
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOsmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells†
Sonia
Infante-Tadeo
 a,
Vanessa
Rodríguez-Fanjul
a,
Abraha
Habtemariam
ab and
Ana M.
Pizarro
*ac
a,
Vanessa
Rodríguez-Fanjul
a,
Abraha
Habtemariam
ab and
Ana M.
Pizarro
*ac
aIMDEA Nanociencia, Faraday 9, 28049 Madrid, Spain. E-mail: ana.pizarro@imdea.org
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
cUnidad Asociada de Nanobiotecnología CNB-CSIC-IMDEA, 28049 Madrid, Spain
First published on 10th June 2021
Abstract
Aquation is often acknowledged as a necessary step for metallodrug activity inside the cell. Hemilabile ligands can be used for reversible metallodrug activation. We report a new family of osmium(II) arene complexes of formula [Os(η6-C6H5(CH2)3OH)(XY)Cl]+/0 (1–13) bearing the hemilabile η6-bound arene 3-phenylpropanol, where XY is a neutral N,N or an anionic N,O− bidentate chelating ligand. Os–Cl bond cleavage in water leads to the formation of the hydroxido/aqua adduct, Os–OH(H). In spite of being considered inert, the hydroxido adduct unexpectedly triggers rapid tether ring formation by attachment of the pendant alcohol–oxygen to the osmium centre, resulting in the alkoxy tethered complex [Os(η6-arene-O-κ1)(XY)]n+. Complexes 1C–13C of formula [Os(η6:κ1-C6H5(CH2)3OH/O)(XY)]+ are fully characterised, including the X-ray structure of cation 3C. Tether-ring formation is reversible and pH dependent. Osmium complexes bearing picolinate N,O-chelates (9–12) catalyse the hydrogenation of pyruvate to lactate. Intracellular lactate production upon co-incubation of complex 11 (XY = 4-Me-picolinate) with formate has been quantified inside MDA-MB-231 and MCF7 breast cancer cells. The tether Os–arene complexes presented here can be exploited for the intracellular conversion of metabolites that are essential in the intricate metabolism of the cancer cell.
Introduction
Cisplatin (cis-diamminedichloridoplatinum(II)) is one of the most well-known anticancer drugs due to its widespread use in the clinic. Its intracellular mechanism of action is believed to involve Pt–Cl hydrolysis prior to target interaction.1,2 Following the success of Pt drugs, a number of new organometallic coordination compounds with labile chlorido ligands have been synthesized and tested for anticancer activity. New complexes of Pt,3,4 Au,5 Ru,6–8 Re,9 Ir,10,11 and Os12–14 have been reported to be highly cytotoxic, even circumventing cisplatin-resistance. Additionally, a search for control over metallodrug activation has resulted in a variety of interesting selectivity-seeking approaches, such as photoactivation,15–17 ligand redox-mediated activation,11,18–20 and pH-dependent activation,21–26 among others.We25–28 and others21–23,29,30 have seen an opportunity in designing organometallic compounds bearing hemilabile ligands for metallodrug controlled activation. Hemilabile ligands are defined as bidentate non-symmetric ligands in which one of the binding groups is firmly attached to the metal centre. The other coordinated group is weakly bound and therefore can easily be dissociated.31 Since the displaced atom with metal-binding capabilities stays in close proximity to the metal, the process of opening and closing is reversible. Some have described hemilabile ligands as “swinging gates” to enable metal reactivity.32,33 A significant number of ruthenium(II) half-sandwich complexes bearing tethered groups containing C,34–36 N,23,25,37 O,26,37–39 S,40–42 and P37,43–46 donors have been reported. However, the only tethered η6-arene–osmium(II) described to date in the literature is the [3 + 2] annulation product of osmium hydrido alkenylcarbyne complex [OsH{![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC(PPh3)
CC(PPh3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh}(PPh3)2Cl2]+ with a substituted allenoate, which affords a typical three-legged piano-stool structure with the η6-benzene, two PPh3, and an alkenyl carbon atom (pendant from the arene) connected to the metal center.47 In addition, octahedral complexes of formula [OsCl2(N^P)2] have shown some cis versus trans fluxional conversion attributable to the hemilability of the N^P ligand in solution.48
CHPh}(PPh3)2Cl2]+ with a substituted allenoate, which affords a typical three-legged piano-stool structure with the η6-benzene, two PPh3, and an alkenyl carbon atom (pendant from the arene) connected to the metal center.47 In addition, octahedral complexes of formula [OsCl2(N^P)2] have shown some cis versus trans fluxional conversion attributable to the hemilability of the N^P ligand in solution.48
Organometallic Os(II) complexes have similar, often almost identical, structures to the analogous ruthenium compounds, yet they are often more inert,49 a useful feature vis-à-vis exerting control over metal reactivity. Os(II) half-sandwich compounds of general formula [Os(η6-arene)(XY)Z]n+ are less reactive towards hydrolysis —the rate of aquation is often ca. 100 times slower than for Ru—50–53 and once aquated the aqua ligand is more acidic, ca. −1.5 pKa units.49,51 Slowing down metal reactivity, which can also be achieved by replacing chlorido by iodido ligand, has indeed been related to high cytotoxic potency.14,18,54 For iridium complexes we have recently reported that stabilising a closed tether structure in complexes [Ir(η5:κ1-C5Me4CH2py)(C,N)]PF6, and thus delaying metal reactivity towards substitution reactions, increases cytotoxicity by up to two orders of magnitude.28
Transfer hydrogenation (TH) by transition metal compounds of Ru(II),8,26,55–57 Rh(III),58 and Ir(III),8,10,27 are attracting much attention in bioinorganic chemistry since they have demonstrated the potential of catalytically consuming or producing important metabolites, such as NADH, that are vital for a number of cellular processes.59–62 In particular, lactate and pyruvate are key cell metabolites that the tumour cell requires to promote cancer invasion as they help circulating tumour cells to survive by enhancing their resistance against oxidative stress.63
Osmium(II) arene [Os(η6-p-cymene)(impy-NMe2)Cl/I]PF6 (impy = iminopyridine) can oxidize NADH to NAD+ by Os-driven hydride abstraction.64 Importantly, chiral 16-electron complex [Os(η6-p-cymene)(TsDPEN)], (TsDPEN = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine), inspired by the Noyori-type of catalysts for asymmetric transfer hydrogenation,65 is capable of catalysing asymmetric reduction of pro-chiral pyruvate to lactate using non-toxic doses of sodium formate as a hydride donor inside cells.66,67
Herein we describe the synthesis of thirteen osmium(II) complexes 1–13, of formula [Os(η6-C6H5(CH2)3OH)(XY)Cl]+/0, bearing the hemilabile η6-bound arene 3-phenylpropanol and where XY is a neutral N,N or an anionic N,O− bidentate chelating ligand. We report for the first time Os(II) tethered half-sandwich complexes in their closed-tether form, 1C–13C, including the X-ray structure of cation 3C, where the hemilabile arene ligand chelates the osmium centre by simultaneous η6-C(arene) and κ1-O(R) binding. We show how the high stability of Os(II) η6-arene and the exceptional lability of alcohol oxygen as a coordinating group appear as the ideal combination to allow for control over metal reactivity in aqueous solution throughout the biologically relevant pH range. Aiming to unravel speciation in aqueous solution we have been surprised by the unexpected reactivity of the hydroxido adduct (the Os–OH bond), and the highly reversible open/close nature of the Os–O(tether) ring. Finally, we evaluate the reactivity of these complexes toward catalytic transfer hydrogenation of pyruvate using formate as hydride-source, finding that compounds reported here are capable of producing quantifiable excess lactate inside cancer cells.
Results and discussion
Synthesis and characterization
While a common synthetic method to introduce variations in Ru(II) arenes involves arene-exchange reactions in [Ru(η6-Et/Me-benzoate)Cl2]2 or other electron withdrawing substituted arene Ru(II) dimers,25,34,43,45 this method proved ineffective in the case of osmium, as our arene-exchange reactions repeatedly failed when using [Os(η6-Et-benzoate)Cl2]2 as a suitable dimer precursor. It was thus deemed necessary to reduce the specified arene ligand to the corresponding cyclohexadiene. To obtain 3-(1,4-cyclohexadien-1-yl)-1-propanol, the Birch reduction of 3-phenylpropanoic acid was carried out, following an experimental procedure similar to that described by Habtemariam et al.68 The reduced cyclohexadiene carboxylic acid was further reduced to the corresponding alcohol by the addition of LiAlH4. Osmium(II) dimer [Os(η6-C6H5(CH2)3OH)(μ-Cl)Cl]2 was then synthesized by the reaction of 3-(1,4-cyclohexadien-1-yl)-1-propanol with OsCl3·3H2O, as reported previously (Chart S1A†).49 Microwave synthetic methods reported by Tönnemann et al.69 were used to improve the yield and reaction times.All chlorido complexes 1–13 were synthesized from the reaction of the dimeric precursor, [Os(η6-C6H5(CH2)3OH)(μ-Cl)Cl]2, with the corresponding chelating ligand using similar procedures to those reported for other half-sandwich arene complexes (Chart S1B†).13,52,64Chart 1 shows the compounds obtained with general formula [Os(η6-C6H5(CH2)3OH)(XY)Cl]+/0, where XY = ethylenediamine, en (1), bipyridine, bipy (2), phenanthroline, phen (3), 1-(2-pyridinyl)methanamine, ampy (4), N-phenyl-1-(pyridin-2-yl)methanimine, Ph-impy (5), N-(4-(tert-butyl)phenyl)-1-(pyridin-2-yl)methanimine, tBuPh-impy (6), N-(2-bromophenyl)-1-(pyridin-2-yl)methanimine, BrPh-impy (7), N-phenethyl-1-(pyridin-2-yl)methanimine, PhEt-impy (8), picolinate, pico (9), 6-methylpicolinate, 6-Me-pico (10), 4-methylpicolinate, 4-Me-pico (11), 4-carboxy-2-pyridinecarboxylate, 4-COOH-pico (12), and 8-quinolinate, quinol (13). Cationic complexes 1–8 were isolated as chloride salts while 9–13 as neutral. All complexes were isolated in good yields (56–94%).
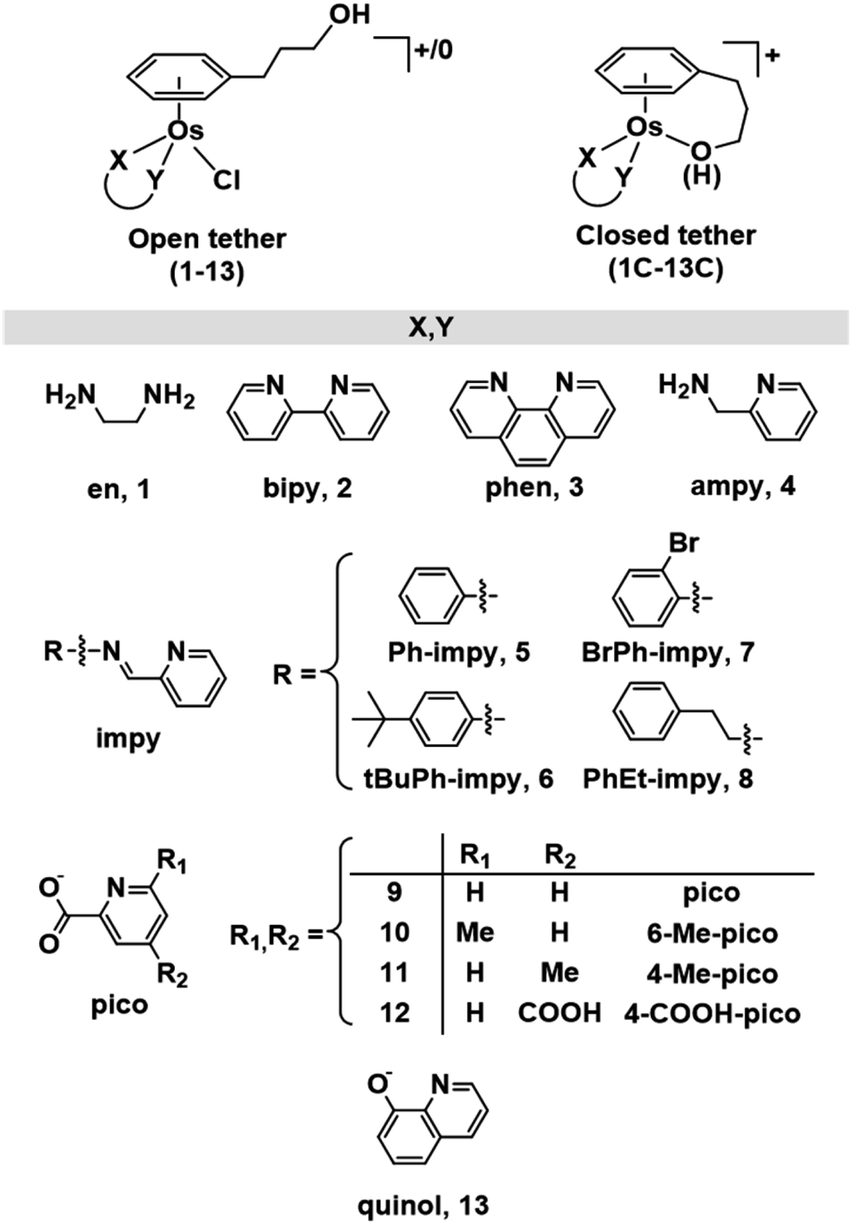 | ||
| Chart 1 Open and closed tethered osmium(II) arene complexes studied in this work of the general formulae [Os(η6-C6H5(CH2)3OH)(XY)Cl]+/0 and [Os(η6:κ1-C6H5(CH2)3OH/O)(XY)]+. | ||
The corresponding closed tether complexes of formula [Os(η6:κ1-C6H5(CH2)3OH/O)(XY)]+ (1C–13C; Chart 1) were synthesised using silver nitrate to abstract the chlorido ligand of the open-tether monomer in water or methanol (Chart S1B†). In several cases it was necessary to modify the pH of the aqueous solution for the intramolecular rearrangement (closure of the tether-ring) to occur (vide infra). Closed-tether complexes 2C, 3C, 4C, 8C, 10C, 11C and 13C were synthesised and obtained as nitrate salts in good yields (56–99%). Complexes 1C, 5C, 6C, 7C, 9C and 12C were characterized in solution as isolation was unsuccessful due to the fast interconversion dynamics with their open-tether counterparts. The 1H NMR spectra of open- and close-tether complexes display in all cases distinct patterns for the resonances of the η6-bound arene protons. In complexes 1–13, Δδ(bound arene) spans 0.14–0.54 ppm, and in the closed-tether counterparts, 1C–13C, spans 0.34–1.02 ppm (Table S1†), that is, the increment of chemical shift between the most deshielded and most shielded η6-bond arene proton signals was larger for closed-tether complexes than that of their open tether counterparts, attributable to restricted η6-bound arene rotation along the Os-centroid axis for closed tether complexes. This is in agreement with the observed differences of 1H NMR shifts corresponding to closed vs. open Ru(II) tether compounds.25,26,37
Ruthenium open and closed tether complexes bearing bipy and phen 2Ru, 2CRu, 3Ru and 3CRu and complex [Ru(η6:κ1-C6H5(CH2)3OH)Cl2] have been reported before,37,70,71 and are included for comparison purposes. The cations were isolated as their chloride or BF4− salts while the dichlorido complex is neutral.
Details of the synthesis and full characterization of all complexes are given in the ESI (Fig. S1–S23†).
Interestingly, while N,N-chelating ligands afforded Os closed-tether complexes with an alkoxy group coordinatively bound to the metal (deprotonated tethered alcohol), complexes bearing N,O-chelating ligands produced complexes with the alcohol attached to the metal. All our closed complexes are therefore +1 cations. Conductivity measurements supported the observation, giving values in the range ca. 100 S cm2 mol−1 corresponding to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolytes.72 The 1H NMR signal of such an alcohol proton can indeed be identified in the N,O closed tether structures when the spectrum is recorded in DMSO-d6 (see for example spectrum of 10C in Fig. S17†), contrariwise the proton signal cannot be found in DMSO-d6 solutions of N,N-chelated complexes.
:1 electrolytes.72 The 1H NMR signal of such an alcohol proton can indeed be identified in the N,O closed tether structures when the spectrum is recorded in DMSO-d6 (see for example spectrum of 10C in Fig. S17†), contrariwise the proton signal cannot be found in DMSO-d6 solutions of N,N-chelated complexes.
The X-ray crystal structures of chlorido complexes 5·PF6, 8·PF6, 9, 10, 11, 13, and closed tether complexes 3C·PF6 and dichlorido [Os(η6:κ1-C6H5(CH2)3OH)Cl2] (Fig. 1), unambiguously supported our structural determination. All complexes present the familiar pseudo-octahedral “three-legged piano-stool” geometry, with osmium(II) π-bonded to the η6-arene ligand. Open tether complexes are σ-bonded to a chlorido and a bidentate chelating ligand, while in closed tether complexes one of the three “legs” is occupied by the σ-bonded alcohol/alkoxy pendant from the η6-arene, affording an η6-Os-κ1 six-membered chelate ring in the structure (Fig. 1G and 1H).
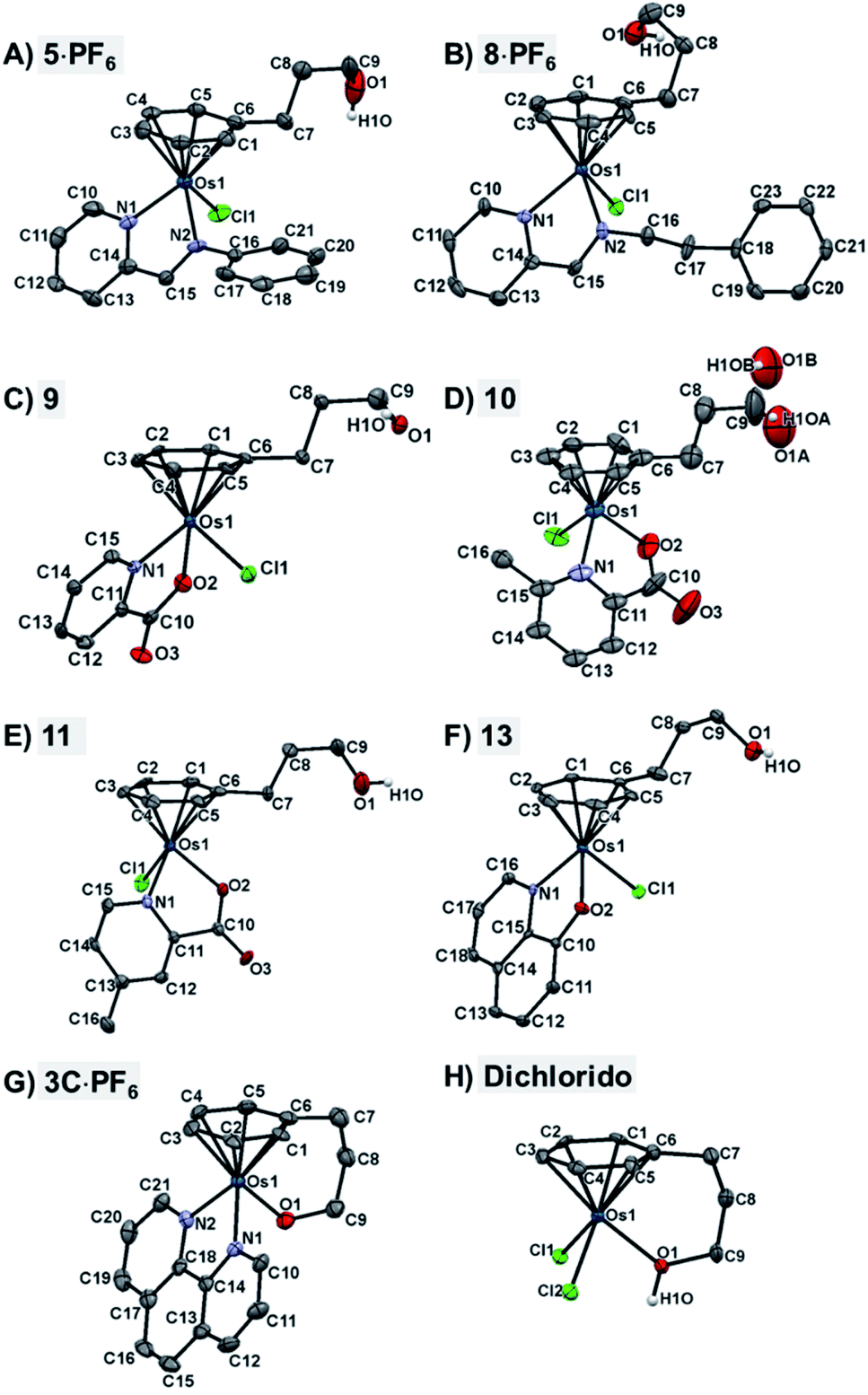 | ||
| Fig. 1 ORTEP diagrams and atom numbering schemes for compounds: (A) 5·PF6, (B) 8·PF6, (C) 9, (D) 10, (E) 11, (F) 13, (G) 3C·PF6, and (H) dichlorido complex [Os(η6:κ1-C6H5(CH2)3OH)Cl2] (50% probability ellipsoids). The H atoms (with the exception of the alcohol hydrogens), the solvent molecules, and the counterions have been omitted for clarity. Complex 10 in (D) shows disorder of the tethered oxygen occupying two different positions, resulting in a lower quality of structural determination. | ||
The crystal structures presented in this work are the first to be reported with a pendant alcohol function attached to the η6-bound arene in complexes of general formula [Os(η6-arene)(XY)Z]n+. Moreover, no X-ray structures of osmium closed tether complexes of formula [Os(η6-arene-O-κ1)(XY)]n+, where a coordinating tethered oxygen is bound to the metal centre, have been previously reported. In fact, tethered η6-arene–osmium(II) compounds are extremely rare; we only found complex [Os(η6-benzene-C-κ1)(PPh3)2]+, with a tethered alkenyl pendant from the derivatised arene,47 in the Cambridge Database.
Closed-tether structures feature a strong offset of C7 (first carbon in the tether chain attached to the arene) toward the osmium atom with regard to the plane that contains the η6-bound arene, with calculated values of 0.138 and 0.193 Å, respectively, for both 3C·PF6 and dichlorido [Os(η6:κ1-C6H5(CH2)3OH)Cl2] (Table S2†). Another interesting feature in all the X-ray structures is the extensive hydrogen bonding of the oxygen pendant from the arene, especially strong in the case of cation 3C between the tethered O-alkoxy and a water molecule of crystallization (Fig. S24 and Table S3†).
Full crystallographic analysis, selected bonds and angles (Table S2†), π–π intermolecular interactions, directionality, angles and distances of hydrogen-bonding interactions (Fig. S24 and Table S3†), and detailed crystallographic data (Table S4†), are included in the ESI† of this manuscript. CCDC 2026997–2027004 contain the ESI crystallographic data for this paper.
Aqueous solution studies
Metallodrug activity inside the cell is believed to be triggered by aquation. The hydrolysis rate of Os(II) arene compounds of the type [Os(η6-arene)(XY)Cl]n+ is highly affected by the nature of the chelating ligand.13,49,52,53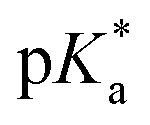 It has been proven that compounds with N,O-chelated ligands display aqueous behaviour intermediate between those bearing N,N- and O,O-chelates, the former being too slow for the complex to have an impact on cell survival, and the latter being too fast, resulting in the loss of the XY ligand and leading to the formation of inert di-osmium OH-bridged species.53 Following Os–Cl cleavage, the aqua adduct is formed. Above the
It has been proven that compounds with N,O-chelated ligands display aqueous behaviour intermediate between those bearing N,N- and O,O-chelates, the former being too slow for the complex to have an impact on cell survival, and the latter being too fast, resulting in the loss of the XY ligand and leading to the formation of inert di-osmium OH-bridged species.53 Following Os–Cl cleavage, the aqua adduct is formed. Above the 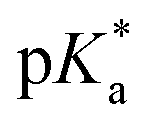 corresponding to the Os–OH2/OH equilibria, the hydroxido Os–OH species will predominate. This is believed to render the complex inactive towards substitution reactions,49 including potential intracellular targets, and thus negatively impacting their biological effectivity.53
corresponding to the Os–OH2/OH equilibria, the hydroxido Os–OH species will predominate. This is believed to render the complex inactive towards substitution reactions,49 including potential intracellular targets, and thus negatively impacting their biological effectivity.53
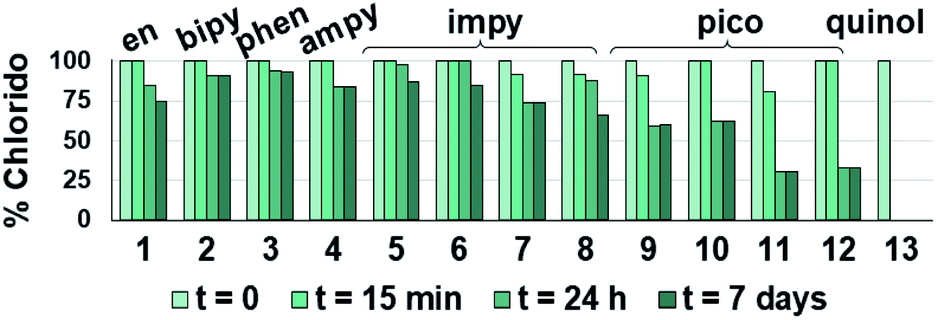 | ||
| Fig. 2 Hydrolysis of the Os–Cl bond for complexes 1–13. The bars represent the percentage of remaining open-tether chlorido complexes 1–13 over time in unbuffered D2O as determined by 1H NMR. Equilibrium is mostly reached in the first 24 h. Complex 13 is fully hydrolysed from the first data recording (t ≤ 15 min upon dissolution). | ||
The relatively faster hydrolysis of the Os–Cl species in complexes 9–12, containing picolinate ligands, is in agreement with previous reports.13,49,53 It may be explained on the basis of an increased electron density at the metal centre due to the anionic chelating ligand, possibly aided by the stabilising interaction between hydrogen atoms of the coordinated water in the aqua species and the oxygen atom of the tethered alcohol group. Indeed, the X-ray structures of complexes 9, 11 and 13 indicate strong hydrogen bonding of the tethered alcohol oxygen with neighbouring Os(II) molecules with strong covalent character (Fig. S24 and Table S3†).74
The rate of hydrolysis in D2O of picolinate compounds 9–12 was monitored by 1H NMR at 300, 310 and 320 K at various time intervals. The percentage of disappearance of chlorido adduct for 9–12 (based on peak integration) was plotted against time and fitted to pseudo-first order kinetics, and their half-life times were calculated. For all four complexes hydrolysis reaches equilibrium within the first hour even at the lowest temperature (Table 1 and Fig. S25–S27†).
| T (K) | k hyd (min−1) | t 1/2 (min) | E a (kJ mol−1) | ΔH‡ (kJ mol−1) | ΔS‡ (J K−1 mol−1) | |
|---|---|---|---|---|---|---|
| 9 | 300 | 0.08 | 8.6 | 67.0 ± 0.9 | 64.4 ± 1.0 | −51.5 ± 3.1 |
| 310 | 0.19 | 3.7 | ||||
| 320 | 0.48 | 1.4 | ||||
| 10 | 300 | 0.06 | 11.9 | 63.9 ± 15.5 | 61.4 ± 15.5 | −65.0 ± 49.9 |
| 310 | 0.09 | 7.5 | ||||
| 320 | 0.29 | 2.4 | ||||
| 11 | 300 | 0.20 | 3.5 | 40.8 ± 11.1 | 38.3 ± 11.0 | −131.7 ± 35.6 |
| 310 | 0.26 | 2.7 | ||||
| 320 | 0.54 | 1.3 | ||||
| 12 | 300 | 0.05 | 13.2 | 67.0 ± 4.7 | 64.4 ± 4.7 | −55.0 ± 15.1 |
| 310 | 0.11 | 6.1 | ||||
| 320 | 0.28 | 2.5 |
Arrhenius activation energies (Ea) and activation enthalpies (ΔH‡) of hydrolysis of compounds 9, 10 and 12 (Table 1 and Fig. S26–S27†) are lower than those previously found for similar picolinate Os–arenes, which are reported to be ca. 90 kJ mol−1 for both parameters.13,53 However, activation entropies (ΔS‡) are similar to those determined for similar complexes varying from −70 to −50 J K−1 mol−1.13,53 The large negative activation entropies (ΔS‡) found for the hydrolysis of 9, 10, 11 and 12 indicate that entropy decreases on forming the transition state, which suggests that an associative mechanism is involved, in which two reaction partners form a single activated complex. The fastest hydrolysing compound, 11, presents the most negative entropy, −131.7 J K−1 mol−1, a value more than two-fold of its analogue [Os(η6-C6H5C6H5)(4-Me-pico)Cl]+, with an entropy of −55.6 ± 1.6 J K−1 mol−1.13 Complex 11 also requires the lowest ΔH‡ towards hydrolysis.
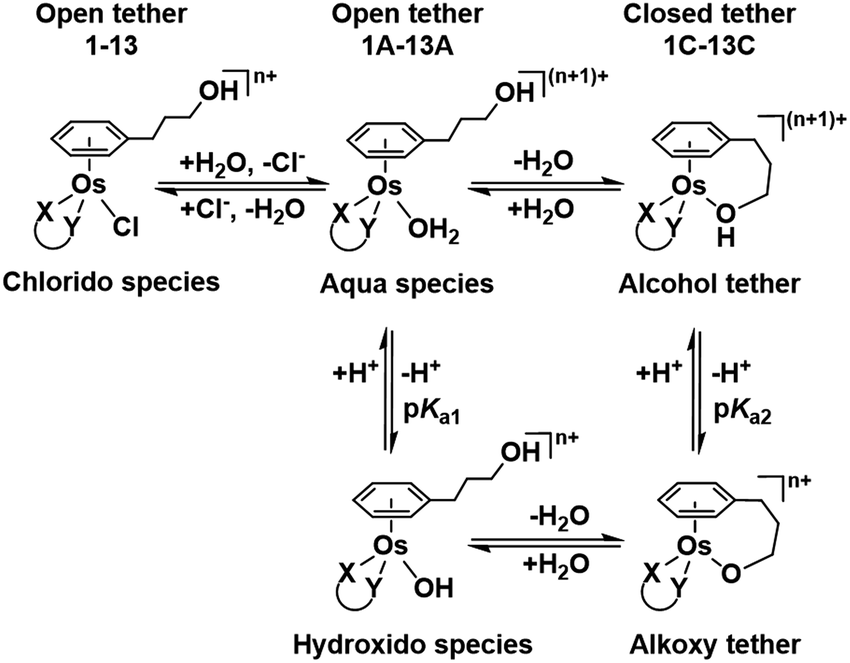 | ||
| Chart 2 Aqueous speciation of open and closed tether complexes, including acid–base equilibria (n = 1 for complexes 1–8; n = 0 for 9–13). | ||
Such a three-way speciation of complexes 1–13 was first detected by 1H NMR in unbuffered D2O (Fig. S28†). Addition of excess sodium chloride to the mixture of species at equilibrium confirmed the assignment of the aqua adduct by 1H NMR (by conversion of the aqua to the chlorido species). Addition of AgNO3 to solutions of complexes 2, 3, 8, 10 and 11 aided the unambiguous characterization of the closed tether species. Furthermore, Ag+-mediated choride sequestration indicated that Os–Cl bond cleavage favours tether-ring closure.
Peacock et al. have shown pH to be important in the aqueous behaviour and thus the biological impact of hydrolysable Os–arenes as potential anticancer agents.13,49,52,53,75 We aimed to assess the effect of pH on interconversion of species in aqueous solution of the Os(II) half-sandwich OH-tethered complexes. Selected compounds 1, 2, 5, 8, 9, 10, 11 and 13 were dissolved in D2O (at [Os] = 5 mM) in four unbuffered pH solutions (pH 1, 4, 7 and 10). Fig. 3 shows how our complexes undergo speciation, whereby the aqua (or hydroxido) species seemingly evolve to the closed tether complex until reaching equilibria where coexistence of the three species (chlorido, aqua/hydroxido and closed tether species) occurred upon 24 h of incubation.
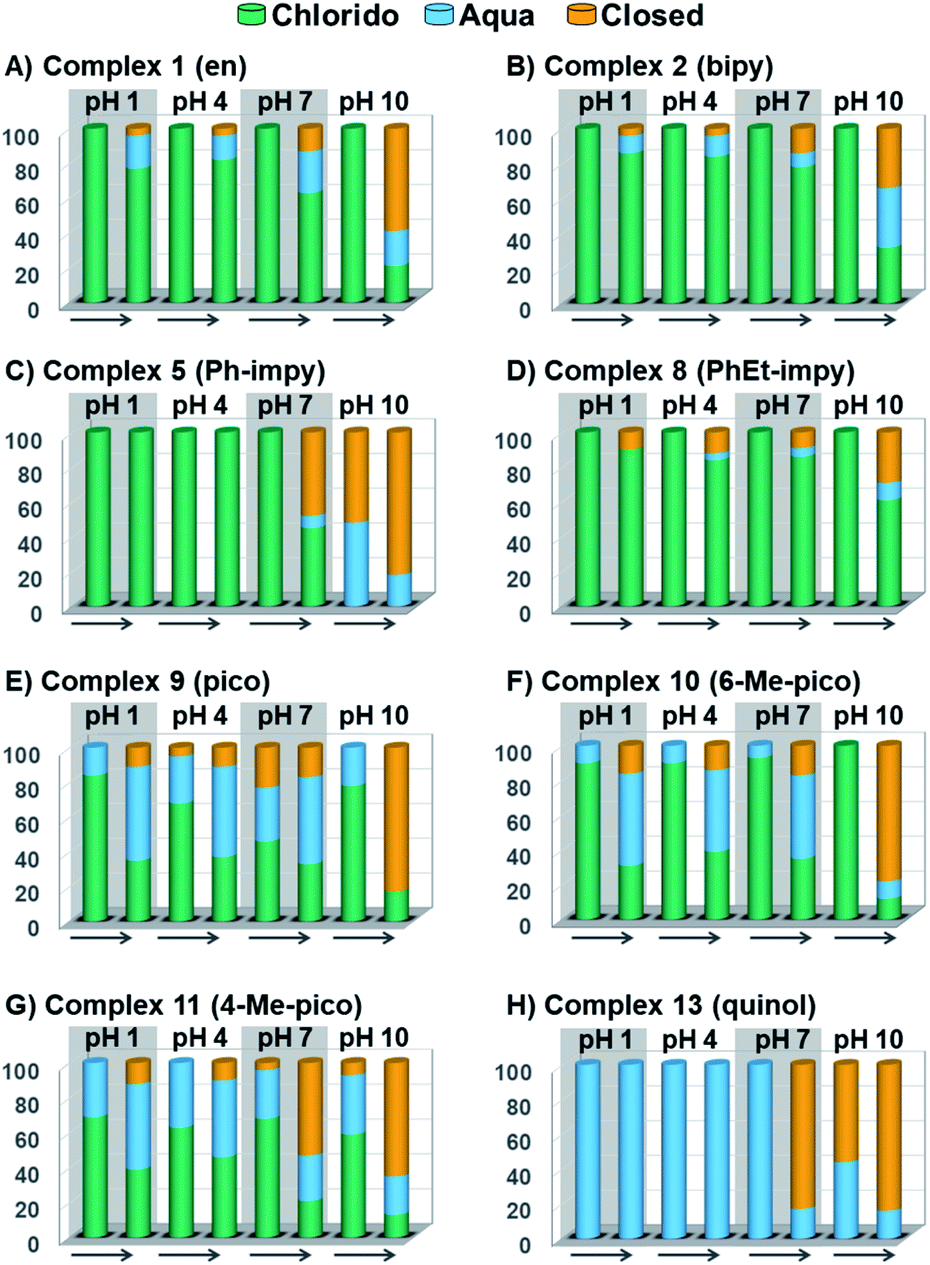 | ||
| Fig. 3 Speciation of chlorido complexes (A) 1, (B) 2, (C) 5, (D) 8, (E) 9, (F) 10, (G) 11, and (H) 13 at time ca. 15 min and after 24 h in unbuffered aqueous solutions at pH 1, 4, 7 and 10. The arrow represents time forward, from t = 15 min to t = 24 h. | ||
The percentage of hydrolysis-triggered species is similar at pH 1 and 4, i.e., pH-variation when the proton concentration is high has no impact in the speciation of these complexes. The predominant species are chlorido and aqua species (Chart 2). At pH 10, however, most complexes show a remarkable preference for intramolecular rearrangement, as seen by the evolution over time towards the closed tether species. All but complexes 2 and 8 show more than 60% of closed tether structure at basic pH after 24 h. Even fully aquated complex 13 shows a clear preference for the closed tether species 13C at pH 10 even at t = 15 min.
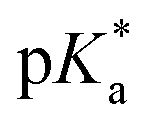 determination.
Following hydrolysis rate determination and finding reversible interconversion of open and closed tether complexes in a pH-dependent manner, we endeavour to determine the pKas of the species in solution. Both aqua (1A–13A) and closed tether (1C–13C) species are subjected to acid–base equilibria (Chart 2). Alcohol coordination to osmium(II) in the closed tether complexes renders the σ-bonded alcohol proton highly acidic, favouring deprotonation of an otherwise largely basic group (estimated pKa of 3-phenyl-1-propanol ca. 16).
determination.
Following hydrolysis rate determination and finding reversible interconversion of open and closed tether complexes in a pH-dependent manner, we endeavour to determine the pKas of the species in solution. Both aqua (1A–13A) and closed tether (1C–13C) species are subjected to acid–base equilibria (Chart 2). Alcohol coordination to osmium(II) in the closed tether complexes renders the σ-bonded alcohol proton highly acidic, favouring deprotonation of an otherwise largely basic group (estimated pKa of 3-phenyl-1-propanol ca. 16).
Complexes 1–13 were treated with AgNO3 in water or alcoholic aqueous solutions to ensure the formation of both the aqua and subsequently the closed tether species. Compounds 2Ru and 3Ru were synthesised37,70 and included for comparison purposes. The  values for the aqua/hydroxido (OH2/OH) adduct of formula [M(η6-C6H5(CH2)3OH)(XY)(OH2/OH)]n+, for complexes 1A, 2A, 2ARu, 3A, 3ARu, 8A, 12A and 13A, and the
values for the aqua/hydroxido (OH2/OH) adduct of formula [M(η6-C6H5(CH2)3OH)(XY)(OH2/OH)]n+, for complexes 1A, 2A, 2ARu, 3A, 3ARu, 8A, 12A and 13A, and the 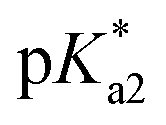 values for the coordinated alcohol/alkoxy group (ROH/RO) in closed tether complexes of formula [M(η6:κ1-C6H5(CH2)3OH/O)(XY)]n+, for complexes 1C, 2C, 2CRu, 3C, 3CRu, 8C and 12C, were determined and are presented in Table 2 and Fig. S29.†1H NMR peaks corresponding to coordinated arene protons in closed and aqua adducts 1C–3C, 3CRu, 1A–3A and 3ARu, or chelating ligand protons in 8A, 8C, 12A, 12C and 13A, were followed as they shifted to high field on increasing pH* (Fig. S30† contains the titration of a mixture containing 3, 3A and 3C, as an example). Changes in the 1H NMR chemical shifts of closed tether and aqua species were recorded at 298 K over the 0–12 pH range by the addition of NaOD or DNO3 as appropriate. The chemical shift-versus-pH plots and
values for the coordinated alcohol/alkoxy group (ROH/RO) in closed tether complexes of formula [M(η6:κ1-C6H5(CH2)3OH/O)(XY)]n+, for complexes 1C, 2C, 2CRu, 3C, 3CRu, 8C and 12C, were determined and are presented in Table 2 and Fig. S29.†1H NMR peaks corresponding to coordinated arene protons in closed and aqua adducts 1C–3C, 3CRu, 1A–3A and 3ARu, or chelating ligand protons in 8A, 8C, 12A, 12C and 13A, were followed as they shifted to high field on increasing pH* (Fig. S30† contains the titration of a mixture containing 3, 3A and 3C, as an example). Changes in the 1H NMR chemical shifts of closed tether and aqua species were recorded at 298 K over the 0–12 pH range by the addition of NaOD or DNO3 as appropriate. The chemical shift-versus-pH plots and  values obtained from the titrations are compiled in Fig. S31.†
values obtained from the titrations are compiled in Fig. S31.†
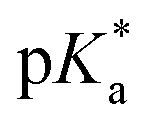 values of the aqua ligand in complexes 1A, 2A, 2ARu, 3A, 3ARu, 8A, 12A and 13A, and the tethered alcohol in complexes 1C, 2C, 2CRu, 3C, 3CRu, 8C and 12C
values of the aqua ligand in complexes 1A, 2A, 2ARu, 3A, 3ARu, 8A, 12A and 13A, and the tethered alcohol in complexes 1C, 2C, 2CRu, 3C, 3CRu, 8C and 12C
|
|
|
|
|---|---|---|
| [Os(η6-C6H5(CH2)3OH)(en)(OH2)]2+ (1A) | 5.83 | — |
| [Os(η6:κ1-C6H5(CH2)3OH)(en)]2+ (1C) | — | 5.46 |
| [Os(η6-C6H5(CH2)3OH)(bipy)(OH2)]2+ (2A) | 5.81 | — |
| [Os(η6:κ1-C6H5(CH2)3OH)(bipy)]2+ (2C) | — | 4.23 |
| [Ru(η6-C6H5(CH2)3OH)(bipy)(OH2)]2+ (2ARu) | 7.28 | — |
| [Ru(η6:κ1-C6H5(CH2)3OH)(bipy)]2+ (2CRu) | — | 5.78 |
| [Os(η6-C6H5(CH2)3OH)(phen)(OH2)]2+ (3A) | 5.62 | — |
| [Os(η6:κ1-C6H5(CH2)3OH)(phen)]2+ (3C) | — | 4.26 |
| [Ru(η6-C6H5(CH2)3OH)(phen)(OH2)]2+ (3ARu) | 7.18 | — |
| [Ru(η6:κ1-C6H5(CH2)3OH)(phen)]2+ (3CRu) | — | 6.39 |
| [Os(η6-C6H5(CH2)3OH)(PhEt-impy)(OH2)]2+ (8A) | 5.69 | — |
| [Os(η6:κ1-C6H5(CH2)3OH)(PhEt-impy)]2+ (8C) | — | 4.41 |
| [Os(η6-C6H5(CH2)3OH)(4-COOH-pico)(OH2)]+ (12A) | 6.64 | — |
| [Os(η6:κ1-C6H5(CH2)3OH)(4-COOH-pico)]+ (12C) | — | 5.63 |
| [Os(η6-C6H5(CH2)3OH)(quinol)(OH2)]+ (13A) | 7.01 | — |
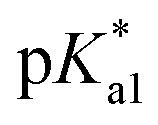
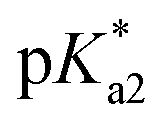
The nature of the chelating ligand (N,N, N,O or O,O) shows a great influence on the 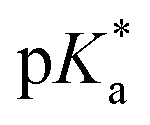 values of the M–OH2 group (basicity increasing N,N < N,O < O,O, with better π-acceptors, such as phen, affording more acidic metal centres, which resulted in lower
values of the M–OH2 group (basicity increasing N,N < N,O < O,O, with better π-acceptors, such as phen, affording more acidic metal centres, which resulted in lower 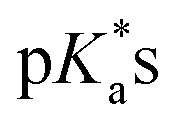 for the aqua adduct).13,26,49,51–53,76 The high acidity of osmium arene aqua complexes has been previously attributed to increased mixing of the δσ* (Os) → σ (OH−) orbitals.77,78 The higher acidity of Os versus Ru can also explain the acidity of the coordinated hemilabile alcohol/alkoxy (Fig. S29†).23,49,77
for the aqua adduct).13,26,49,51–53,76 The high acidity of osmium arene aqua complexes has been previously attributed to increased mixing of the δσ* (Os) → σ (OH−) orbitals.77,78 The higher acidity of Os versus Ru can also explain the acidity of the coordinated hemilabile alcohol/alkoxy (Fig. S29†).23,49,77
Closed-tether and aqua species identification was confirmed by the 1H NMR pH titration associated with both  The chlorido species are devoid of chemical shift variation due to the lack of (de)protonatable positions in the structure, with the exception of complex 12, which bears a carboxylic acid in the picolinate ligand (Fig. S31F†).
The chlorido species are devoid of chemical shift variation due to the lack of (de)protonatable positions in the structure, with the exception of complex 12, which bears a carboxylic acid in the picolinate ligand (Fig. S31F†).
Since the  data is below 7.4 (physiological pH) for all Os–arenes, in a live cell environment, the complexes are expected to be present in the hydroxido form rather than the aqua species. Yet in our complexes the hydroxido species readily evolves to the closed tether complex. Contrary to previous reports on Os–arene aquation,49,53,75 inert hydroxido-bridged dimers have not been detected throughout this work. In fact, the species reported here are interconvertible in a reversible manner over the pH range. The formation of closed tether species presents a very exciting turn of events due to their strong predominance at
data is below 7.4 (physiological pH) for all Os–arenes, in a live cell environment, the complexes are expected to be present in the hydroxido form rather than the aqua species. Yet in our complexes the hydroxido species readily evolves to the closed tether complex. Contrary to previous reports on Os–arene aquation,49,53,75 inert hydroxido-bridged dimers have not been detected throughout this work. In fact, the species reported here are interconvertible in a reversible manner over the pH range. The formation of closed tether species presents a very exciting turn of events due to their strong predominance at 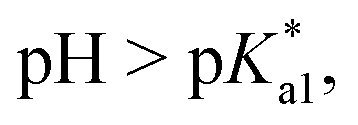 preventing deactivation of the Os(II) compound.
preventing deactivation of the Os(II) compound.
Speciation proved indeed highly dependent on pH (Fig. 3), clearly favouring tether ring closure even at 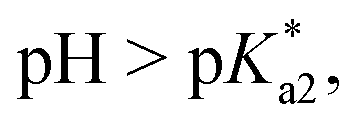 that is, the
that is, the 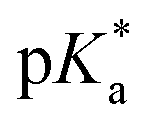 of the Os-bound alcohol. We postulate a concerted associative mechanism for intramolecular rearrangement resulting in tether ring formation by which an incoming ligand, the tethered alcohol, likely assisted by H-bonding, transfers its proton to the hydroxido ligand (favoured by
of the Os-bound alcohol. We postulate a concerted associative mechanism for intramolecular rearrangement resulting in tether ring formation by which an incoming ligand, the tethered alcohol, likely assisted by H-bonding, transfers its proton to the hydroxido ligand (favoured by 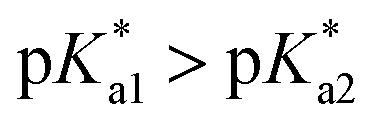 ), thereby facilitating the release of a water molecule from the osmium first coordination sphere (Chart 3). This is supported by extensive hydrogen-bonding by the pendant oxygen as observed upon X-ray analysis, especially strong for closed tether cation 3C (Fig. S24 and Table S3†).
), thereby facilitating the release of a water molecule from the osmium first coordination sphere (Chart 3). This is supported by extensive hydrogen-bonding by the pendant oxygen as observed upon X-ray analysis, especially strong for closed tether cation 3C (Fig. S24 and Table S3†).
 | ||
Chart 3 Suggested mechanism of tether-ring closure as dependent on pH, given that 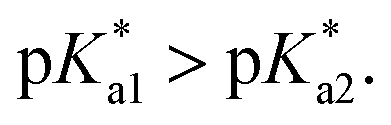 | ||
Speciation of closed tether complexes 2C, 10C and 13C in the presence of chloride ions was also investigated. Since water-mediated speciation is highly dependent on pH, the behaviour of 2C, 10C and 13C was evaluated in 0.1 M DCl and 0.1 M DClO4 solutions (Fig. S33†) at 310 K over 24 h. These solutions were selected because the concentration of protons is equivalent in both, yet the chloride concentration varies from 100 mM Cl− in DCl (similar to that in plasma) to nil in DClO4. The results indicate that the tether-ring opening occurs to approximately the same extent, clarifying that pH dominates speciation over chloride concentration.
Catalytic reduction of pyruvate to lactate
Osmium(II) arenes have been reported to carry out catalytic transfer hydrogenation reactions.64,66 A screening of selected complexes 1, 3, 5, 6, 9, 10, 11, 12, 13 was carried out to evaluate the conversion of pyruvate (pyr) to lactate using formate (for) as the hydride source, at ratios 1:2:100 (Os catalyst/pyr/for) at pH 4 and 310 K, by means of 1H NMR.
Complexes 1, 3, 5 and 6 with N,N ligands did not transform pyruvate to lactate over 48 h, even after formation of the Os-formate adduct for complexes 1 and 5 was observed by 1H NMR. The formate adduct is believed to precede hydride abstraction by the metal centre during the catalytic cycle.55,56,66,79 It has been reported that chlorido removal increases the activity of catalytic metal complexes by generating species that can bind to formate.56 Hydrolysis of complexes 9–13, bearing N,O-chelating ligands, could thus be in their favour to mediate the transformation of pyruvate to lactate. In the 1H NMR spectra of the hydrogenation reactions by 9, 11, 12 and 13 two new sets of peaks were attributed to the Os–aqua (Os–OH2) and the Os–formate (Os–OOCH) adducts. A sharp singlet at ca. 7.9 ppm was attributed to the Os-bound formate (free formate appears at ca. 8.4 ppm), in accordance with analogous Ru(II) arene formate complexes (Fig. S34†).26,80 Although the species (η6-arene)Os–H in TH catalysis has been reported to appear around −4 ppm by 1H NMR,64 in our experiments the hydrido species was elusive.26,81 The intensity of the signals of free pyruvate (3H, singlet, 2.36 ppm) decreased and a new peak assignable to lactate (3H, doublet, 1.33 ppm) appeared. Compound 11 mediated total reduction of pyruvate to lactate after 18 h (Fig. S35†) whereas 13 barely achieved stoichiometric conversion after 2 days (Fig. S36A†). Since more than one mol equiv. of pyruvate was reduced per mol equiv. of osmium complexes 9, 11 and 12, a catalytic mechanism is implied.
We further explored the catalytic capabilities of complex 9, bearing the unsubstituted picolinate, by varying the pH as well as increasing the amount of H-source and the temperature. TONt and TOFmax data are compiled in Fig. S36B.† The most favourable pH in the range 3–7 was 5 (reaction almost 4× faster than at pH 3), which could be rationalized by considering that the pKa of formic acid is 3.75 and that the formation of the formate adduct holds a central role in the catalytic transformation.55,56 Lack of stability of pyruvate at low pH82,83 could be affecting the efficiency of our catalysts in acidic solutions. Both an increase in the amount of formate and of temperature resulted in higher TONt and TOF values as anticipated.
Complex 11, selected on the basis of its fast hydrolysis and catalytic performance was probed as a catalyst at three different temperatures (300, 310, and 320 K) and concentrations of hydride source (100, 200 and 400 mM sodium formate; Table 3). TOF values increased with temperature. For example, an increment of 20 K resulted in ca. 11-fold increase in TH rate. This is a similar increment to that reported for other Ru–arene complexes bearing N,O-chelates, in which a similar variation of temperature (increase of 23 K) resulted in an increment of 7-fold in the TOF value.84 Compound 11 has a maximum TOF value of 0.20 h−1 at pH 4 (Os/pyr/for; 1:2:100), which is comparable to pyruvate reduction by the reported 16-electron species [Os(η6-p-cymene)(TsDPEN)], for which the TOFmax in PBS is 1.5 h−1 (for Os/pyr/for, 1:200:400).85 The percentage of conversion from pyruvate to lactate after 1 h incubation shows that an increment of 10 K doubled the percentage of conversion (Fig. S37†).
| Compound | T (K) | HCOONa (mM) | TOFmax (h−1) | R 2 |
|---|---|---|---|---|
| 9 | 310 | 100 | 0.049 | 0.992 |
| 11 | 300 | 100 | 0.037 | 0.989 |
| 310 | 100 | 0.200 | 0.990 | |
| 320 | 100 | 0.417 | 0.985 | |
| 300 | 200 | 0.052 | 0.998 | |
| 300 | 400 | 0.061 | 0.983 | |
| 12 | 310 | 100 | 0.050 | 0.967 |
| 13 | 310 | 100 | 0.140 | 0.970 |
Complexes 9–13 are, to the best of our knowledge, the first 18-electron Os arene complexes to achieve catalytic TH of pyruvate. Such an activity, in conjunction with the high solubility in aqueous media and the protective nature of the dynamic tether ring formation, prompted us to investigate the potential of carrying out TH inside cells by selected complex 11.
Production of lactate inside cells
The majority of the Os(II) complexes reported here had no impact on cell survival of MDA-MB-231 and HCT116 cells up to a concentration of 200 μM (Fig. S38 and Table S5†), largely in agreement with data reported for similar Os–arenes in other cancer cell lines.12,49,52,64 Only complexes 11 and 11C appear to be an exception regarding cytotoxic potential, presenting the lowest IC50 value in the series in colon cancer cells HCT116 (IC50 values 30.5 ± 3.3 and 19.5 ± 0.6 μM, respectively). Intracellular accumulation experiments in MDA-MB-231 and HCT116 cells showed that uptake for 11C was high, as determined by ICP-MS (Table S6†). These results, together with the catalytic capabilities of 11 for transfer hydrogenation, encouraged us to probe this complex for osmium intracellular reactivity as a potential metabolite-modulating tool.We investigated whether direct detection of Os-mediated lactate production was possible inside cancer cells. One concern was the lack of structural compromise that could promote enantiomeric enrichment in the reduction of pro-chiral pyruvate by 11. Our complex is synthesized as a racemic mixture, and it is only reasonable to think that the product of the reaction, lactate, would also be obtained as a racemic mixture.
We first optimized a UV-vis-based assay for lactate determination, examining whether complex 11 is capable of producing a valid substrate for enzymatic quantification from pro-chiral pyruvate (Fig. S39†). We used in this experiment L-lactate deshydrogenase (L-LDH) as L-lactate is produced from pyruvate during anaerobic glycolysis and is present in humans at concentrations 100 times greater than D-lactate. L-Lactate generated by 11 was confirmed by reaction with L-LDH enzyme, a process accompanied by concomitant production of UV-vis-active NADH (Fig. S39†). As anticipated, osmium-produced lactate increased with increase in reaction time from 0 to 4 h in the presence of formate (Fig. S39B†).
Next we carried out lactate determination in cells. The choice of cell line to demonstrate whether it would be possible for 11 to produce measurable lactate inside cancer cells was carefully considered. MDA-MB-231 and MCF7 were selected on the basis of their growth media composition (non-pyruvate and pyruvate containing, respectively). The low cytotoxic effect of 11 in these cell lines (IC50 90.6 ± 4.0 and 105.1 ± 10.6 μM in MDA-MB-231 and MCF7, respectively) was also advantageous as it allowed us to expose the cells to a relatively high osmium concentration (300 μM). Pyruvate (2 mM) and formate (10 mM) were added to the MDA-MB-231 cells media whereas only formate (10 mM) was added to the MCF7 cells (media from commercial sources already contain 1 mM pyruvate). Controls were stimulated only with the osmium compound (we also included double-negative controls; detailed protocol in the ESI†). Upon exposing the drugs to 11 for either 12 or 24 h, the cells were washed, lysed, deproteinated and centrifuged. The lysate supernatants were then tested for L-lactate determination and quantification. The results are shown in Fig. 4. Lactate increases when cells are exposed to 11 and formate, in comparison to those exposed to osmium alone. The impact of formate and Os in lactate generation in MCF7 at 24 h is the most noticeable and the differences are significate (p-value < 0.01) when compared to the same experiment lacking formate, and despite being exposed to half of the amount of exogenous pyruvate (1 mM in MCF7 and 2 mM in MDA-MB-231). We tentatively attribute the different results in both cell lines to the readiness of MCF7 cells to internalize pyruvate, since it is a media component.
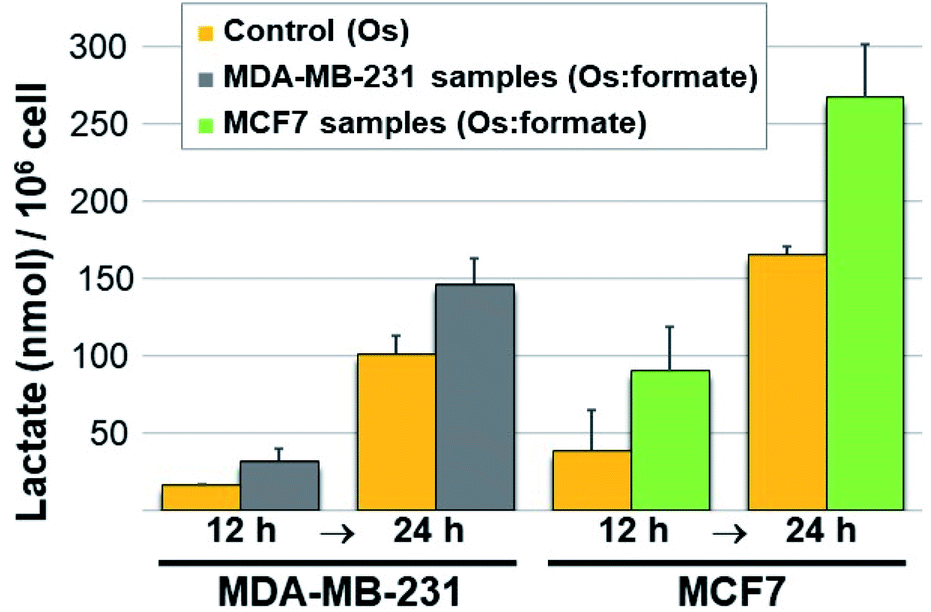 | ||
| Fig. 4 Lactate generated (nmol) per million cells determined in MDA-MB-231 and MCF7 breast cancer cell lines at 12 and 24 h. In yellow, lactate in cells exposed to 11 (lactate background level resulting from Os-stimulated cell metabolism only). In grey, lactate produced upon co-incubation of osmium, formate and pyruvate (0.3, 10, and 2 mM, respectively) in MDA-MB-231 cells. In green, lactate determined upon co-incubation of osmium, formate and pyruvate (0.3, 10, and 1 mM, respectively) in MCF7 cells. | ||
Our results indicate that the tether Os–arene complexes presented here can be exploited for the conversion of metabolites that are essential in the demanding metabolism of the cancer cell. Depletion of the enantiomeric excess of the hydrogenated product (racemic lactate as opposed to L-lactate) might also have an impact on intracellular activity and warrants further exploration. Additionally, artificial non-enzymatic TH of pyruvate inside cells can affect the activity of lactate dehydrogenase (LDH) in converting pyruvate to lactate and vice versa, which in turn is intimately ligated to the generation of NAD+ and NADH for a number of intracellular processes. An organometallic complex capable of modulating metabolites pyruvate/lactate (and consequently NADH/NAD+) can be a powerful intracellular tool to target the metabolic plasticity of the cancer cell.
Conclusions
Water-mediated processes in Os–arenes (such as conversion of the Os–chlorido bond into the Os–aqua bond) are either inefficient or tend to result in species with low reactivity. We have overcome this issue by introducing a pendant arm primed with a terminal alcohol functionality attached to the arene ligand. The η6-bound arene behaves now as hemilabile, with a weakly κ1-bound alcohol group. Strikingly, this ligand furnishes the Os complex with an entirely new reactivity profile. Osmium–arenes often generate Os–OH inactive species due to the acidity of the metal.52,53 In our compounds, the hydroxido adduct (Os–OH) triggers intramolecular rearrangement culminating in the binding of the pendant oxygen to the metal centre (formation of a closed tether complex), protecting the complex towards irreversible inactivation. The ligand exchange fluxionality of the hemilabile ligand thus ensures the kinetic reactivation of the metal centre. We reported here reversible formation of Os–arene tether complexes in water. Our results imply the unprecedented reactivation of the otherwise inert hydroxido Os(II)–arene complexes.Finding metallodrugs that can exert catalytic activity inside cells is a challenging task. Alcohol-tethered Os(II)–arene complexes have demonstrated to carry out transfer hydrogenation reactions inside cells. The combination of tether-ring protection towards excessive metal reactivity in the intracellular nucleophile-rich microenvironment (protection towards catalyst poisoning), their high-water solubility, and the readiness towards catalytic transformations when exposed to the appropriate sacrificial agent, makes them strong contenders as intracellular catalysts. Tumour cell metabolism is considered an exploitable vulnerability in cancer therapy,86 where lactate and pyruvate have central, yet not completely unveiled, roles.87 We have demonstrated that catalytic tethered Os–arenes can be extraordinary candidates for modulating such an aspect of cancer progression in foreseeable therapies.
As whether the pH-dependent aqueous dynamics of this new class of Os compounds will have an impact on the anticancer capabilities of these complexes, further work is necessary to understand such subtleties. Internal pH (pHi) in heterogeneous cancer cells varies in comparison to normal cells as a result of different stages of metabolic reprogramming, and also varies from tumour to tumour depending on malignity.63,88 We hypothesize that the cancer cell exceptional metabolism, which results in cytosolic and intra-organelle pH disruption will impact metallodrug speciation and thus tune its anticancer effect. In other words, different drugs will exert differing degrees of lethality to pre-selected tumours based on their pH-responsiveness, highlighting an additional structural advantage by the Os-tether design presented here.
Data availability
Crystallographic data for chlorido complexes 5·PF6, 8·PF6, 9, 10, 11, 13, and closed tether complexes 3C·PF6 and dichlorido [Os(η6:κ1-C6H5(CH2)3OH)Cl2] has been deposited at the Cambridge Crystallographic Data Centre under the accession numbers CCDC 2026997–2027004 and can be obtained from https://ccdc.cam.ac.uk. Additional data for this paper, including NMR, UV-vis and ICP-MS datasets, are available at IMDEA Nanociencia Repository at http://hdl.handle.net/20.500.12614/2629.Author contributions
A. M. P. conceived and designed the study. S. I.-T. and A. H. developed the synthetic methodology. S. I.-T. synthesized the complexes and carried out the aqueous studies. S. I.-T. and V. R.-F. carried out the experiments in cells. S. I.-T, V. R.-F. and A. M. P. analysed the data. S. I.-T., V. R.-F., A. H and A. M. P discussed the findings and contributed towards writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr J. Perles and Dr M. Ramírez (Universidad Autónoma de Madrid) for the analysis of X-ray data. Dr Z. Pardo and Dr A. Arnáiz and Ms C. Leis (IMDEA Nanociencia) are gratefully acknowledged for assistance with NMR experiments and cell culture techniques, respectively. We acknowledge funding from the EC (FP7-PEOPLE-2013-CIG, no. 631396), from the Spanish MINECO (RYC-2012-11231, CTQ2014-60100-R, SEV-2016-0686, and CTQ2017-84932-P), and the Comunidad Autónoma de Madrid (Scholarship PEJD-2016/IND-2608). Dr A. Habtemariam was supported by the Comunidad Autónoma de Madrid (Professorship of Excellence 2016-T3/IND-2054).References
- J. Reedijck, Chem. Commun., 1996, 801–806 RSC.
- L. Cubo, A. G. Quiroga, J. Zhang, D. S. Thomas, A. Carnero, C. Navarro-Ranninger and S. J. Berners-Price, Dalton Trans., 2009, 3457–3466, 10.1039/b819301k.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- A. G. Quiroga, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef CAS PubMed.
- S. Carboni, A. Zucca, S. Stoccoro, L. Maiore, M. Arca, F. Ortu, C. Artner, B. K. Keppler, S. M. Meier-Menches, A. Casini and M. A. Cinellu, Inorg. Chem., 2018, 57, 14852–14865 CrossRef CAS PubMed.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- H. Chen, J. A. Parkinson, O. Novakova, J. Bella, F. Wang, A. Dawson, R. Gould, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14623–14628 CrossRef CAS PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS PubMed.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed.
- W.-Y. Zhang, S. Banerjee, G. M. Hughes, H. E. Bridgewater, J.-I. Song, B. G. Breeze, G. J. Clarkson, J. P. C. Coverdale, C. Sanchez-Cano, F. Ponte, E. Sicilia and P. J. Sadler, Chem. Sci., 2020, 11, 5466–5480 RSC.
- H. Kostrhunova, J. Florian, O. Novakova, A. F. A. Peacock, P. J. Sadler and V. Brabec, J. Med. Chem., 2008, 51, 3635–3643 CrossRef CAS PubMed.
- S. H. van Rijt, A. F. A. Peacock, R. D. L. Johnstone, S. Parsons and P. J. Sadler, Inorg. Chem., 2009, 48, 1753–1762 CrossRef CAS PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743 CrossRef CAS PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- S. Alonso-deCastro, A. L. Cortajarena, F. López-Gallego and L. Salassa, Angew. Chem., Int. Ed., 2018, 57, 3143–3147 CrossRef CAS PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- C. A. Riedl, M. Hejl, M. H. M. Klose, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Dalton Trans., 2018, 47, 4625–4638 RSC.
- S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628–11633 CrossRef CAS PubMed.
- S. M. Valiahdi, A. E. Egger, W. Miklos, U. Jungwirth, K. Meelich, P. Nock, W. Berger, C. G. Hartinger, M. Galanski, M. A. Jakupec and B. K. Keppler, J. Biol. Inorg Chem., 2013, 18, 249–260 CrossRef CAS PubMed.
- A. Habtemariam, B. Watchman, B. S. Potter, R. Palmer, S. Parsons, A. Parkin and P. J. Sadler, Dalton Trans., 2001, 1306–1318, 10.1039/b009117k.
- T. J. Prior, H. Savoie, R. W. Boyle and B. S. Murray, Organometallics, 2018, 37, 294–297 CrossRef CAS.
- A. M. Pizarro, M. Melchart, A. Habtemariam, L. Salassa, F. P. A. Fabbiani, S. Parsons and P. J. Sadler, Inorg. Chem., 2010, 49, 3310–3319 CrossRef CAS PubMed.
- F. Martínez-Peña and A. M. Pizarro, Chem.–Eur. J., 2017, 23, 16231–16241 CrossRef PubMed.
- F. Martínez-Peña, S. Infante-Tadeo, A. Habtemariam and A. M. Pizarro, Inorg. Chem., 2018, 57, 5657–5668 CrossRef PubMed.
- A. C. Carrasco, V. Rodriguez-Fanjul and A. M. Pizarro, Inorg. Chem., 2020, 59, 16454–16466 CrossRef CAS PubMed.
- A. C. Carrasco, V. Rodriguez-Fanjul, A. Habtemariam and A. M. Pizarro, J. Med. Chem., 2020, 63, 4005–4021 CrossRef CAS PubMed.
- T. G. Scrase, M. J. O'Neill, A. J. Peel, P. W. Senior, P. D. Matthews, H. Shi, S. R. Boss and P. D. Barker, Inorg. Chem., 2015, 54, 3118–3124 CrossRef CAS PubMed.
- J. Molas Saborit, A. Caubet, R. F. Brissos, L. Korrodi-Gregorio, R. Perez-Tomas, M. Martinez and P. Gamez, Dalton Trans., 2017, 46, 11214–11222 RSC.
- J. C. Jeffrey and T. B. Rauchfuss, Inorg. Chem., 1979, 18, 2658–2666 CrossRef CAS.
- W. G. Rohly and K. B. Mertes, J. Am. Chem. Soc., 1980, 102, 7939–7940 CrossRef CAS.
- C. S. Slone, D. A. Weinberger and C. A. Mirkin, in Prog. Inorg. Chem., ed. K. D. Karlin, John Wiley & Sons. Inc., 1999, vol. 48, pp. 233–350 Search PubMed.
- B. Cetinkaya, S. Demir, I. Ozdemir, L. Toupet, D. Semeril, C. Bruneau and P. H. Dixneuf, Chem.–Eur. J., 2003, 9, 2323–2330 CrossRef CAS PubMed.
- T. J. Geldbach, G. Laurenczy, R. Scopelliti and P. J. Dyson, Organometallics, 2006, 25, 733–742 CrossRef CAS.
- N. Kaloglu, I. Ozdemir, N. Gurbuz, H. Arslan and P. H. Dixneuf, Molecules, 2018, 23, 647 CrossRef PubMed.
- Y. Miyaki, T. Onishi and H. Kurosawa, Inorg. Chim. Acta, 2000, 300, 369–377 CrossRef.
- G. Marconi, H. Baier, F. W. Heinemann, P. c. Pinto, H. Pritzkow and U. Zenneck, Inorg. Chim. Acta, 2003, 352, 188–200 CrossRef CAS.
- B. Lastra-Barreira, J. Díez, P. Crochet and I. Fernández, Dalton Trans., 2013, 42, 5412–5420 RSC.
- T. Stahl, H. F. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248–1251 CrossRef CAS PubMed.
- S. Wubbolt, M. S. Maji, E. Irran and M. Oestreich, Chem.–Eur. J., 2017, 23, 6213–6219 CrossRef PubMed.
- M. Draganjac, C. J. Rufflng and T. B. Rauchfuss, Organometallics, 1985, 4, 1909–1911 CrossRef CAS.
- R. Aznar, A. Grabulosa, A. Mannu, G. Muller, D. Sainz, V. Moreno, M. Font-Bardia, T. Calvet and J. Lorenzo, Organometallics, 2013, 32, 2344–2362 CrossRef CAS.
- J. W. Faller and P. P. Fontaine, Organometallics, 2005, 24, 4132–4138 CrossRef CAS.
- R. Gonzalez-Fernandez, P. Crochet and V. Cadierno, Dalton Trans., 2020, 49, 210–222 RSC.
- L. Rafols, S. Torrente, D. Aguilà, V. Soto-Cerrato, R. Pérez-Tomás, P. Gamez and A. Grabulosa, Organometallics, 2020, 39, 2959–2971 CrossRef CAS.
- X. Zhou, X. He, J. Lin, Q. Zhuo, Z. Chen, H. Zhang, J. Wang and H. Xia, Organometallics, 2015, 34, 1742–1750 CrossRef CAS.
- S. L. Dabb, B. A. Messerle, M. K. Smith and A. C. Willis, Inorg. Chem., 2008, 47, 3034–3044 CrossRef CAS PubMed.
- A. F. A. Peacock, A. Habtemariam, R. Fernandez, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- A. M. Pizarro, A. Habtemariam and P. J. Sadler, Top. Organomet. Chem., 2010, 32, 21–56 CrossRef CAS.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- A. F. A. Peacock, A. Habtemariam, S. A. Moggach, A. Prescimone, S. Parsons and P. J. Sadler, Inorg. Chem., 2007, 46, 4049–4059 CrossRef CAS PubMed.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS PubMed.
- R. J. Needham, H. E. Bridgewater, I. Romero-Canelón, A. Habtemariam, G. J. Clarkson and P. J. Sadler, J. Inorg. Biochem., 2020, 210, 111154 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam, A. F. A. Peacock and P. J. Sadler, J. Biol. Inorg Chem., 2006, 11, 483–488 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, P. C. A. Bruijnincx, A. Habtemariam, G. J. Clarkson, R. J. Deeth and P. J. Sadler, Organometallics, 2012, 31, 5958–5967 CrossRef CAS.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, A. Habtemariam, I. Romero-Canelon and P. J. Sadler, J. Inorg. Biochem., 2015, 153, 322–333 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- M. Martínez-Calvo and J. L. Mascareñas, Coord. Chem. Rev., 2018, 359, 57–79 CrossRef.
- S. Alonso de Castro, A. Terenzi, J. Gurruchaga Pereda and L. Salassa, Chem.–Eur. J., 2019, 25, 6651–6660 CrossRef CAS PubMed.
- S. Banerjee and P. J. Sadler, RSC Chem. Biol., 2021, 2, 12–29 RSC.
- G. Bergers and S.-M. Fendt, Nat. Rev. Cancer, 2021, 21, 162–180 CrossRef CAS PubMed.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- E. M. Bolitho, J. P. C. Coverdale, H. E. Bridgewater, G. J. Clarkson, P. D. Quinn, C. Sanchez-Cano and P. J. Sadler, Angew. Chem., Int. Ed., 2021, 60, 6462–6472 CrossRef CAS PubMed.
- A. Habtemariam, S. Betanzos-Lara and P. J. Sadler, in Inorganic Synthesis, ed. T. B. Rauchfuss, John Wiley & Sons, 2010, ch. 160-163 Search PubMed.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 CrossRef.
- T. Ohnishi, Y. Miyaki, H. Asano and H. Kurosawa, Chem. Lett., 1999, 28, 809–810 CrossRef.
- J. Čubrilo, I. Hartenbach, T. Schleid and R. F. Winter, Z. Anorg. Allg. Chem., 2006, 632, 400–408 CrossRef.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- S. H. van Rijt, A. J. Hebden, T. Amaresekera, R. J. Deeth, G. J. Clarkson, S. Parsons, P. C. McGowan and P. J. Sadler, J. Med. Chem., 2009, 52, 7753–7764 CrossRef CAS PubMed.
- T. Reiner, M. Waibel, A. N. Marziale, D. Jantke, F. J. Kiefer, T. F. Fässler and J. Eppinger, J. Organomet. Chem., 2010, 695, 2667–2672 CrossRef CAS.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- S. H. van Rijt, A. F. A. Peacock and P. J. Sadler, in Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy, ed. A. Bonetti, R. Leone, F. Muggia and S. B. Howel, Springer Science, 2009, pp. 73–79, DOI:10.1007/978-1-60327-459-3_10.
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 1845–1851 CrossRef CAS.
- Y. Hung, W.-J. Kung and H. Taube, Inorg. Chem., 1981, 20, 457–463 CrossRef CAS.
- F. Chen, J. J. Soldevila-Barreda, I. Romero-Canelón, J. P. C. Coverdale, J.-I. Song, G. J. Clarkson, J. Kasparkova, A. Habtemariam, V. Brabec, J. A. Wolny, V. Schünemann and P. J. Sadler, Dalton Trans., 2018, 47, 7178–7189 RSC.
- T. Koike and T. Ikariya, Adv. Synth. Catal., 2004, 346, 37–41 CrossRef CAS.
- J. M. Gichumbi, B. Omondi and H. B. Friedrich, Eur. J. Inorg. Chem., 2017, 2017, 915–924 CrossRef CAS.
- V. S. Griffiths and G. Socrates, Trans. Faraday Soc., 1967, 63, 673–677 RSC.
- J. Damitio, G. Smith, J. E. Meany and Y. Pocker, J. Am. Chem. Soc., 1992, 114, 3081–3087 CrossRef CAS.
- M. M. Haghdoost, J. Guard, G. Golbaghi and A. Castonguay, Inorg. Chem., 2018, 57, 7558–7567 CrossRef CAS PubMed.
- J. P. C. Coverdale, PhD thesis, University of Warwick, 2017.
- G. Kroemer and J. Pouyssegur, Cancer Cell, 2008, 13, 472–482 CrossRef CAS PubMed.
- I. San-Millán and G. A. Brooks, Carcinogenesis, 2017, 38, 119–133 Search PubMed.
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2026997–2027004. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01939b |
| This journal is © The Royal Society of Chemistry 2021 |