
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(II) ketimides in sp3 C–H amination†
Isuri U.
Jayasooriya
 ,
Abolghasem (Gus)
Bakhoda‡
,
Rachel
Palmer
,
Kristi
Ng
,
Nour L.
Khachemoune
,
Jeffery A.
Bertke
and
Timothy H.
Warren§
*
,
Abolghasem (Gus)
Bakhoda‡
,
Rachel
Palmer
,
Kristi
Ng
,
Nour L.
Khachemoune
,
Jeffery A.
Bertke
and
Timothy H.
Warren§
*
Department of Chemistry, Georgetown University, Box 571227-1227, Washington, DC 20057, USA. E-mail: warre155@msu.edu
First published on 5th November 2021
Abstract
Commercially available benzophenone imine (HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh2) reacts with β-diketiminato copper(II) tert-butoxide complexes [CuII]–OtBu to form isolable copper(II) ketimides [CuII]–NCPh2. Structural characterization of the three coordinate copper(II) ketimide [Me3NN]Cu–NCPh2 reveals a short Cu-Nketimide distance (1.700(2) Å) with a nearly linear Cu–N–C linkage (178.9(2)°). Copper(II) ketimides [CuII]–NCPh2 readily capture alkyl radicals R˙ (PhCH(˙)Me and Cy˙) to form the corresponding R–NCPh2 products in a process that competes with N–N coupling of copper(II) ketimides [CuII]–NCPh2 to form the azine Ph2CN–NCPh2. Copper(II) ketimides [CuII]–NCAr2 serve as intermediates in catalytic sp3 C–H amination of substrates R–H with ketimines HNCAr2 and tBuOOtBu as oxidant to form N-alkyl ketimines R–NCAr2. This protocol enables the use of unactivated sp3 C–H bonds to give R–NCAr2 products easily converted to primary amines R–NH2via simple acidic deprotection.
CPh2) reacts with β-diketiminato copper(II) tert-butoxide complexes [CuII]–OtBu to form isolable copper(II) ketimides [CuII]–NCPh2. Structural characterization of the three coordinate copper(II) ketimide [Me3NN]Cu–NCPh2 reveals a short Cu-Nketimide distance (1.700(2) Å) with a nearly linear Cu–N–C linkage (178.9(2)°). Copper(II) ketimides [CuII]–NCPh2 readily capture alkyl radicals R˙ (PhCH(˙)Me and Cy˙) to form the corresponding R–NCPh2 products in a process that competes with N–N coupling of copper(II) ketimides [CuII]–NCPh2 to form the azine Ph2CN–NCPh2. Copper(II) ketimides [CuII]–NCAr2 serve as intermediates in catalytic sp3 C–H amination of substrates R–H with ketimines HNCAr2 and tBuOOtBu as oxidant to form N-alkyl ketimines R–NCAr2. This protocol enables the use of unactivated sp3 C–H bonds to give R–NCAr2 products easily converted to primary amines R–NH2via simple acidic deprotection.
Introduction
Transition metal-catalysed sp3 C–H amination protocols have gained immense attention in the synthetic community over the past couple of decades.1–4 A majority of these protocols proceed via metal–nitrene2,5 [M]NR′ or metal–amide [M]–NR′R′′ intermediates.1,6 Extensive studies on such intermediates and underlying mechanisms have paved the way towards more efficient sp3 C–H amination protocols.1
Related metal–ketimide [M]–NCR′R′′ intermediates, however, have received less attention in C–H amination chemistry. The strong metal–Nketimide interaction makes ketimides effective spectator ligands. For instance, ketimides stabilize high valent homoleptic Mn(IV),7 Fe(IV)8 and Co(IV)9 complexes (Fig. 1a). In some cases, ketimides can also form via nickel and copper arylimido/nitrene intermediates [M]NAr via C–C coupling at the para-position of the aryl nitrene ligand (Fig. 1b). While this reactivity was initially uncovered with nickel β-diketiminato complexes,10 reversible C–C bond formation/cleavage in related copper complexes provides access to terminal copper nitrenes [Cu]NAr that participate in sp3 C–H amination.11,12
 | ||
| Fig. 1 Transition metal–ketimide complexes. | ||
Fewer examples of ketimides exist, however, in which the ketimide ligand serves as a reactive functional group in discrete transition metal complexes.13 Metal ketimide intermediates have been proposed in several Pd-catalysed cross-coupling reactions of aryl (Fig. 1c)14 and alkyl halides (Fig. 1d)15 with benzophenone imine. Cu-catalysed photoredox cross-coupling reactions of redox-active alkyl esters (Fig. 1e)16 and Cu-catalysed benzylic sp3 C–H amination with benzophenone imine (Fig. 1f)17 are among other examples that may be mediated by metal–ketimide intermediates. Moreover, Stahl and colleagues have proposed copper(II) ketimides in the N–N oxidative coupling of imines Ar2CNH to azines Ar2CN–NCAr2 under aerobic or electrocatalytic conditions (Fig. 1g).18,19
Herein we describe discrete first-row transition metal–ketimide complexes intimately involved in C–H amination chemistry. Building upon the Kharasch–Sosnovsky reaction,20–22 we previously demonstrated that copper(II) alkyl amides [CuII]–NHR′,23 anilides [CuII]–NHAr,6,24 and aryloxides [CuII]–OAr25 serve as key intermediates in a radical relay protocol for sp3 C–H functionalisation (Fig. 2). Formed via acid–base6,23,24 or transesterification25 reactions between [CuII]–OtBu with H-FG or Ac-FG reagents, these copper(II) complexes [CuII]–FG capture sp3-C radicals R˙ generated via H-atom abstraction from R–H to furnish the functionalized product R-FG. We anticipated that the relatively high acidity of the imine N–H bond26 coupled with a preference for binding at copper with softer N-donors should enable the formation of [CuII]–NCAr2 species from [CuII]–OtBu complexes and HNCPh2 allow for an examination of copper(II) ketimides in C–H amination catalysis.
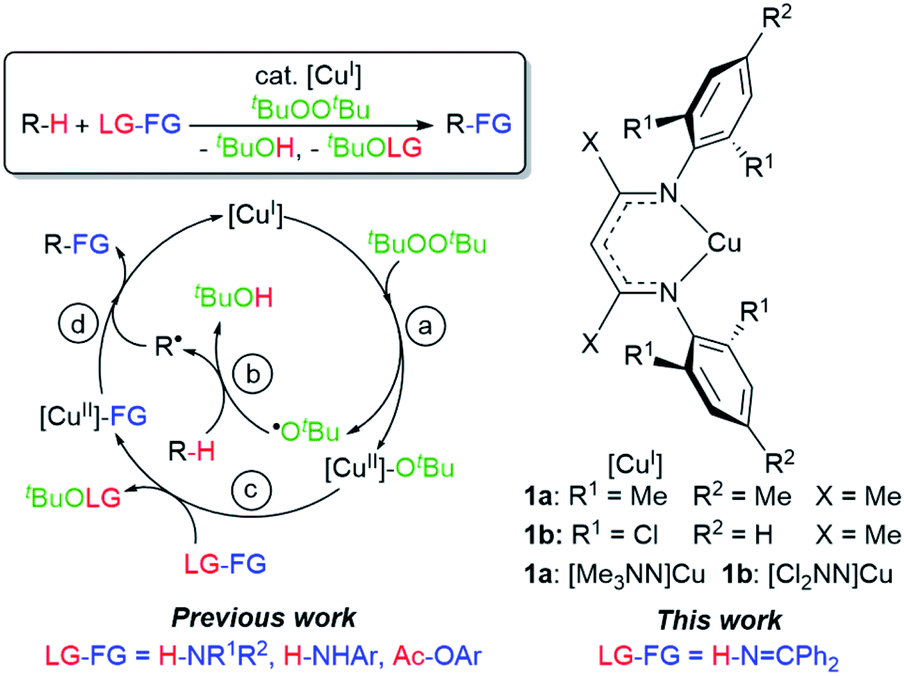 | ||
| Fig. 2 Mechanism of C–H functionalisation via β-diketiminato copper(II) intermediates [CuII]–FG. | ||
Results and discussion
Synthesis and characterization of copper(II) ketimides
Monitored by UV-vis spectroscopy, addition of benzophenone imine (1 equiv.) to a solution of [Me3NN]Cu-OtBu (2a) in toluene at −80 °C results in decay of the characteristic UV-vis absorption of 2a at 470 nm with growth of a new band at 570 nm (Fig. S2†). Performed on a preparative scale, this new species [Me3NN]Cu–NCPh2 (3a) may be isolated as dark purple crystals from pentane at −35 °C in 78% yield (Fig. 3a).
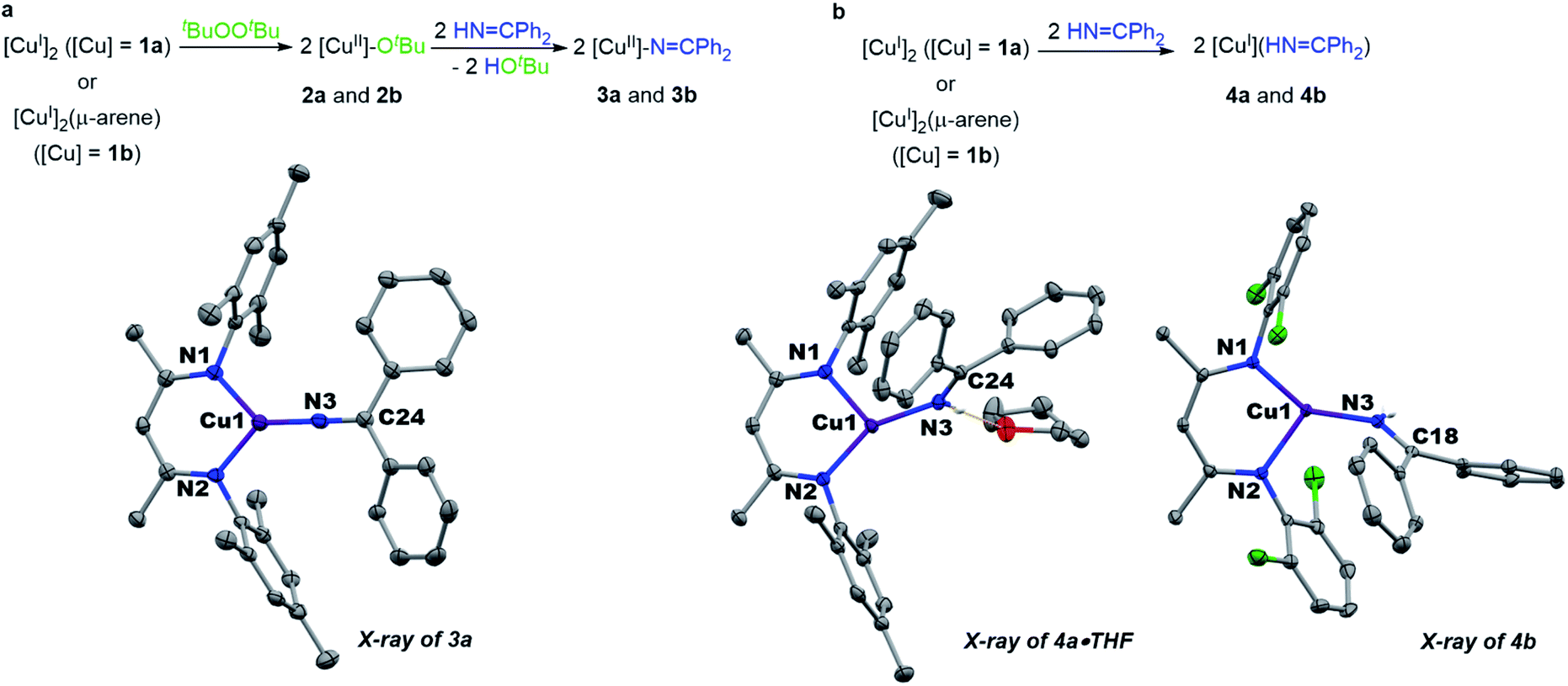 | ||
| Fig. 3 (a) Synthesis and structure of copper(II) ketimides. (b) Synthesis and structure of copper(I) imine adducts. | ||
The X-ray crystal structure of [Me3NN]Cu–NCPh2 (3a) (Fig. 3a) reveals the Cu–Nketimide distance of 1.700(2) Å, significantly shorter than the Cu–N bond found in the copper(II) amide [Cl2NN]Cu–NHAd (1.839(9) Å)23 and copper(II) anilide [Cl2NN]Cu–NHArCl3 (1.847(3) Å).6 Copper(II) ketimide 3a possesses a nearly linear Cu–N3–C24 angle of 178.9(2)°. The short Cu–Nketimide distance and linear Cu–N3–C24 angle support effective sp hybridization at the ketimide N atom. These values remarkably differ from those in the homoleptic copper(I) ketimide [Cu–NCPh2]4 with bridging ketimide ligands that lead to a square-like tetrameric structure with Cu–N distances 1.847(2)–1.861(2) Å and Cu–N–Cu angles of 94.17(9)–98.25(9).27 To outline differences between coordination of anionic ketimide ligands and their neutral ketimine counterparts, we prepared the corresponding benzophenone imine adducts [Me3NN]Cu(NHCPh2) (4a) and [Cl2NN]Cu(NHCPh2) (4b) (Fig. 3b). These copper(I) complexes feature substantially longer Cu–Nketimine distances of 1.8940(14) and 1.8937(14) Å. These ketimine adducts 4a and 4b each exhibit a pronounced bend in the Cu–ketimide linkage with Cu–N–C angles of 132.68(12) and 130.25(12)° consistent with sp2 hybridization at N.
UV-vis analysis of copper(II) ketimide [Me3NN]Cu–NCPh2 (3a) reveals the presence of a single low energy absorption band at 570 nm (ε = 1910 M−1 cm−1) in toluene at room temperature. The EPR spectrum of 3a in a mixture of toluene and pentane at room temperature shows a signal centred at giso = 2.081 with very well resolved coupling to 63/65Cu (ACu = 298.0 MHz) and additional hyperfine modelled with three equivalent 14N nuclei (AN = 35.0 MHz) (Fig. S13†). The related copper(II) ketimide [Cl2NN]Cu–NCPh2 (3b) prepared from [Cl2NN]Cu-OtBu (2b) and HNCPh2 exhibits a similar spectroscopic profile. The UV-vis spectrum of [Cl2NN]Cu–NPh2 (3b) exhibits a single absorption at 520 nm (ε = 3120 M−1 cm−1) in toluene at room temperature and possesses a similar isotropic EPR spectrum to that of 3a (Fig. S14†). Unfortunately, the greater thermal sensitivity of [Cl2NN]Cu–NPh2 (3b) has precluded its crystallographic characterization.
DFT calculations reveal remarkably high unpaired electron density on the ketimide N atom of both 3a (0.58) and 3b (0.61) (Fig. 4 and S23†). These values are significantly higher than values reported for related three coordinate β-diketiminato Cu(II) anilides [CuII]–NHAr (0.23–0.25)6 and a copper(II) amide [CuII]–NHAd (0.49).23 We rationalize this as a result of a 2-center 3-electron π interaction between the highest energy d orbital at the copper(II) center destabilized by the β-diketiminato N-donors and a p orbital of the sp-hybridized ketimide N atom (Fig. 4a). In addition, the orthogonal orientation of the Cu–Nketimide π-interaction relative to the conjugated ketimide NCPh2 π system further limits the delocalization of unpaired electron density away from the ketimide N atom (Fig. 4b and c).
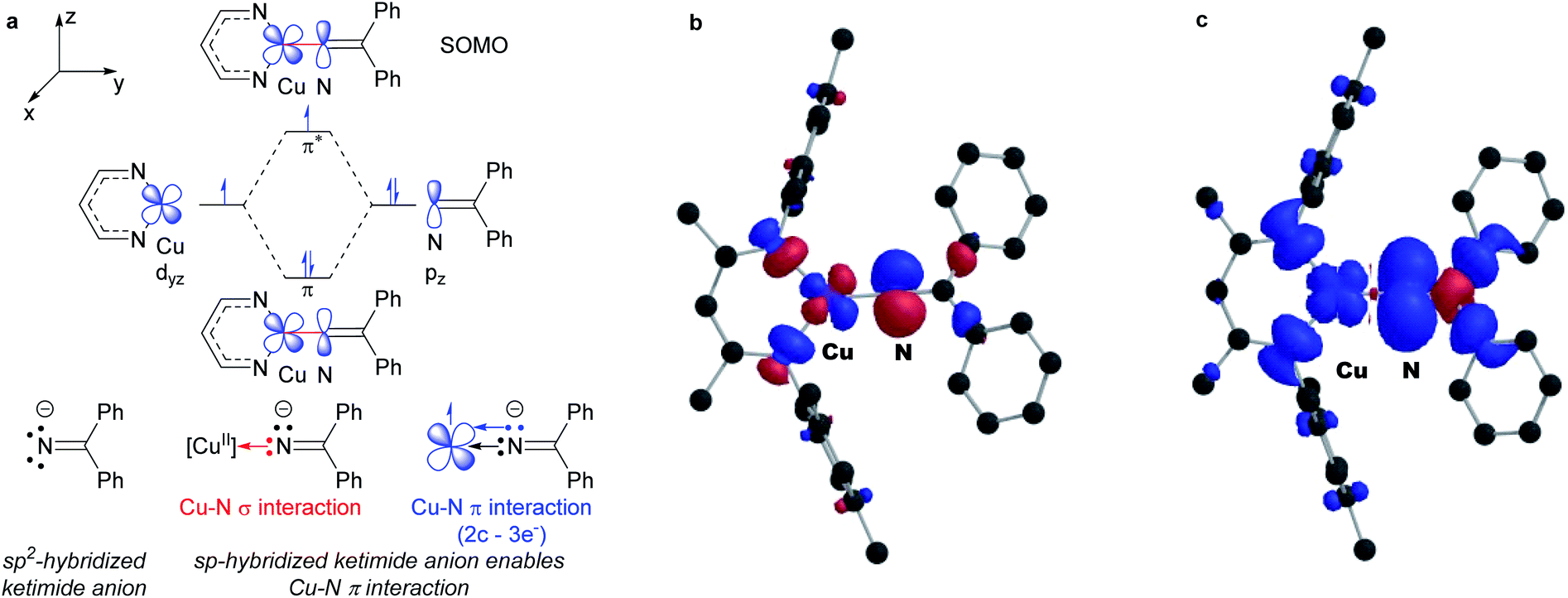 | ||
| Fig. 4 (a) Electronic structure of copper(II) ketimides. (b) SOMO and (c) spin density plot of copper(II) ketamide 3a (net spin α: blue, net spin β: red, 0.001 isospin value). | ||
Copper(II) ketimide reactivity: radical capture and N–N bond formation
The ability of many β-diketiminato copper(II) complexes to participate in catalytic sp3 C–H functionalisation via radical relay (Fig. 2) encouraged us to assess the reactivity of copper(II) ketimides 3 towards alkyl radicals. We find that [CuII]–NCPh2 species 3a and 3b capture alkyl radicals R˙ to provide the corresponding R–NCPh2 products (Fig. 5a). [CuI] is anticipated to form in these radical capture reactions that correspond to step d in the radical relay catalytic cycle (Fig. 2). For instance, reaction of 3a and 3b with (E/Z)-azobis(α-phenylethane) at 90 °C that generates the benzylic radical PhCH(˙)Me upon heating provides the alkylated imine PhCH(NCPh2)Me in 40% and 74% yields, respectively. Generation of Cy˙ radicals in the presence of 3a and 3b by heating tBuOOtBu in cyclohexane (via H-atom abstraction by tBuO˙ radicals) provides Cy-NCPh2 in 58% and 41% yields, respectively.
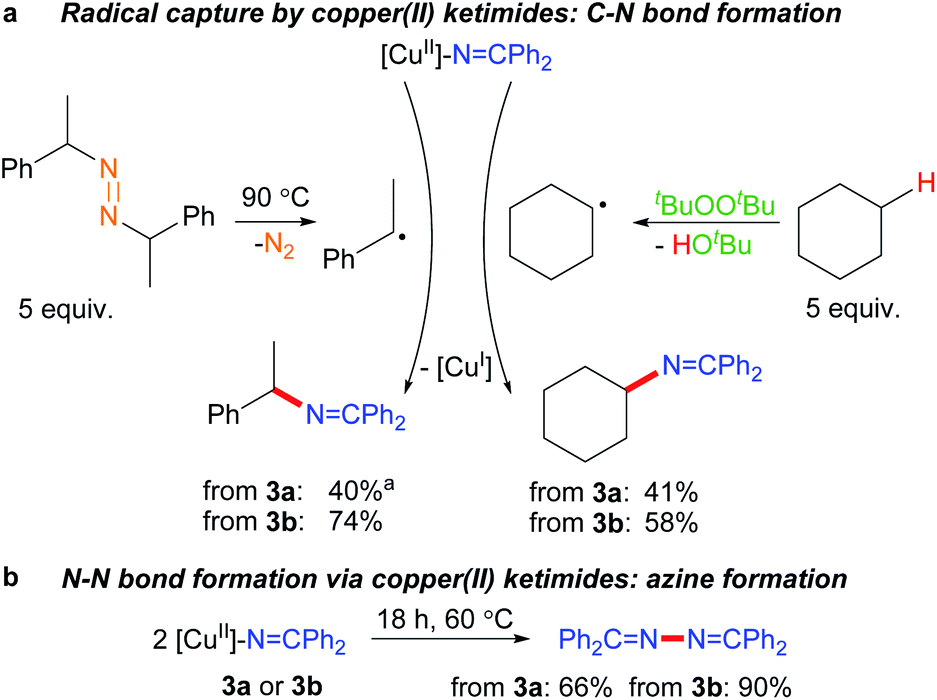 | ||
| Fig. 5 Reactivity of copper(II) ketimides. a 2 equiv. diazene radical precursor. | ||
Upon heating to 60 °C, copper(II) ketimides 3a and 3b undergo N–N coupling to form benzophenone azine Ph2CN–NCPh2 isolated in 66% and 90% yields, respectively (Fig. 5b). This represents a competing reaction for radical capture at copper(II) ketimides 3a and 3b.
Copper(II) ketimides in sp3 C–H amination
With a fundamental understanding of copper(II) ketimide formation and reactivity, we explored these complexes in catalytic C–H amination via radical relay. Using ethylbenzene as a model R–H substrate, we screened a modest range of copper(I) β-diketiminato catalysts 1 that possess different electronic and steric properties (Table 1). The catalyst [Cl2NN]Cu (1b) provides the highest yield compared to more electron-rich (1a and 1c) and electron-poor (1d) catalysts. Increasing the tBuOOtBu oxidant amount does not significantly improve the yield. Lowering the temperature from 90 °C reduces the yield drastically (Table S1†), possibly due to binding of the ketimine HNCAr2 to the copper(I) catalyst (Fig. 3b) that inhibits tBuOOtBu activation.28
|
|
|||
|---|---|---|---|
| Entry | Catalyst | (X, R1, R2) | Yield (%) |
| a Conditions: 50 equiv. R–H. All yields determined by 1H NMR | |||
| 1 | [Me3NN]Cu 1a | (Me, Me, Me) | 34 |
| 2 | [Cl2NN]Cu 1b | (Me, Cl, H) | 65 |
| 3 | [tPr2NN]Cu 1c | (Me, tPr, H) | 30 |
| 4 | [Cl2NNF6]Cu 1d | (CF3, Cl, H) | 42 |
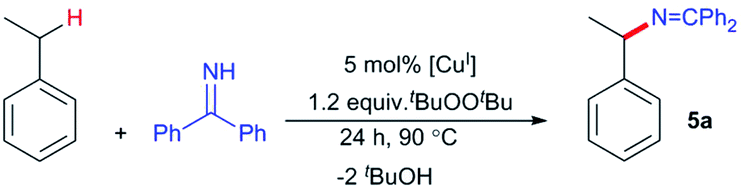
While (1-(tert-butoxy)ethyl)benzene forms in trace amounts via C–H etherification,28 the azine Ph2CN–NCPh2 is the main byproduct in these catalytic C–H amination reactions, representing non-productive consumption of H–NCPh2. In a previous study of C–H amination with anilines H2NAr employing the [Cl2NN]Cu/tBuOOtBu catalyst system, electron-poor anilines provided the highest yields in the face of competing diazene ArNNAr formation.24 Copper(II) anilido intermediates [CuII]–NHAr serve as intermediates in C–H amination with anilines H2NAr; those derived from electron-poor anilines H2NAr (e.g. Ar = 2,4,6-Cl3C6H2) proved more resistant to reductive bimolecular N–N bond formation.6,24
To examine whether similar electronic changes in the ketimine H–NCAr2 could similarly promote more efficient catalysis, we explored two electron-poor ketimine derivatives H–NCAr2 (Ar = 4-CF3C6H4 and 4-FC6H4) in C–H amination (Table 2). Although the p-CF3 substituted imine provides a higher C–H amination yield with cyclohexane (C–H BDE = 97 kcal mol−1),29 the increase in yield is modest with the benzylic substrate ethylbenzene (C–H BDE = 87 kcal mol−1).29 No significant differences were observed between benzophenone imine and the p-F substituted analogue.
|
|
|||
|---|---|---|---|
| Entry | Ar | Yield (%) | |
|
|
|
||
| a Conditions: 10 equiv. R–H, 1.2 equiv. tBuOOtBu, 1 mol% [Cl2NN]Cu, 90 °C, 24 h. Yields are determined by 1H NMR. | |||
| 1 |
|
44 (5a) | 40 (5b) |
| 2 |
|
51 (5a-CF3) | 56 (5b-CF3) |
| 3 |
|
36 (5a-F) | 39 (5b-F) |

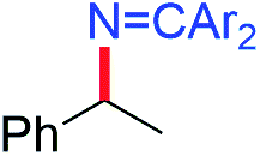
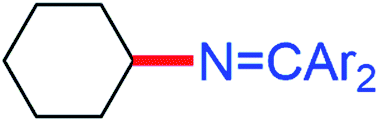
While electron-poor imines can give somewhat higher C–H amination yields, we most broadly examined the commercially available H–NCPh2 to survey the scope of R–H substrates in sp3 C–H amination (Table 3). Ethers such as THF, 1,4-dioxane, or even 12-crown-4 undergo C–H amination at the α-carbon in relatively high yields (6a–6d). Amination of the benzylic secondary C–H bonds in heteroaromatic substrates occurs (6f–6g), though yields may be lower due to the possibility of coordination of these substrates and/or products to the copper(I) centre that can decrease the rate of reoxidation with tBuOOtBu.28 Aromatic substrates with benzylic C–H bonds undergo C–H amination in moderate to high yields (6h–6k). Cycloalkanes with stronger, unactivated sp3 C–H bonds give moderate yields with electron-poor ketimine HNCAr′2 (Ar′ = 4-CF3C6H4) (6l–6o). The bicyclic eucalyptol undergoes C–H amination in 32% yield (6e). These aminated products may be isolated either as synthetically versatile protected primary amines R–NCPh2via column chromatography (6a–6g) or as the primary ammonium salts [R–NH3]Cl via deprotection upon simple acidic work up (6h–6o) under mild conditions. The potential to use recovered benzophenone from deprotection of ketimine products and azine byproducts to regenerate the Ph2CNH starting material30 enhances the overall atom economy of this amination protocol.
CAr2a


Conclusions
The isolation of mononuclear copper(II) ketimides [CuII]–NCPh2 reveals the role that they play as intermediates in sp3 C–H amination. These reactive intermediates readily form via acid–base exchange between [CuII]–OtBu and HNCPh2, amenable to spectroscopic and structural investigation. Importantly, [CuII]–NCPh2 complexes efficiently intercept alkyl radicals R˙ generated via H-atom abstraction by tBuO˙ from substrates R–H that ultimately enable the C–H amination of unactivated sp3 C–H substrates. DFT analysis reveals a significant amount of unpaired electron density at the ketimide N atom of 0.58 and 0.61 e− for [Me3NN]Cu–NCPh2 (3a) and [Cl2NN]Cu–NCPh2 (3b) (Fig. 4 and S23†), respectively, opening a facile pathway for C–N bond formation with radicals R˙ to form R–NCPh2 products (Fig. 5a). Moreover, this spin density at the ketimide N-atom likely facilitates N–N bond formation via copper(II) ketimides [CuII]–NCPh2 to give the azine Ph2CN–NCPh2 (Fig. 5b), a competing pathway in sp3 C–H functionalisation. Use of the more electron-poor ketimine HNCAr′ (Ar′ = 4-CF3C6H4) extends the scope of catalysis to unactivated sp3 C–H bonds in cycloalkanes (Table 3; entries 6l–6o). Nonetheless, facile N–N bond formation also by copper(II) ketimides [CuII]–NCAr2 underscores the role that they may play in the (electro)catalytic copper(II) promoted oxidative N–N coupling of benzophenone imine to form benzophenone azine (Fig. 1g).18
Experimental section
Detailed experimental procedures are provided in the ESI.†Data availability
All synthetic procedures, characterization data, spectroscopic data, computational data, supplementary figures and tables, and detailed crystallographic information can be found in the ESI.† Crystallographic data are available via the Cambridge Crystallographic Data Centre (CCDC): 1940417, 1945374, 1940418, 1945375, 1940420, 2035780.Author contributions
I. U. J. and A. B. prepared and characterized the metal complexes, I. U. J. performed reactivity and computational studies with metal complexes, I. U. J., R. P., K. N., and N. L. K. carried out catalytic amination experiments, isolating and characterizing organic products, J. A. B. solved and refined X-ray diffraction data, T. H. W. guided the research and assisted with data analysis, I. U. J. and T. H. W. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSF (CHE-1665348 and CHE-1955942) for support of this work.Notes and references
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- R. T. Gephart III and T. H. Warren, Organometallics, 2012, 31, 7728–7752 CrossRef.
- P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440–11453 RSC.
- E. S. Jang, C. L. McMullin, M. Kass, K. Meyer, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2014, 136, 10930–10940 CrossRef CAS PubMed.
- R. A. Lewis, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 4660–4668 CrossRef CAS PubMed.
- R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 12814–12816 CrossRef CAS PubMed.
- R. A. Lewis, S. P. George, A. Chapovetsky, G. Wu, J. S. Figueroa and T. W. Hayton, Chem. Commun., 2013, 49, 2888–2890 RSC.
- G. Bai and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 1856–1859 CrossRef CAS PubMed.
- A. G. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 CrossRef CAS PubMed.
- K. M. Carsch, I. M. Dimucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138–1143 CrossRef CAS PubMed.
- Y. Kondo, H. Morimoto and T. Ohshima, Chem. Lett., 2020, 49, 497–504 CrossRef CAS.
- J. P. Wolfe, J. Åhman, J. P. Sadighi, R. A. Singer and S. L. Buchwald, Tetrahedron Lett., 1997, 38, 6367–6370 CrossRef CAS.
- D. M. Peacock, C. B. Roos and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 647–652 CrossRef CAS PubMed.
- R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 9501–9504 CrossRef CAS PubMed.
- S. Kramer, Org. Lett., 2019, 21, 65–69 CrossRef CAS PubMed.
- M. C. Ryan, Y. J. Kim, J. B. Gerken, F. Wang, M. M. Aristov, J. R. Martinelli and S. S. Stahl, Chem. Sci., 2020, 11, 1170–1175 RSC.
- F. Wang, J. B. Gerken, D. M. Bates, Y. J. Kim and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 12349–12356 CrossRef CAS PubMed.
- M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS.
- M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819–5824 CrossRef CAS.
- D. J. Rawlinson and G. Sosnovsky, Synthesis, 1972, 1, 1–28 CrossRef.
- S. Wiese, Y. M. Badiei, R. T. Gephart, S. Mossin, M. S. Varonka, M. M. Melzer, K. Meyer, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2010, 49, 8850–8855 CrossRef CAS PubMed.
- R. T. Gephart III, D. L. Huang, M. J. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488–6492 CrossRef PubMed.
- T. K. Salvador, C. H. Arnett, S. Kundu, N. G. Sapiezynski, J. A. Bertke, M. Raghibi Boroujeni and T. H. Warren, J. Am. Chem. Soc., 2016, 138, 16580–16583 CrossRef CAS PubMed.
- F. G. Bordwell and G. Z. Ji, J. Am. Chem. Soc., 1991, 113, 8398–8401 CrossRef CAS.
- R. A. D. Soriaga, S. Javed and D. M. Hoffman, J. Cluster Sci., 2010, 21, 567–575 CrossRef CAS.
- R. T. Gephart, 3rd, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350–17353 CrossRef PubMed.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, 2002 Search PubMed.
- A. K. H. Hayashi, M. Katayama, K. Kawasaki and T. Okazaki, Ind. Eng. Chem. Prod. Res. Dev., 1976, 15, 299–303 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1940417, 1940418, 1940420, 1945374, 1945375 and 2035780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01990b |
| ‡ Current Address: Department of Chemistry, Towson University, 8000 York Road, Towson, MD, 21252, USA. |
| § Current Address: Department of Chemistry, Michigan State University, 578 S. Shaw Lane, East Lansing, MI 48824, USA. |
| This journal is © The Royal Society of Chemistry 2021 |