
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHit-optimization using target-directed dynamic combinatorial chemistry: development of inhibitors of the anti-infective target 1-deoxy-D-xylulose-5-phosphate synthase†
Ravindra P.
Jumde
 a,
Melissa
Guardigni
ad,
Robin M.
Gierse
abc,
Alaa
Alhayek
ab,
Di
Zhu
abc,
Zhoor
Hamid
ab,
Sandra
Johannsen
ab,
Walid A. M.
Elgaher
a,
Philipp J.
Neusens
ab,
Christian
Nehls
e,
Jörg
Haupenthal
a,
Norbert
Reiling
fg and
Anna K. H.
Hirsch
*ab
a,
Melissa
Guardigni
ad,
Robin M.
Gierse
abc,
Alaa
Alhayek
ab,
Di
Zhu
abc,
Zhoor
Hamid
ab,
Sandra
Johannsen
ab,
Walid A. M.
Elgaher
a,
Philipp J.
Neusens
ab,
Christian
Nehls
e,
Jörg
Haupenthal
a,
Norbert
Reiling
fg and
Anna K. H.
Hirsch
*ab
aDepartment of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS) – Helmholtz Centre for Infection Research (HZI), Campus Building E8.1, 66123 Saarbrücken, Germany. E-mail: Anna.Hirsch@helmholtz-hips.de
bDepartment of Pharmacy, Saarland University, Campus Building E8.1, 66123 Saarbrücken, Germany
cStratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
dD3-PharmaChemistry, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genoa, Italy
eRG Biophysics, Research Center Borstel, Leibniz Lung Center, Borstel, Germany
fRG Microbial Interface Biology, Research Center Borstel, Leibniz Lung Center, Borstel, Germany
gGerman Center for Infection Research (DZIF), Partner site Hamburg-Lübeck-Borstel-Riems, Borstel, Germany
First published on 28th April 2021
Abstract
Target-directed dynamic combinatorial chemistry (tdDCC) enables identification, as well as optimization of ligands for un(der)explored targets such as the anti-infective target 1-deoxy-D-xylulose-5-phosphate synthase (DXPS). We report the use of tdDCC to first identify and subsequently optimize binders/inhibitors of the anti-infective target DXPS. The initial hits were also optimized for their antibacterial activity against E. coli and M. tuberculosis during subsequent tdDCC runs. Using tdDCC, we were able to generate acylhydrazone-based inhibitors of DXPS. The tailored tdDCC runs also provided insights into the structure–activity relationship of this novel class of DXPS inhibitors. The competition tdDCC runs provided important information about the mode of inhibition of acylhydrazone-based inhibitors. This approach holds the potential to expedite the drug-discovery process and should be applicable to a range of biological targets.
Introduction
In dynamic combinatorial chemistry (DCC), compound libraries can be generated under thermodynamic control from a range of appropriate building blocks. The addition of external stimuli or targets can alter this thermodynamic equilibrium and change the composition of these libraries. In target-directed dynamic combinatorial chemistry (tdDCC), the target protein selects its binders and favors their formation over non-binders, resulting in their amplification in the library pool. Since the first report of tdDCC by Lehn and coworkers,1,2 it has emerged as a powerful tool to identify new ligands of biological targets.3–9 This self-screening approach reduces the synthetic efforts given that only the amplified hits need to be synthesized. Ultimately, it results in acceleration of the discovery of hit compounds in the early stages of drug discovery.Discovered in 1993, the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway is an alternative biosynthetic route to generate the universal isoprenoid precursors and corresponding essential metabolites for cell survival.10 The enzyme 1-deoxy-D-xylulose-5-phosphate synthase (DXPS) catalyzes the first, rate-limiting step for the production of isoprenoid precursors of this pathway. It is also involved in the biosynthesis of thiamine (vitamin B1) and pyridoxal (vitamin B6) in bacteria.11,12 The absence of the MEP pathway in humans makes it a promising new target for the development of novel medicines against many life-threatening diseases like tuberculosis and malaria. Despite substantial efforts dedicated to the discovery of inhibitors for DXPS, to date, very few active compounds are known, which fulfill the requirements as an ideal candidate for further development.13–17 The scarcity of inhibitors and X-ray crystal structures of the anti-infective target DXPS makes tdDCC a particularly attractive approach for the discovery of new inhibitors.
To date, the use of tdDCC in drug discovery has been limited to the initial hit-identification process. We decided to explore the possibility of using tdDCC both for hit-identification and hit-optimization. In the traditional medicinal-chemistry approach of hit-optimization, iterative cycles of ‘design–synthesis–test–analysis’ are carried out until the suitable candidate for further development has emerged.18 This process involves cumbersome synthetic campaigns, followed by purification, characterization, and biological evaluation of every candidate. In tdDCC, the target selects and amplifies its best binders/inhibitors from the library. Thus, it can be used for further optimization of the initial hits in subsequently tailored libraries inspired by the initial hits. Moreover, as only amplified hits need to be synthesized and analyzed for their biological activities, this approach has the potential to reduce the time and cost associated with the traditional workflow of hit-optimization.
Results and discussion
For this study, we used acylhydrazone formation as a reversible reaction of choice and DXPS enzyme from Deinococcus radiodurans (drDXPS) as a target protein. Considering the stability issues of the DXPS protein at lower pH and the slow acylhydrazone formation at neutral pH, the use of aniline as a nucleophilic catalyst was required.8,19 The template effect of drDXPS on the dynamic combinatorial library (DCL) was observed by comparing it with the blank DCC experiment (without protein) using LC-MS/MS analysis.Design of the initial dynamic combinatorial library
The choice of the first DCL was based on structural similarities of building blocks with the cofactor thiamine diphosphate (ThDP) of the enzyme DXPS (Fig. 1a). We selected three different aldehydes containing pyrimidine and pyridine moieties and seven different hydrazides mostly featuring various heterocyclic cores. The docking study of the 21 possible acylhydrazones products from DCL-1 using LeadIT20 and SeeSAR21 showed that they are accommodated in the ThDP binding pocket and occupy a part of the substrate binding pocket of the enzyme drDXPS (Fig. 1 and ESI, Fig. 1–6†).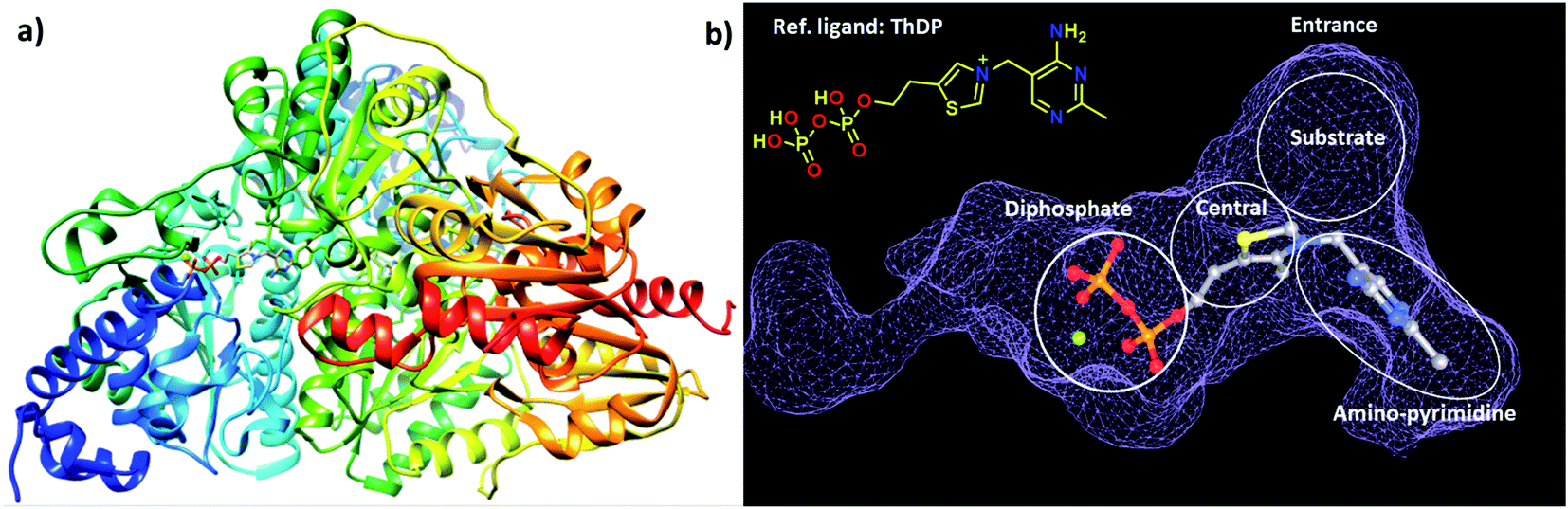 | ||
| Fig. 1 (a) Crystal structure of drDXPS (PDB code: 2O1X) this image is created using UCSF Chimera.30 (b) active site of drDXPS with bound cofactor thiamine diphosphate (ThDP) and the unoccupied substrate binding pocket, this image is created using online DoGSiteScorer tool.31 | ||
tdDCC-1
We performed the first DCC experiment by reacting three aldehydes (A1–A3) with seven hydrazides (H1–H7) in phosphate buffer (pH 6.25) with an excess of aniline and 5% DMSO (Fig. 2a). We evaluated the composition of DCL-1 in the blank reaction after it had reached equilibrium; after 6 h, no considerable changes in the relative peak areas (RPAs) of products were visible (Fig. 2b). A comparison of an adaptive DCC experiment in presence of drDXPS protein (40 mol%) with the blank DCL-1 revealed the amplification of five acylhydrazones (1–5) (Fig. 2c and ESI, Fig. 8†). Subsequently, we evaluated the effect of protein concentration on the amplification of these hits in an adaptive DCL, where 20 mol% of drDXPS were used, and the effect of pre-equilibration, where drDXPS (20 mol%) was added to the pre-equilibrated library (after 6 h). The systematic effect of protein concentration on the amplification is rarely studied,22,23 as the use of a large amount of protein is often considered necessary for tdDCC. Comparison of the composition of these DCLs with the blank DCLs still revealed the template effect, although the amplification of all five hits (1–5) was substantially reduced when 20 mol% of drDXPS were used as compared to 40 mol% of drDXPS (Fig. 2e and ESI, Fig. 9†). When drDXPS (20 mol%) was added to the pre-equilibrated library, and the composition of the DCL was checked after an additional 6 h, the amplification of these hits was further reduced. Nonetheless, in these three experiments, all five acylhydrazones (1–5) were amplified (Fig. 2e and ESI, Fig. 10†). These data suggest that even a minute amount of the protein drDXPS can alter the equilibrium of the library and induce the template effect. These findings are particularly important for future DCC experiments with precious proteins.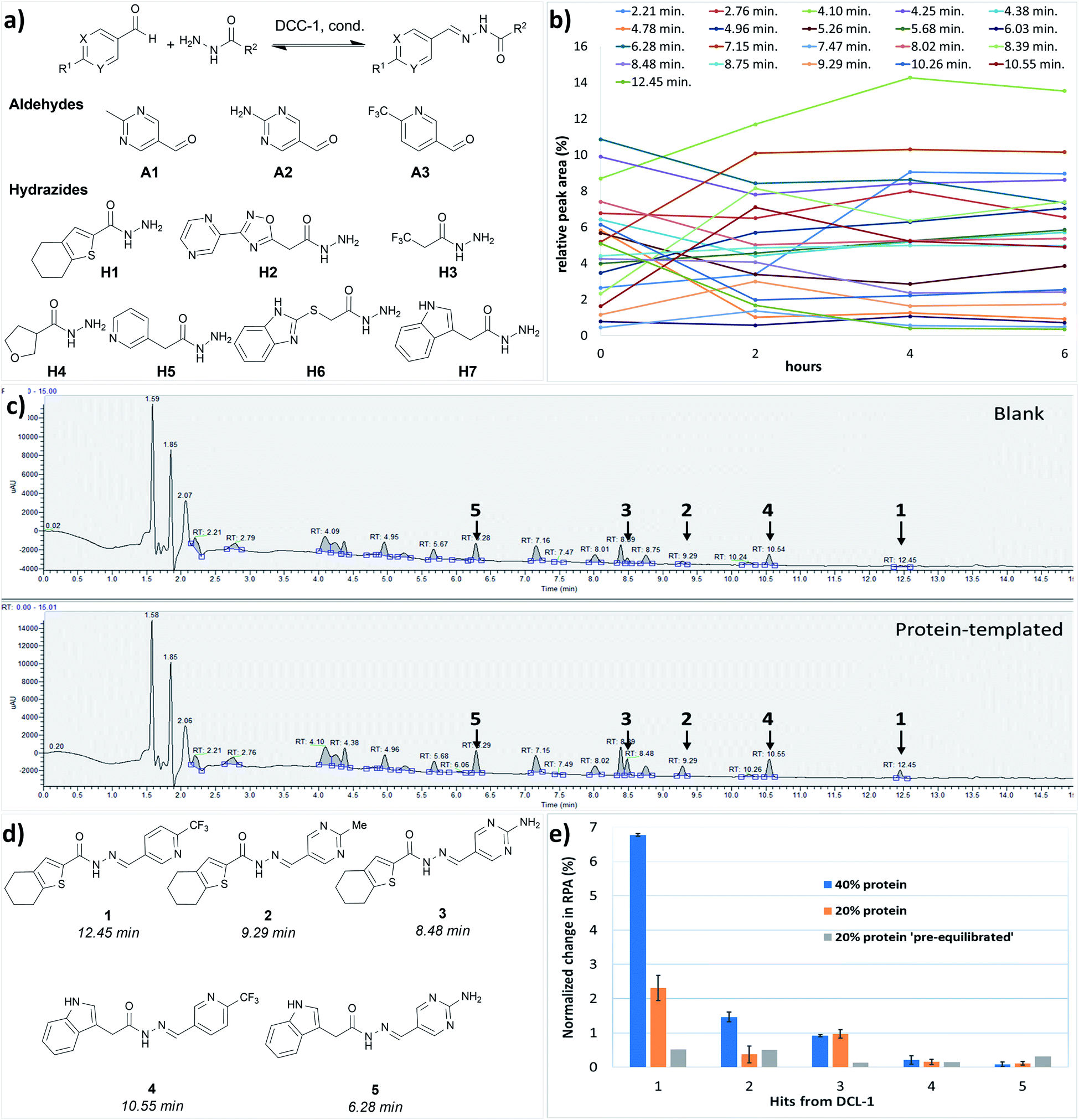 | ||
| Fig. 2 (a) Dynamic combinatorial library-1, conditions: experiment was run in phosphate buffer (pH 6.25) and 5% DMSO, aldehydes (100 μM each, in DMSO), hydrazides (300 μM each, in DMSO), aniline (10 mM in DMSO), drDXPS protein (20–40 μM in phosphate buffer); (b) evaluation of the equilibrium state of DCL-1, lines represent the formation of 21 acylhydrazone products over time; (c) UV-chromatogram of the blank and protein-templated adaptive DCL (40 mol% protein), see ESI, Fig. 8d and e† for peaks associated with different acylhydrazones; (d) amplified acylhydrazone hits (1–5) in DCL-1 and other experiments; (e) effect of protein concentration in the adaptive DCL run in duplicate and pre-equilibration on amplification of hits. | ||
Biological evaluation of hits from tdDCC-1
We synthesized all five hits (1–5) from the tdDCC-1 and evaluated their binding affinities for drDXPS and the truncated homolog from Mycobacterium tuberculosis (ΔmtDXPS)24 using surface plasmon resonance (SPR, Table 1). The five hits showed moderate binding affinity (KD), ranging from 55–270 μM for drDXPS and 40–250 μM for ΔmtDXPS. Interestingly, the trend of binding affinities of these hits mostly correlates with their amplification in the tdDCC. Furthermore, we evaluated the inhibitory activity of these hits against drDXPS and the homologous target in M. tuberculosis mtDXPS, which showed a similar trend, the most amplified hit was the most active one (1, IC50: drDXPS = 51 ± 3 μM, Table 1, entry 1). Unfortunately, none of these hits showed considerable antibacterial activity against Escherichia coli TolC (<25% inhibition at 50 μM), which may be ascribed to permeability issues. Nonetheless, using tdDCC, we were able to find a new class of inhibitors of the anti-infective target DXPS, which provides a good starting point for further optimization. Instead of the traditional medicinal-chemistry approach, we employed tdDCC for this purpose, letting the target protein DXPS choose its best binders.| Entry | Compound | K D (μM) | IC50 (μM)/inhib. (%) | MIC (μM)/inhib. (%)E. coli TolC | ||
|---|---|---|---|---|---|---|
| drDXPS | ΔmtDXPS | drDXPS | mtDXPS | |||
| a Inhibition of drDXPS was determined at a concentration of 120 μM. Inhibition of mtDXPS was determined at 200 μM. n.d. = not determined. n.i. = no inhibition. Percent (%) growth inhibition of E. coli TolC was determined at 50 μM. | ||||||
| 1 | 1 | 70 ± 5 | 40 ± 10 | IC50: 51 ± 3 | IC50: 78 ± 3 | n.i. |
| 2 | 2 | 55 ± 5 | 40 ± 10 | Inhib.: 31 ± 4 | IC50: 71 ± 1 | Inhib.: 24 ± 0 |
| 3 | 3 | 64 ± 2 | 40 ± 10 | Inhib.: 15 ± 9 | n.d. | n.i. |
| 4 | 4 | n.d. | 80 ± 10 | n.i. | Inhib.: 25 ± 5 | n.d. |
| 5 | 5 | 270 ± 40 | 250 ± 20 | Inhib.: 14 ± 23 | Inhib.: 47 | Inhib.: 18 ± 12 |
tdDCC-2
We assumed that using the common structural motifs from the first hits as building blocks in the second tdDCC, along with new aldehyde and/or hydrazide counterparts would provide better chances of identifying improved hits. From the initial hits (1–5), tetrahydrobenzothiophene and indole emerged as two privileged structural motifs, which are present in hits 1–3 and hits 4 and 5, respectively. We wanted to investigate the effect of inverting the position of these two groups in the acylhydrazone. To do so, we included their corresponding aldehydes A4 and A5 in DCL-2 instead of hydrazides H1 and H7. In DCL-2, along with A4 and A5, we included A6 as an additional aldehyde to extend the structural diversity of the possible hits, and four new hydrazides H8–H11 along with the three old ones (H2, H5, and H6, Fig. 3a). To improve the protein stability (see ESI, Table 1†) in the tdDCC conditions, we decided to use near-neutral pH from the second tdDCC experiments onwards. Analysis of DCL-2 revealed the amplification of five new hits (6–10, Fig. 3b and ESI, Fig. 12†).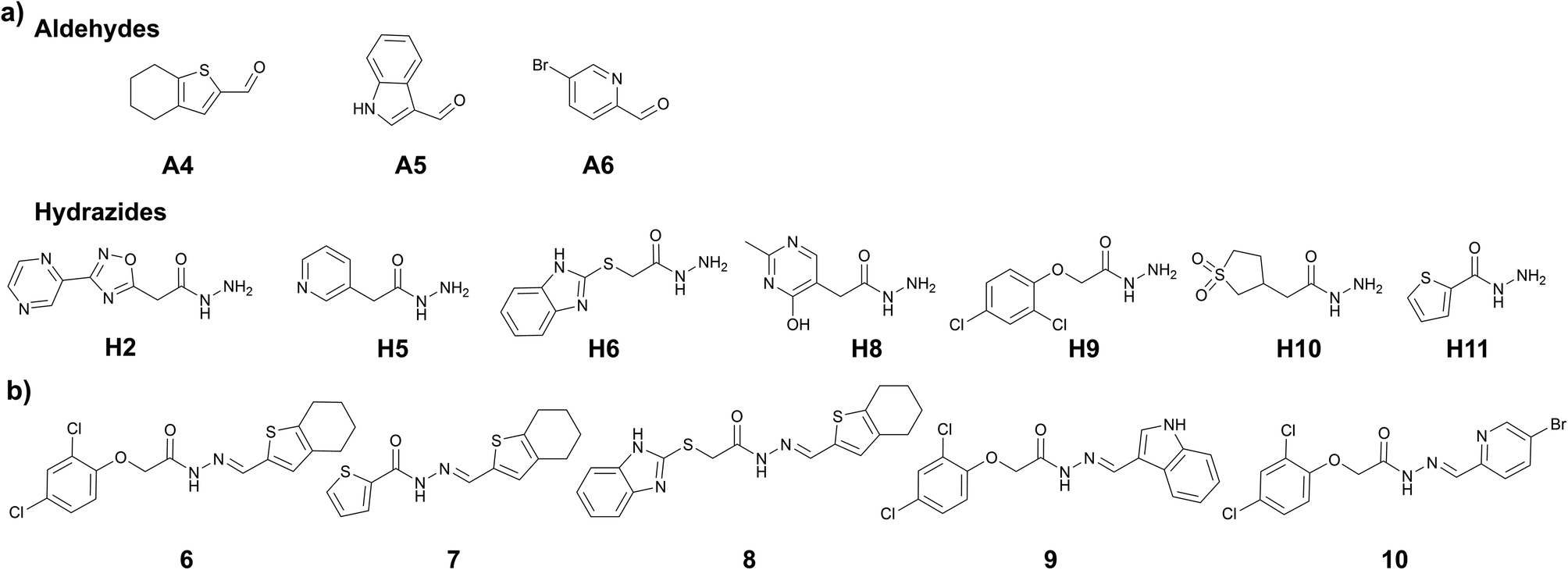 | ||
| Fig. 3 (a) DCC-2, building blocks, conditions: experiment was run in phosphate buffer (pH 7.04) and 5% DMSO, aldehyde (100 μM each, in DMSO), hydrazides (300 μM each, in DMSO), aniline (10 mM in DMSO), drDXPS protein (40 μM in phosphate buffer); (b) amplified acylhydrazone hits in DCL-2. | ||
Biological evaluation of hits from tdDCC-2
We synthesized all five new hits (6–10) and evaluated their binding affinities for drDXPS and ΔmtDXPS by SPR (Table 2). The KD values of these five hits were in the range of 7–150 μM for drDXPS and 13–230 μM for ΔmtDXPS, a significant increase compared to the first five hits from tdDCC-1. However, when tested against drDXPS and mtDXPS, these new hits (6–10) showed no significant improvement in their inhibitory activity. The most amplified hit 6 was most active against drDXPS (IC50 = 101 ± 15 μM) and hit 7 was the most active against mtDXPS (65 ± 4% inhibition at 200 μM). Interestingly, hits (7–10) from DCC-2 showed an improvement in antibacterial activity against E. coli TolC (21–100% growth inhibition at 50 μM) compared to the hits from DCC-1. The most active compound 9 showed an MIC value of 19 ± 7 μM. These improvements in binding affinity and antibacterial activity confirmed our hypothesis that tdDCC can be used for the optimization of inhibitors/hits.| Entry | Compound | K D (μM) | IC50 (μM)/inhib. (%) | MIC (μM)/inhib. (%) E. coli TolC | ||
|---|---|---|---|---|---|---|
| drDXPS | ΔmtDXPS | drDXPS | mtDXPS | |||
| a Inhibition of drDXPS was determined at a concentration of 120 μM. Inhibition of mtDXPS was determined at 200 μM. * = inhibition of mtDXPS was determined at 120 μM. n.d. = not determined. Percent (%) growth inhibition of E. coli TolC was determined at 50 μM. MIC values were determined only for the best compounds. # = growth inhibition of E. coli TolC was determined at 25 μM. | ||||||
| 1 | 6 | n.d. | 21 ± 3 | IC50: 101 ± 15 | Inhib: 46 ± 3* | n.i.# |
| 2 | 7 | 40 ± 10 | 13 ± 3 | Inhib: 37 ± 9 | Inhib: 65 ± 4* | Inhib: 57 ± 2 |
| 3 | 8 | n.d. | 230 ± 20 | Inhib: 13 ± 8 | Inhib: 44 ± 2* | Inhib: 73 ± 6 |
| 4 | 9 | 150 ± 30 | 140 ± 50 | Inhib: 38 ± 4 | Inhib: 15 ± 3 | MIC: 19 ± 7 |
| 5 | 10 | 7 ± 1 | n.d. | Inhib: 14 ± 12 | Inhib: 32 ± 6 | Inhib: 21 ± 7 |
tdDCC-3 & -4
To further support our hypothesis, we performed another two tailored tdDCC runs using the common structural motifs from the potent hits from DCL-2. We selected thiophenyl from hit 7 and 2,4-dichlorophenyl from hit 9 and included the corresponding hydrazides H11 and H12 in the next tdDCC-3 (Fig. 4). We also sought to unravel the influence of flexible linkers on the activity of the hits and thus included H12. In tdDCC-3 along with hydrazides H11 and H12, we selected 22 commercially available aldehydes (A1, A2, A7–A26, see Fig. 4a and b). For operational simplicity, we divided tdDCC-3 into two groups tdDCC-3a and tdDCC-3b, where two hydrazides H11 and H12 were reacted with two separate groups of eleven aldehydes each (Fig. 4a and b).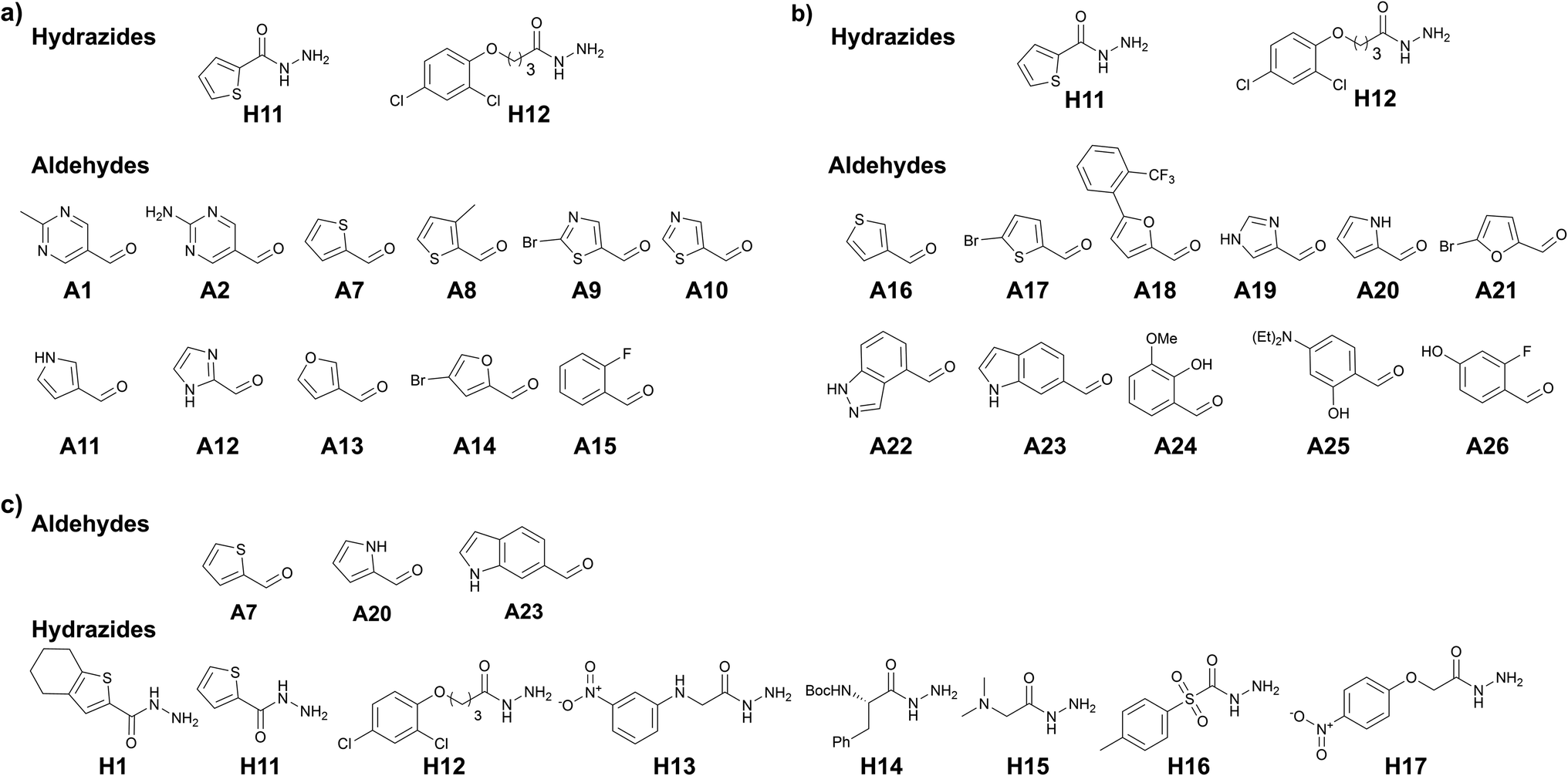 | ||
| Fig. 4 (a) DCC-3a, building blocks; (b) DCC-3b, building blocks, conditions: experiment was run in phosphate buffer (pH 7.04) and 5% DMSO, aldehyde (100 μM each, in DMSO), hydrazides (2000 μM each, in DMSO), aniline (10 mM in DMSO), drDXPS protein (40 μM in phosphate buffer); (c) DCC-4, building blocks, conditions: experiment was run in phosphate buffer (pH 7.04) and 5% DMSO, aldehyde (100 μM each, in DMSO), hydrazides (300 μM each, in DMSO), aniline (10 mM in DMSO), drDXPS protein (40 μM in phosphate buffer). | ||
For tdDCC-4, another common structural motif (indolyl) from the active hits of DCC-1 and DCC-2 was included as a building block (Fig. 4c). Here, we used 1H-indole-6-carbaldehyde (A23) instead of 1H-indole-3-carbaldehyde (A5) to understand the effect of substitution patterns of ligands on the binding to the protein and eventually in the activity of the acylhydrazones. Similarly, to understand the effect of inverting the position of the thiophenyl moiety in the acylhydrazone, we included the corresponding aldehyde A7. Along with these two aldehydes we also included 1H-pyrrole-2-carbaldehyde (A20) and eight hydrazides, including five new hydrazides (H13–H17). Here, using DCC, and including a range of heterocyclic building blocks to cover a wide chemical space with good structural diversity, we aimed at studying to some extent the traditional medicinal chemistry approach driven by structure–activity relationships. Contrary to the traditional medicinal chemistry approach, where all possible 68 acylhydrazone products from these three DCLs should be synthesized and tested for their biological activity, we let the protein select its best binders and synthesized only the amplified derivatives.
The analysis of these target-directed DCLs revealed the amplification of ten hits each for DCL 3a and 3b and nine hits for DCL 4 (Fig. 5, and ESI, Fig. 14, 16, and 18). Carefully inspecting the hits from all three DCLs revealed that the 2,4-dichlorophenyl motif and five-membered heterocycles (thiophene, furan, imidazole, and pyrrole) are favored. As hypothesized, the use of 1H-indole-6-carbaldehyde instead of 1H-indole-3-carbaldehyde was well-tolerated given that five hits containing this structural motif were amplified in these DCLs (26, 31, 32, 35, and 37). Moreover, several hits containing different phenyl rings (20, 21, 23, 25, and 30) were also amplified.
 | ||
| Fig. 5 (a) Amplified acylhydrazone hits in DCL-3a; (b) in DCL-3b; (c) in DCL-4. | ||
Biological evaluation of hits from tdDCC-3a, -3b and -4
We decided to synthesize a total of eleven out of 29 hits, based on their representative class (five-membered and bicyclic heterocycles, functionalized phenyl rings, and chiral compounds) and/or their amplification in the DCL and evaluated their binding affinity for drDXPS and ΔmtDXPS by SPR (Table 3). Most of the hits show substantial improvements in binding affinity compared to the hits from the first and second round of tdDCC (Table 3). Hits 11, 12, 21, 26, and 37 showed single-digit micromolar affinities (2–8 μM) and hits 22, and 35 displayed binding affinity of 15 ± 3 μM and 32 ± 3 μM against drDXPS, respectively. Similarly, the binding affinities of these hits were also improved for ΔmtDXPS, compound 22 which showed a single-digit micromolar affinity (KD = 6 ± 1 μM), and compound 21, 25, and 37 showed binding affinities in the range of 18–25 μM. When we evaluated these eleven hits for their inhibitory activities, six compounds (22, 23, 25, 26, 35, and 37) emerged as moderate inhibitors of drDXPS (40–62% activity inhibition at 120 μM) similar to the hits from DCL-1 and -2, out of which compound 23 showed the best IC50 value of 34 ± 4 μM. In general, these hits turn out to be less potent inhibitors of mtDXPS and compound 35 showed the highest inhibition value of 30 ± 1% at 120 μM in the series. However, five hits (21, 23, 25, 26, and 35) showed improved antibacterial activities against E. coli (TolC), out of which compound 26 and 35 featured the best MIC values (MIC = 14 ± 4 μM and MIC = 16 ± 5 μM, respectively). Overall, the hits from the last three rationally designed DCLs showed significant improvements in the binding affinity and antibacterial activity compared to the hits from tdDCC-1 and tdDCC-2 and compound 23 emerged as the most potent inhibitor of drDXPS.| Entry | Compound | K D (μM) | IC50 (μM)/inhib. (%) | MIC (μM)/inhib. (%) E. coli TolC | ||
|---|---|---|---|---|---|---|
| drDXPS | ΔmtDXPS | drDXPS | mtDXPS | |||
| a Inhibition of drDXPS was determined at a concentration of 120 μM concentration of compounds. Percent (%) growth inhibition of E. coli TolC was determined at 50 μM. MIC values were determined only for the best compounds. n.d. = not determined. n.i. = no inhibition. | ||||||
| 1 | 11 | 8 ± 2 | 90 ± 30 | Inhib.: 24 ± 10 | Inhib.: 16 ± 4 | n.i. |
| 2 | 12 | 2 ± 1 | 60 ± 20 | Inhib.: 33 ± 16 | Inhib.: 9 ± 11 | Inhib.: 30 ± 17 |
| 3 | 16 | n.d. | n.d. | Inhib.: 4 ± 9 | n.i. | Inhib.: 28 ± 17 |
| 4 | 21 | 3 ± 1 | 25 ± 8 | Inhib.: 6 ± 14 | Inhib.: 11 ± 9 | MIC: 70 ± 14 |
| 5 | 22 | 15 ± 3 | 6 ± 1 | Inhib.: 47 ± 10 | n.i. | n.i. |
| 6 | 23 | 160 ± 50 | 160 ± 40 | IC50: 34 ± 4 | n.i. | MIC: 45 ± 0 |
| 7 | 24 | 260 ± 80 | 250 ± 60 | Inhib.: 14 ± 14 | n.i. | Inhib.: 67 ± 23 |
| 8 | 25 | 90 ± 20 | 20 ± 4 | Inhib.: 45 ± 10 | n.i. | MIC: 60 ± 14 |
| 9 | 26 | 5 ± 2 | 110 ± 40 | Inhib.: 58 | Inhib.: 5 ± 1 | MIC: 14 ± 4 |
| 10 | 35 | 32 ± 8 | 60 ± 20 | Inhib.: 62 ± 6 | Inhib.: 30 ± 1 | MIC: 16 ± 5 |
| 11 | 37 | 3 ± 1 | 18 ± 3 | Inhib.: 40 ± 10 | Inhib.: 3 ± 6 | Inhib.: 60 ± 24 |
Antitubercular activity of DCC hits
We further evaluated the antitubercular (anti-TB) activity of 14 selected hit compounds from the three rounds of DCC (Fig. 6). These hit compounds were selected based on their inhibitory potency (1, 2, 6, 7, 23), binding affinities (11, 12, 21, 22, 26, 37), and antibacterial activity against E. coli TolC (9, 23–26, 35). Prior to anti-TB activity tests, dynamic light scattering (DLS) analyses were performed to establish whether the addition of the compounds to complex test culture media would lead to aggregate formation and subsequent precipitation, which would prevent functional analyses in biological systems. The DLS analyses revealed that a total of six compounds showed a concentration-dependent and up to 20–25 fold increase in the derived count rate, which was used as a parameter to measure aggregate formation in our test culture media. This allowed the determination of the highest sample concentration at which the respective compound was not yet present with an increased degree of aggregate formation (ESI, Fig. 194†). Based on these data we performed a head-to-head comparative analysis of anti-TB activity for all 14 compounds at 16 μM. These analyses identified compounds 7 and 35 from the second and third round of DCC, respectively, as the most active of the 14 compounds tested (Fig. 6). As both compounds did not induce aggregate formation when added at higher concentrations, MIC values could be determined to be 64 μM for 35 and slightly above 64 μM for 7 (ESI, Fig. 195 and 196†).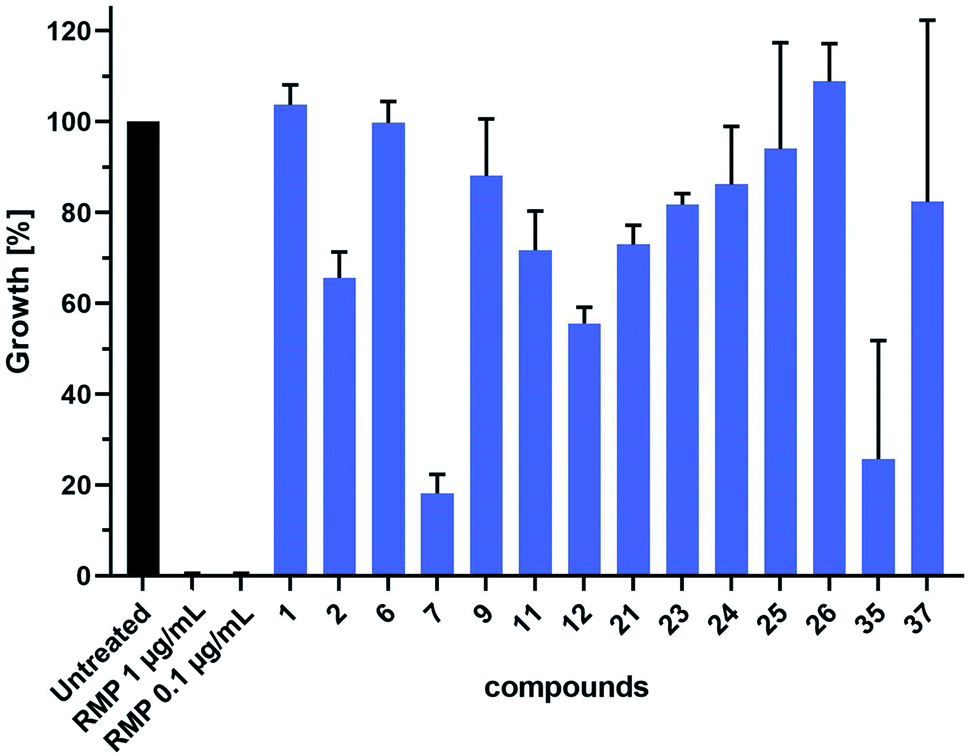 | ||
| Fig. 6 Head-to-head comparative analysis of the antiTB activity of DCC hits. mCherry-expressing M. tuberculosis H37Rv bacteria were cultured in the absence or presence of DCC hits (16 μM) or rifampicin (RMP, pos. ctrl). Shown are data of two independent experiments performed in triplicates. | ||
As the DLS data suggested that six compounds showed some degree of aggregation, we decided to use Triton X-100 or Tween20 in the enzyme-activity assay and binding assay respectively, to rule out any non-specific inhibition of the enzyme activity or binding to the protein by colloidal aggregates.
Mode-of-inhibition studies
To obtain information about the binding site of the inhibitors, we selected representative compound 1 for the mode-of-inhibition studies. We performed competition enzyme activity assays, where inhibition of drDXPS was measured with varying substrate and cofactor concentrations (curves are shown in ESI, Fig. 191–193†).The concentration of cofactor ThDP had a strong influence on the inhibitory activity, shifting the IC50 value of compound 1 from 35 ± 10 μM at 0.3 μM ThDP to only 20% inhibition of drDXPS by compound 1 at 5 μM ThDP. Pyruvate concentrations showed a similar effect, increasing the IC50 value from 19 ± 5μM at 0.1 mM pyruvate to 86 ± 15 μM at 0.4 mM and just 32% inhibition at the highest concentration of 0.6 mM. Concerning D/L-GAP, no influence of the substrate concentration on inhibition could be observed, all measured IC50 values are in the range of 50 μM. These first findings show that compound 1 is ThDP- and pyruvate-competitive and non-competitive with respect to D/L-GAP.
In the first step of the reaction catalyzed by DXPS, ThDP and pyruvate are forming a covalent intermediate in the active site, while the binding of D/L-GAP to its charged pocket is accelerated 600-fold after the formation of this covalent intermediate.25 A compound binding to the ThDP binding site is likely to also occupy parts of the pyruvate pocket, as both are located adjacent to each other. To bind to the cationic binding site of D/L-GAP, compounds with negatively charged functional groups, enabling ionic interactions, would be beneficial. The type of inhibition characteristics observed for compound 1 fits well with the initial docking study of acylhydrazones products from DCL-1 in the ThDP binding site (ESI, Fig. 1–6†). In particular, the docked compound 1 is accommodated well in the ThDP binding pocket and occupies a part of the substrate pocket (Fig. 7).
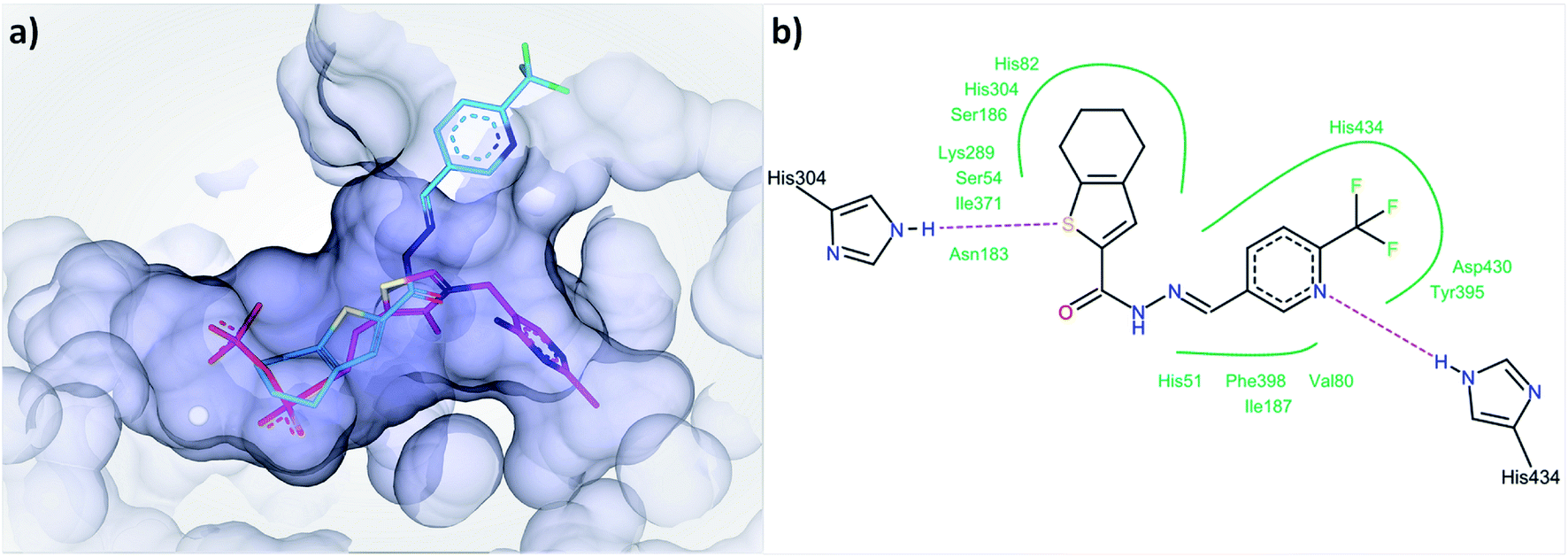 | ||
| Fig. 7 (a) Docked binding mode of compound 1 (cyan) compared to ThDP (pink) in the active site of drDXPS (PDB code: 2O1X), this figure is generated using SeeSAR 10.1 (BioSolveIT),21 (b) interaction of compound 1 with the residues lining the active site of drDXPS, this figure is generated with Pose View as implemented in the LeadIT suite.20 | ||
The reported inhibitor butylacetylphosphonate (BAP), containing an acetylphosphonate mimic of pyruvate,26 binds in the active site of DXPS and forms a covalent complex with the cofactor ThDP and extends into the substrate pocket of the active site (for the active site of drDXPS, see Fig. 1b).27 The presence of ThDP and/or BAP in the protein-templated DCL can influence the amplification of acylhydrazones negatively if they are ThDP- and substrate-competitive inhibitors.
To provide support for this notion, two competition DCC experiments using DCL-1 were performed in the presence of ThDP (DCC-5) and ThDP + BAP (DCC-6). The composition of tdDCC-5 and -6 was analyzed (ESI, Fig. 19 and 20†) and compared with the composition of tdDCC-1 (Fig. 8). The addition of ThDP leads to a considerable decrease in the amplification of compounds 1, 2, 4, and 5, while compound 3 showed no change in the amplification (tdDCC-5, Fig. 8). Comparing the results from tdDCC-6 (ThDP + BAP), with tdDCC-1 and tdDCC-5, we observed a further decrease in amplification of compounds 1 and 2, while compound 3 and 5 showed a minor or no decrease in amplification, and compound 4 showed a slight increase in amplification (Fig. 8). These findings suggest that most of the identified hits from DCL-1 compete with ThDP and substrate(s) and supports the results from the MOI study for compound 1.
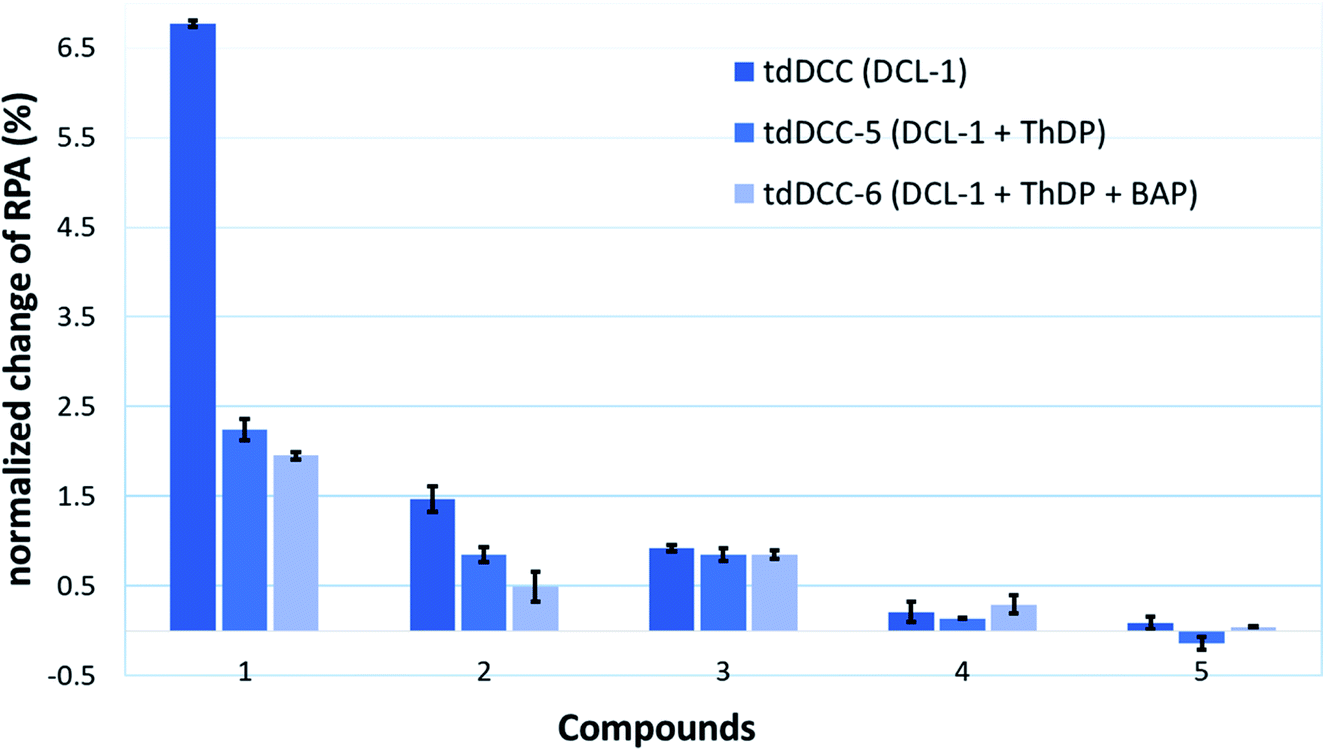 | ||
| Fig. 8 Comparison of amplification of acyhydrazones in tdDCC-1, -5, and -6. | ||
Bioisosteric replacement of the acylhydrazone moiety
Next, we designed and synthesized amide and 1,3,4-oxadiazoles as potential bioisosteres of the acylhydrazones 1 and 23. As compared to acylhydrazones, amides are known to be less prone to hydrolysis and subsequent liberation of potentially toxic hydrazides.28 Moreover, the milder and simple conditions required for the synthesis of amides make them ideal candidates as bioisosteres of acylhydrazones. Similarly, structurally more rigid and hydrolytically stable 1,3,4-oxadiazoles can directly be synthesized from the building blocks of acylhydrazone formation. We synthesized amide bioisosteres 38 and 39 in moderate yield, using peptide-coupling conditions (see ESI†). The bioisosteres 40 and 41 featuring a 1,3,4-oxadiazole linker were prepared by condensation of hydrazide with the corresponding carboxylic acid followed by in situ dehydrative cyclizations of the resulting diacyl hydrazide intermediate in moderate to good yields (see ESI†).29The biological evaluation of these four bioisosteres (Table 4) reveals that amide 38 retains the affinity of the parent acylhydrazone 1 for drDXPS (KD = 40 ± 10 μM vs. 70 ± 5 μM) and ΔmtDXPS (KD = 60 ± 20 μM vs. 40 ± 10 μM). The amide bioisostere 39 also showed binding to drDXPS (KD = 290 ± 30 μM) and ΔmtDXPS (KD = 260 ± 20 μM), although with an almost 2- and 1.5-fold decrease in binding affinities, respectively as compared to the parent acylhydrazone 23 (drDXPS: KD = 160 ± 50 μM and ΔmtDXPS: KD = 160 ± 40 μM). Regarding the enzymatic activity, both amide bioisosteres 38 and 39 showed a reduction in activity (Table 4, entries 1 and 2) compared to the parent acylhydrazones 1 and 23, respectively. The 1,3,4-oxadiazole bioisostere 40 lost affinity and activity completely for DXPS enzymes (Table 4, entry 3), which could be attributed to a more rigid structure as compared to the parent acylhydrazone 1. However, this dramatic loss of activity was not observed for a more flexible 1,3,4-oxadiazole bioisostere 41, which shows a similar affinity as the amide bioisostere 39 (Table 4, entry 4). Interestingly, the 1,3,4-oxadiazole bioisostere 41 showed better inhibitory potency against drDXPS as compared to its amide counterpart (inhib.: 48% at 60 μM vs. 37% at 120 μM) and a three-fold decrease in the IC50 value of the parent acylhydrazone 23 (IC50 (estimated) = 99 ± 2 μM vs. 34 ± 4 μM). Concerning the antibacterial activity against E. coli TolC, the amide bioisostere 38 showed a slight gain in activity compared to the parent acylhydrazone 1 (inhib.: 19% vs. no inhib. at 50 μM), while 1,3,4-oxadiazole bioisostere 40 showed no inhibition. The amide bioisostere 39 largely retains the activity of the parent acylhydrazone 23 (83% inhib. at 50 μM vs. 45 μM MIC), but 1,3,4-oxadiazole bioisostere 41 showed almost a two-fold decrease in the antibacterial activity against E. coli TolC (47% inhib. at 50 μM).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | K D (μM) | IC50 (μM)/inhib. (%) | MIC (μM)/inhib. (%) E. coli TolC | |
| drDXPS | ΔmtDXPS | drDXPS | |||
| a Inhibition of drDXPS was determined at 120 μM. * = inhibition of drDXPS at 60 μM. n.b. = no binding. Percent (%) growth inhibition of E. coli TolC was determined at 50 μM. # = estimated IC50. | |||||
| 1 | 38 | 40 ± 10 | 60 ± 20 | Inhib.: 25.8 ± 2.9 | Inhib.: 19 ± 7 |
| 2 | 39 | 290 ± 30 | 260 ± 20 | Inhib.: 37.3 ± 3.8 | Inhib.: 83 ± 27 |
| 3 | 40 | n.b. | n.b. | n.i. | n.i. |
| 4 | 41 | 290 ± 40 | 210 ± 50 | Inhib.: 48 ± 3 at 60 μM (IC50#: 99 ± 2) | Inhib.: 47 ± 1 |
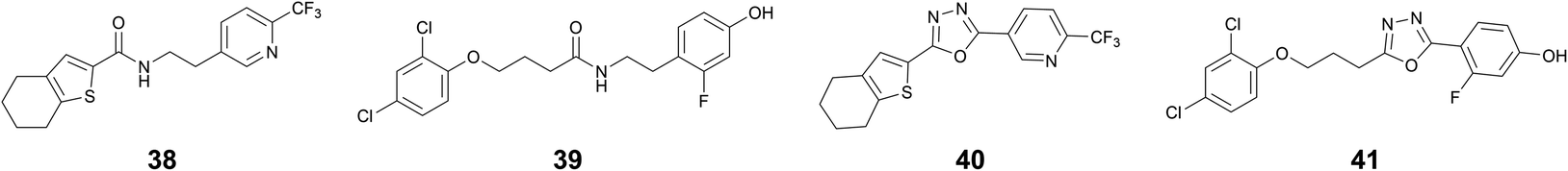
This study reveals that the acylhydrazone motif can be replaced by a more stable amide bioisostere without losing the affinity of the parent acylhydrazone, at least in the case of compound 38. Moreover, the 1,3,4-oxadiazole bioisostere 41, to some extent, was able to retain the enzymatic activity of the parent acylhydrazone 23.
Conclusions
Our study demonstrates that tdDCC can be effectively used as a hit-optimization technique along with its well-established application for hit-identification. The process exhibits the potential of identifying potent hits in less time compared to the traditional medicinal-chemistry approach. Importantly, using tdDCC, no prior knowledge of inhibitors is required, which allows generating new classes of hit compounds for important drug targets including underexplored ones like DXPS with little or no structural information. In the first round, using tdDCC, we were able to identify a new class of hits for the anti-infective target DXPS displaying good binding affinity (hits 1–3) and enzymatic-activity (hits 1 and 2). We simultaneously carried out multiparameter hit optimization using tdDCC to optimize the initial hits for their binding affinity (KD ≤ 5 μM), enzymatic activity (IC50 ≤ 50 μM), and antibacterial activity (MIC ≤ 20 μM). These tailored tdDCC experiments also allowed to shed light on the structure–activity relationships of this new class, highlightingstructural requirements for enhanced biological activity. Compounds bearing dichlorophenoxy (12, 26) thiophenyl (21), and N-Boc phenylalanine (37) moieties and indolyl, 1H-imidazolyl, and substituted phenyl moieties in the hydrazide and aldehyde part, respectively, showed significant improvement in the binding affinities. Compound 23 bearing dichlorophenoxy and 3-flurophenol moieties in the hydrazide and aldehyde part, respectively, showed a slight increase in inhibitory potency. Compounds 9, 26, and 35 bearing a dichlorophenoxy and tetrahydrobenzothiophyl moiety in the hydrazide part and an indolyl in the aldehyde part showed a substantial improvement in the antibacterial activity against E. coli. TolC. Moreover, the initial hits from tdDCC-1 were also optimized for their anti-TB activity using DCC, as compounds 7 and 35 from the second and third round of DCC, respectively, inhibited the growth of M. tuberculosis and thereby qualified for further optimization studies. Additionally, a docking study, competition enzyme activity assays, and competition DCC experiments provided important evidence regarding the mode-of-inhibition of compound 1. We were also able to replace the acylhydrazone from hits 1 and 23 with more stable amide (38, 39) and 1,3,4-oxadiazolyl (40, 41) bioisosteres. Out of these four bioisosteres, compound 38 was able to retain the affinity of the parent acylhydrazone 1, and compounds 41 and 39 were able to largely retain the enzymatic, and antibacterial activity of parent acylhydrazone 23, respectively. Nevertheless, a more focused study to find the ideal bioisostere, which can retain binding, enzymatic and antibacterial activity is needed. Altogether, this proof-of-concept study demonstrates a translational potential of tdDCC in the drug-discovery process.Experimental
Detailed experimental section including materials and methods have been described in ESI.†Author contributions
R. P. J. and M. G. designed and performed the tdDCC experiments and synthesized the hits. R. M. G. expressed and purified the mtDXPS and ΔmtDXPS enzymes, and A. A. together with R. M. G. drDXPS enzyme. R. M. G., A. A., and Z. H. performed enzyme activity assays. D. Z. and R. M. G. performed the competition enzyme activity assays. R. P. J. performed the competition tdDCC experiments. S. J. and R. P. J. performed the binding affinity assays by SPR. W. A. M. E. analyzed the SPR data. J. H. coordinated the tests for antibacterial activity (E. coli TolC) and carried out the evaluation. C. N. performed the DLS experiments. N. R. conducted the antiTB activity assays and carried out the evaluation. P. J. N. synthesized the bioisostere 41. A. K. H. H. and R.P.J. conceived the study and A. K. H. H. supervised the research. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Commission is acknowledged for an Intra-European Marie Skłodowska-Curie actions fellowship under Horizon-2020 (796089-NovInDXS, R. P. J.). Work by Z. H. was (partially) supported by the Schlumberger Foundation Faculty for the Future program. A. K. H. H. gratefully acknowledges funding from the European Research Council (ERC Starting grant 757913), the Netherlands Organisation for Scientific Research (LIFT grant: 731.015.414 and VIDI grant: 723.014.008), and from the Helmholtz Association's Initiative and Networking Fund. We gratefully acknowledge Dr Boris Illarionov from the University of Hamburg for reconfirming the enzymatic inhibition data of the bioisosteres. We gratefully acknowledge Jannine Jung and Dennis Jener from HIPS-HZI, Lisa Niwinski, and Jasmin Scharnberg from the Research Center Borstel for their expert technical support. We gratefully acknowledge Antoine Lacour for designing a TOC image.Notes and references
- I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- R. Huang and I. K. Leung, Molecules, 2016, 21, 910 CrossRef PubMed.
- A. Canal-Martín, J. Sastre, M. J. Sánchez-Barrena, A. Canales, S. Baldominos, N. Pascual, L. Martínez-González, D. Molero, M. E. Fernández-Valle, E. Sáez, P. Blanco-Gabella, E. Gómez-Rubio, S. Martín-Santamaría, A. Sáiz, A. Mansilla, F. J. Cañada, J. Jiménez-Barbero, A. Martínez and R. Pérez-Fernández, Nat. Commun., 2019, 10, 2798 CrossRef PubMed.
- A. M. Hartman, W. A. M. Elgaher, N. Hertrich, S. A. Andrei, C. Ottmann and A. K. H. Hirsch, ACS Med. Chem. Lett., 2020, 11, 1041–1046 CrossRef CAS PubMed.
- O. Ramström, T. Bunyapaiboonsri, S. Lohmann and J. M. Lehn, Biochim. Biophys. Acta, 2002, 1572, 178–186 CrossRef.
- P. Frei, R. Hevey and B. Ernst, Chem.–Eur. J., 2019, 25, 60–73 CrossRef CAS PubMed.
- A. Canal-Martín and R. Pérez-Fernández, ACS Omega, 2020, 5, 26307–26315 CrossRef PubMed.
- M. Rohmer, M. Knani, P. Simonin, B. Sutter and H. Sahm, Biochem. J., 1993, 295, 517–524 CrossRef CAS PubMed.
- Q. Du, H. Wang and J. Xie, Int. J. Biol. Sci., 2011, 7, 41–52 CrossRef CAS PubMed.
- R. E. Hill, K. Himmeldirk, I. A. Kennedy, R. M. Pauloski, B. G. Sayer, E. Wolf and I. D. Spenser, J. Biol. Chem., 1996, 271, 30426–30435 CrossRef CAS PubMed.
- I. Hale, P. M. O'Neill, N. G. Berry, A. Odom and R. Sharma, MedChemComm, 2012, 3, 418–433 RSC.
- J. M. Smith, N. V. Warrington, R. J. Vierling, M. L. Kuhn, W. F. Anderson, A. T. Koppisch and C. L. Freel Meyers, J. Antibiot., 2014, 67, 77–83 CrossRef CAS PubMed.
- D. Bartee and C. L. Freel Meyers, Biochem, 2018, 57, 4349–4356 CrossRef CAS PubMed.
- D. Bartee, S. Sanders, P. D. Phillips, M. J. Harrison, A. T. Koppisch and C. L. Freel Meyers, ACS Infect. Dis., 2019, 5, 406–417 CrossRef CAS PubMed.
- T. Masini, B. Lacy, L. Monjas, D. Hawksley, A. R. de Voogd, B. Illarionov, A. Iqbal, F. J. Leeper, M. Fischer, M. Kontoyianni and A. K. H. Hirsch, Org. Biomol. Chem., 2015, 13, 11263–11277 RSC.
- S. S. Wesolowski and D. G. Brown, in Lead Generation, 2016, pp. 487–512 Search PubMed.
- A. M. Hartman, R. M. Gierse and A. K. H. Hirsch, Eur. J. Org. Chem., 2019, 2019, 3581–3590 CrossRef CAS PubMed.
- LeadIT version 2.3.2, BioSolveIT GmbH, Sankt Augustin, Germany, www.biosolveit.de/LeadIT, 2017 Search PubMed.
- SeeSAR version 10.1, BioSolveIT GmbH, Sankt Augustin, Germany, www.biosolveit.de/SeeSAR, 2020 Search PubMed.
- P. Frei, L. Pang, M. Silbermann, D. Eriş, T. Mühlethaler, O. Schwardt and B. Ernst, Chem.–Eur. J., 2017, 23, 11570–11577 CrossRef CAS PubMed.
- P. Frei, M. Silbermann, T. Muehlethaler, X. Jiang, O. Schwardt, R. Hevey and B. Ernst, ARKIVOC, 2019, 2019, 143 Search PubMed.
- A publication on the details of the truncation and enzymatic properties of mtDXS is in preparation, the protein structure of truncated mtDXS will be published with PDB codes 7A9G and 7A9H..
- H. Patel, N. S. Nemeria, L. A. Brammer, C. L. Freel Meyers and F. Jordan, J. Am. Chem. Soc., 2012, 134, 18374–18379 CrossRef CAS PubMed.
- T. A. O'Brien, R. Kluger, D. C. Pike and R. B. Gennis, Biochim. Biophys. Acta, 1980, 613, 10–17 CrossRef.
- J. M. Smith, R. J. Vierling and C. F. Meyers, MedChemComm, 2012, 3, 65–67 RSC.
- V. R. Jumde, M. Mondal, R. M. Gierse, M. Y. Unver, F. Magari, R. C. W. van Lier, A. Heine, G. Klebe and A. K. H. Hirsch, ChemMedChem, 2018, 13, 2266–2270 CrossRef CAS PubMed.
- N. N. Farshori, A. Rauf, M. A. Siddiqui, E. S. Al-Sheddi and M. M. Al-Oqail, Arabian J. Chem., 2017, 10, S2853–S2861 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- A. Volkamer, D. Kuhn, F. Rippmann and M. Rarey, Bioinformatics, 2012, 28, 2074–2075 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00330e |
| This journal is © The Royal Society of Chemistry 2021 |