Catal., 2015, 5, 2982–2988 CrossRef CAS.
- C. Wang, L. Zhang, X. Huang, Y. Zhu, G. Li, Q. Gu, J. Chen, L. Ma, X. Li, Q. He, J. Xu, Q. Sun, C. Song, M. Peng, J. Sun and D. Ma, Nat. Commun., 2019, 10, 4348 CrossRef PubMed.
- Z. Huang, S. Wang, F. Qin, L. Huang, Y. Yue, W. Hua, M. Qiao, H. He, W. Shen and H. Xu, ChemCatChem, 2018, 10, 4519–4524 CrossRef CAS.
- J. Yang, K. Gong, D. Miao, F. Jiao, X. Pan, X. Meng, F. Xiao and X. Bao, J. Energy Chem., 2019, 35, 44–48 CrossRef.
- S. Z. A. Gilani, L. Lu, M. T. Arslan, B. Ali, Q. Wang and F. Wei, Catal. Sci. Technol., 2020, 10, 3366–3375 RSC.
- M. T. Arslan, B. A. Qureshi, S. Z. A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9, 2203–2212 CrossRef CAS.
- S. Wang, Z. Huang, Y. Luo, J. Wang, Y. Fang, W. Hua, Y. Yue, H. Xu and W. Shen, Catal. Sci. Technol., 2020, 10, 6562–6572 RSC.
- A. Biscardi and E. Iglesia, Catal. Today, 1996, 31, 207–231 CrossRef.
- I. Nowak, J. Quartararo, E. G. Derouane and J. C. Védrine, Appl. Catal., A, 2003, 251, 107–120 CrossRef CAS.
- E. A. Uslamin, B. Luna-Murillo, N. Kosinov, P. C. A. Bruijnincx, E. A. Pidko, B. M. Weckhuysen and E. J. M. Hensen, Chem. Eng. Sci., 2019, 198, 305–316 CrossRef CAS.
- Z. Li, A. W. Lepore, M. F. Salazar, G. S. Foo, B. H. Davison, Z. Wu and C. K. Narula, Green Chem., 2017, 19, 4344–4352 RSC.
- W. Dai, L. Yang, C. Wang, X. Wang, G. Wu, N. Guan, U. Obenaus, M. Hunger and L. Li, ACS Catal., 2018, 8, 1352–1362 CrossRef CAS.
- N. Rane, A. R. Overweg, V. B. Kazansky, R. A. van Santen and E. J. M. Hensen, J. Catal., 2006, 239, 478–485 CrossRef CAS.
- P. Gao, Q. Wang, J. Xu, G. Qi, C. Wang, X. Zhou, X. Zhao, N. Feng, X. Liu and F. Deng, ACS Catal., 2018, 8, 69–74 CrossRef CAS.
- Z. Chen, Y. Ni, Y. Zhi, F. Wen, Z. Zhou, Y. Wei, W. Zhu and Z. Liu, Angew. Chem., Int. Ed., 2018, 130, 12729–12733 CrossRef.
- R. Yang, Y. Fu, Y. Zhang and N. Tsubaki, J. Catal., 2004, 228, 23–35 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- L. Palumbo, F. Bonino, P. Beato, M. Bjørgen, A. Zecchina and S. Bordiga, J. Phys. Chem. C, 2008, 112, 9710–9716 CrossRef CAS.
- Z. S. B. Sousa, D. V. Cesar, C. A. Henriques and V. Teixeira Da Silva, Catal. Today, 2014, 234, 182–191 CrossRef CAS.
- P. N. Plessow and F. Studt, ACS Catal., 2017, 7, 7987–7994 CrossRef CAS.
- L. Yang, T. Yan, C. Wang, W. Dai, G. Wu, M. Hunger, W. Fan, Z. Xie, N. Guan and L. Li, ACS Catal., 2019, 9, 6491–6501 CrossRef CAS.
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 8095–8099 CrossRef CAS PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 128, 16072–16077 CrossRef.
- Y. Liu, S. Müller, D. Berger, J. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 128, 5817–5820 CrossRef.
- W. Wang and M. Hunger, Acc. Chem. Res., 2008, 41, 895–904 CrossRef CAS PubMed.
- J. Li, Z. Wei, Y. Chen, B. Jing, Y. He, M. Dong, H. Jiao, X. Li, Z. Qin, J. Wang and W. Fan, J. Catal., 2014, 317, 277–283 CrossRef CAS.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125, 15260–15267 CrossRef CAS PubMed.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128, 11679–11692 CrossRef CAS PubMed.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- E. Gomez, X. Nie, J. H. Lee, Z. Xie and J. G. Chen, J. Am. Chem. Soc., 2019, 141, 17771–17782 CrossRef CAS PubMed.
- P. Gao, J. Xu, G. Qi, C. Wang, Q. Wang, Y. Zhao, Y. Zhang, N. Feng, X. Zhao, J. Li and F. Deng, ACS Catal., 2018, 8, 9809–9820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterization details. See DOI: 10.1039/d1sc01859k |
| This journal is © The Royal Society of Chemistry 2021 |