
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed cross-electrophile Coupling/C–H alkylation reaction enabled by a mediator generated via C(sp3)–H activation†
Zhuo
Wu
,
Hang
Jiang
and
Yanghui
Zhang
 *
*
School of Chemical Science and Engineering, Shanghai Key Laboratory of Chemical Assessment and Sustainability, Tongji University, 1239 Siping Road, Shanghai 200092, China. E-mail: zhangyanghui@tongji.edu.cn
First published on 19th May 2021
Abstract
Transition-metal-catalyzed cross-electrophile C(sp2)–(sp3) coupling and C–H alkylation reactions represent two efficient methods for the incorporation of an alkyl group into aromatic rings. Herein, we report a Pd-catalyzed cascade cross-electrophile coupling and C–H alkylation reaction of 2-iodo-alkoxylarenes with alkyl chlorides. Methoxy and benzyloxy groups, which are ubiquitous functional groups and common protecting groups, were utilized as crucial mediators via primary or secondary C(sp3)–H activation. The reaction provides an innovative and convenient access for the synthesis of alkylated phenol derivatives, which are widely found in bioactive compounds and organic functional materials.
Introduction
The introduction of alkyl moieties into aromatic rings is an essential transformation in organic synthesis. Nowadays, transition-metal-catalyzed cross-coupling reactions between an electrophile and a nucleophile, generally an organic (pseudo)halide and an organometallic reagent, have been a powerful tool for this transformation.1 However, organometallic reagents used in these reactions need presynthesis and careful handling, which limits their applications.The coupling between two electrophiles avoids the use of organometallic reagents and thus becomes a promising alternative strategy for C(sp2)–C(sp3) bond formation.2 However, there are several obstacles for developing cross-electrophile coupling reactions. Firstly, because of electrophilic properties of both halides, achieving the selective formation of cross-products over two potential symmetric dimers is challenging. Secondly, unlike the traditional coupling between an electrophile and a nucleophile, the catalyst loses electrons in total after coupling two electrophiles, and thus appropriate reductants are required to regenerate the catalyst. Moreover, the coupling becomes even more challenging, considering the great tendency of β-H elimination of alkyl-metal species. Despite these difficulties, in the past few decades, the cross-electrophile C(sp2)–C(sp3) coupling reactions have been successfully developed with Ni3 or Co4 catalysts (Fig. 1a). These reactions proceeded through an alkyl radical mechanism. Typically, alkyl iodides or bromides were reactive agents, whereas alkyl chlorides, which are more inexpensive and less toxic, were usually unreactive.3f A stoichiometric amount of metals, such as Zn or Mn, were frequently employed as reductants in these reactions, which produced a large amount of metal waste.5
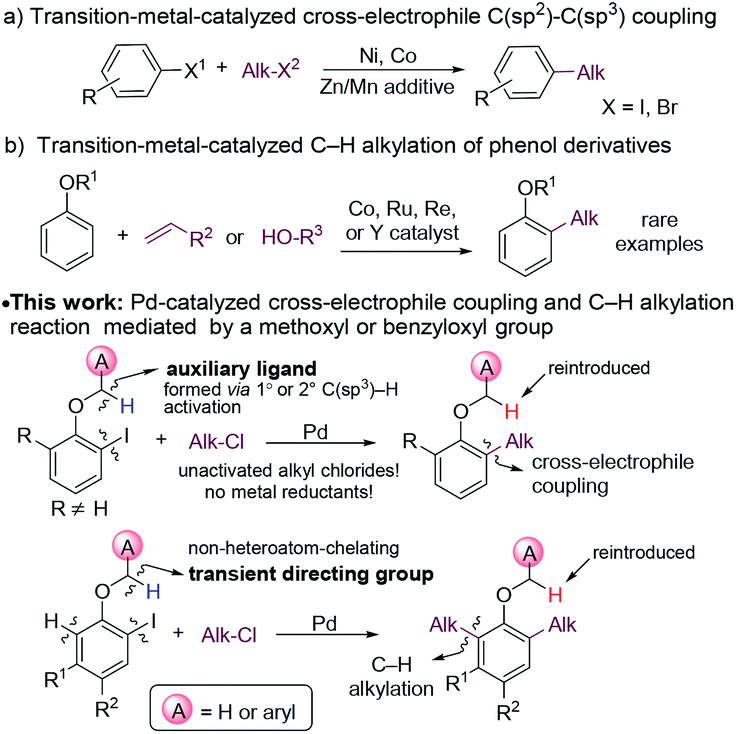 | ||
| Fig. 1 Cross-electrophile C(sp2)–C(sp3) coupling and C–H alkylation of phenol derivatives. | ||
The alkylation of arenes via C–H bond activation represents an even more efficient strategy for the incorporation of an aliphatic moiety. In recent years, great progress has been made in transition-metal-catalyzed alkylation reactions of arenes with alkyl halides, alkenes, or alkylmetal reagents.6 In most cases, an ortho-directing group was required, and the majority of the directing groups were derived from carbonyl,7 carboxyl,8 amido,9 or N-heteroaryl10 groups. By contrast, the hydroxyl group of phenols and their derivatives was much less frequently utilized as a directing group for C–H alkylation. In the current C–H alkylation reactions of phenol derivatives, transition metal catalysts are primarily limited to Co, Ru, Re and Y (Fig. 1b).11 Furthermore, for directing groups in transition metal-catalyzed C–H activation, almost all of them rely on the coordination of catalysts to heteroatoms they contain to assist C–H cleavage. It is desirable to develop new directing strategies for C–H functionalization.
Herein, we report an unusual Pd-catalyzed cascade cross-electrophile coupling and C–H alkylation reaction between ortho-iodophenol derivatives and alkyl chlorides by utilizing a methoxy or benzyloxy group as a mediator. Pd catalysts are much less frequently applied in cross-electrophile C(sp2)–C(sp3) coupling than Ni catalysts,12 because they tend not to undergo a radical mechanism,13 which sets the cross-selectivity difficult to achieve. In this reaction, C(sp3)–H bond activation of the ortho-methoxy and benzyloxy groups plays a crucial role for the cross-selectivity.14 For substrates with the positions ortho to the alkoxyl group unsubstituted, C–H alkylation was initiated. The methoxy and benzyloxy groups served as directing groups herein. The methoxy and benzyloxy groups are different from traditional directing groups relying on the coordination of heteroatoms and represent a new class of directing groups for transition-metal-catalyzed C–H activation.
Results and discussion
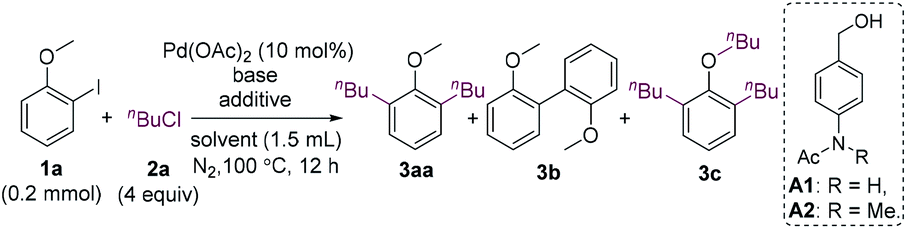
We commenced our research by investigating the reaction of 2-iodoanisole (1a) with 1-chlorobutane (2a). The 2-iodoanisole substrates can be readily synthesized via iodination of anisoles or phenols. Cheap and low-toxicity alkyl chlorides are also ideal alkylating reagents. It should be mentioned that 2-iodoanisoles could undergo homocoupling using a methoxy group as the directing group.15 The homocoupling should be overcome to develop reactions with external reagents. Surprisingly, the expected monoalkylated product was not formed, whereas dialkylated product 3aa and homocoupling product 3b were obtained in low yields in the presence of K3PO4 (Table 1, entry 1).16 When K2CO3 was used as a base, the homocoupling product was suppressed (entry 2). The addition of n-Bu4NBr and benzyl alcohol improved the yield of 3aa greatly, and the yield was further increased to 73% when NMP was used as the solvent (entries 3 and 4). Tetraalkylammonium halides can promote the coupling reactions of aryl halides by stabilizing nano-sized palladium colloids.17 The reaction remained almost unaffected when carried out at 85 °C (entry 5). Although benzyl alcohol was beneficial to the reaction, its oxidized and O-alkylated products caused difficulty for the isolation of the desired product. Thus, a range of alcohols with high polarity were surveyed, and A2 was found to be an effective reductant (entries 6 and 7). When n-Bu4NCl was used instead of n-Bu4NBr, the yield decreased to 27% (entry 8). 1-Bromo or 1-iodobutane was also a suitable alkylating agent (entries 9 and 10). However, the yields were much lower. It should be mentioned that the addition of an alcohol is necessary for the reaction. In the absence of an alcohol, the repeatability of the reaction became low, and an undesired trialkylated product 3c was formed in 5–10% yield.|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (equiv.) | Additive (equiv.) | Solvent | 3aa (%) | 3b (%) |
| a The yields were determined by 1H NMR analysis of the crude reaction mixture using CHCl2CHCl2 as the internal standard. b Isolated yield. c 85 °C. d n-BuBr. e n-BuI. | |||||
| 1 | K3PO4 (3) | — | DMF | 9 | 10 |
| 2 | K2CO3 (3) | — | DMF | 7 | 0 |
| 3 | K2CO3 (5) | n Bu4NBr (3), BnOH (1) | DMF | 64 | 0 |
| 4 | K2CO3 (5) | n Bu4NBr (3), BnOH (1) | NMP | 73 | 0 |
| 5c | K2CO3 (5) | n Bu4NBr (3), BnOH (1) | NMP | 71 | 0 |
| 6c | K2CO3 (5) | n Bu4NBr (3), A1 (1) | NMP | 66 | 0 |
| 7c | K2CO3 (5) | n Bu4NBr (3), A2 (1) | NMP | 72 (68b) | 0 |
| 8c | K2CO3 (5) | n Bu4NCl (3), A2 (1) | NMP | 27 | 0 |
| 9c,d | K2CO3 (5) | n Bu4NBr (3), A2 (1) | NMP | 45 | 0 |
| 10c,e | K2CO3 (5) | n Bu4NBr (3), A2 (1) | NMP | 14 | 0 |
Next, we studied the substrate scope of the dialkylation reaction. The ortho-iodoanisole scope was first probed. The functional group compatibility was investigated by examining the reactions of ortho-iodoanisoles bearing various substituents para to the methoxy group. The reaction has very high functional group compatibility. A wide range of functionalities, including electron-donating and -withdrawing ones, were compatible (Scheme 1, 3ba–3ia), and halo groups were well-tolerated (3ja–3la). The performance of meta-substituted ortho-iodoanisoles was also investigated. Notably, even in the presence of a meta-substituent, ortho-iodoanisoles were dialkylated by overcoming the steric hindrance imposed by the meta-substituents, forming tetrasubstituted arenes (3ma–3ra). However, for the substrates bearing a meta-ester or -tert-butyl group, only monoalkylated products were formed due to the steric hindrance (3sa and 3ta). Heteroaryl groups including pyridyl and pyrrolyl groups were compatible (3ua and 3va), and heteroarenes and its derivative could also undergo the dialkylation reaction (3wa–3ya).
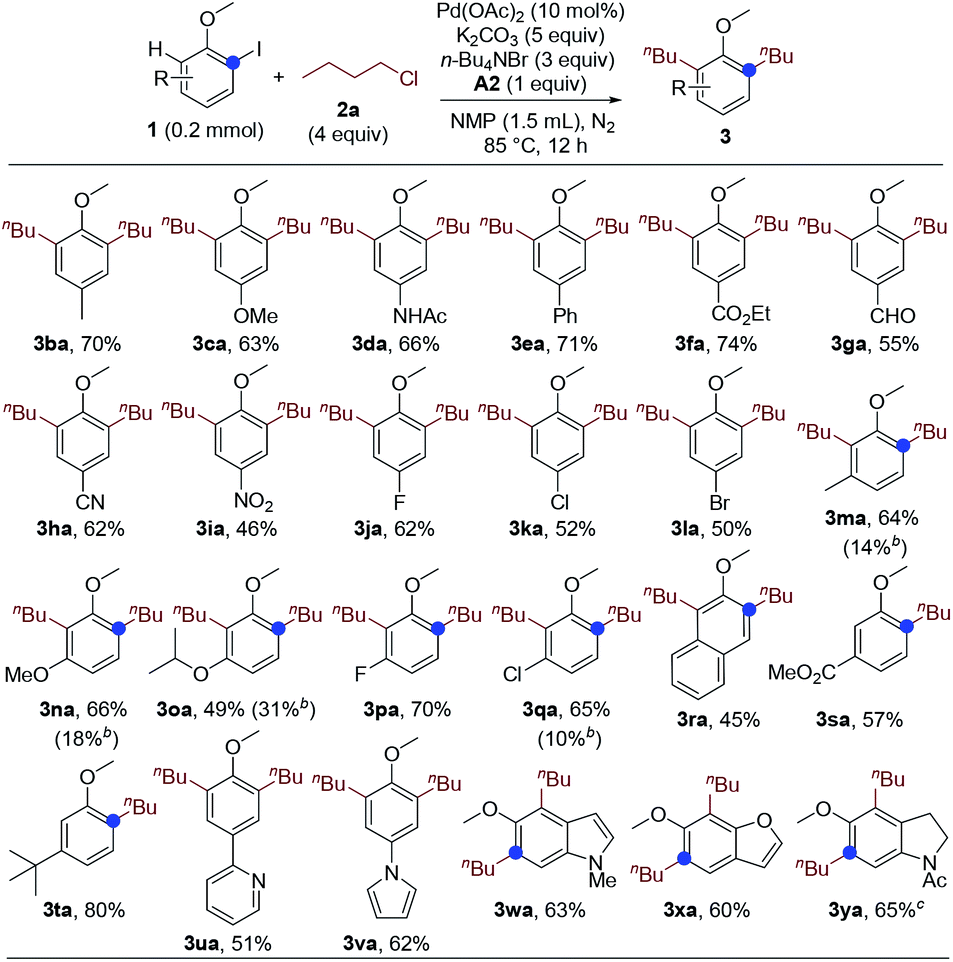 | ||
| Scheme 1 Ortho-Iodoanisole scope. aIsolated yields. bMonoalkylated product. c100 °C. | ||
The first step of the dialkylation reaction should be the alkylation of aryl iodides, which represents a new Pd-catalyzed cross-electrophile coupling. Therefore, we sought to study the reactions by using ortho-iodoanisoles without an ortho-hydrogen. As shown in Scheme 2, a range of ortho-substituted ortho-iodoanisoles cross-coupled with n-butyl chloride to form alkylated anisole products (5aa–5ia). Intriguingly, for 2-iodo-1-methoxynaphthalene, the dehydrogenative coupling reaction occurred after the alkylation to form cyclized product 5ja.
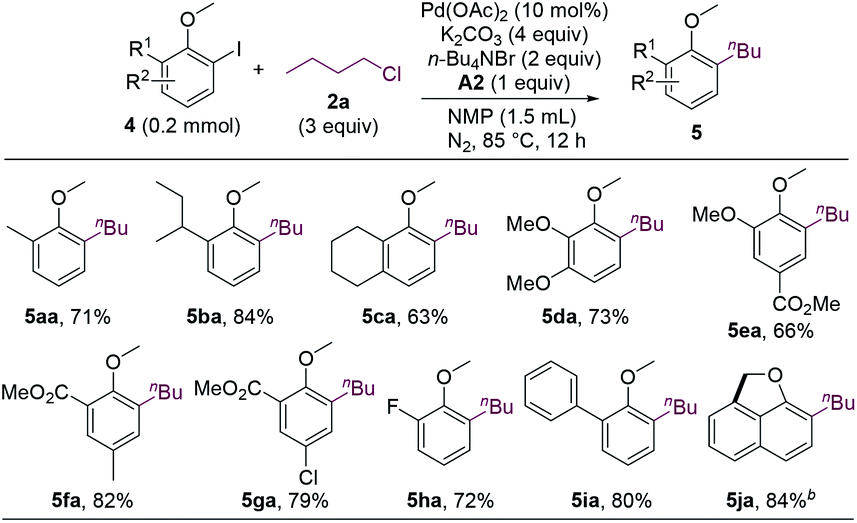 | ||
| Scheme 2 Ortho-Substituted 2-iodoanisoles scope. aIsolated yields. b10 mol% of Pd(OAc)2, 5 equiv. of K2CO3, 3 equiv. of KOAc, 2 equiv. of n-Bu4NBr, and 4 equiv. of n-BuCl were used. A2 was not added. | ||
The alkyl chloride scope was then probed. A wide array of alkyl chlorides containing various functionalities and with different alkyl-chain lengths were effective alkylating reagents, and a range of dialkylated anisoles were formed (Scheme 3, 3ab–3al). Sterically hindered alkyl chlorides could also couple with 1a, albeit in lower yields (3am–3ao).
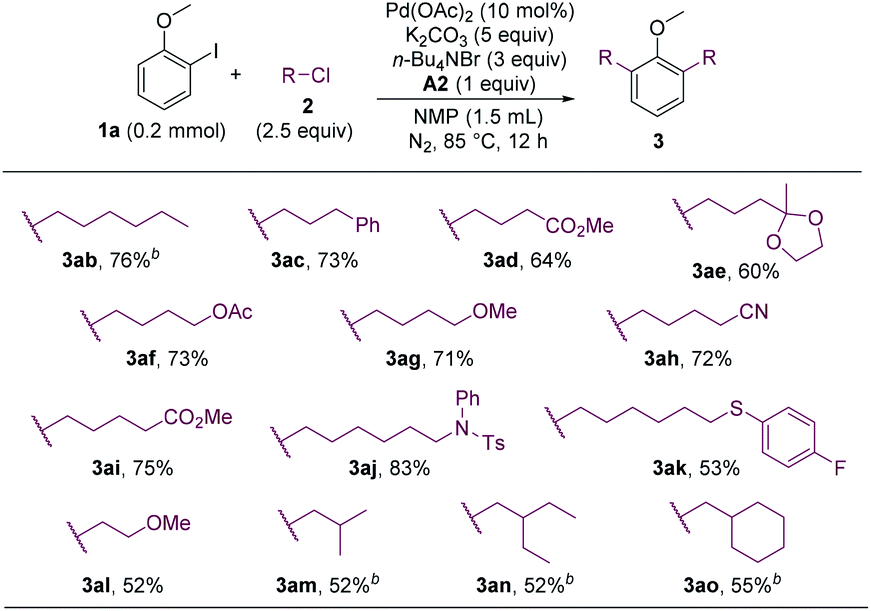 | ||
| Scheme 3 Alkyl chlorides scope. aIsolated yields. b4 equiv. of alkyl chloride. | ||
The cross-coupling involving secondary alkyl halides is usually challenging.18 The reactivity of secondary alkyl halides in the coupling reaction were examined. Whereas 2-chloropropane failed to alkylate 4b, 2-bromopropane could couple with 4b in a moderate yield (Scheme 4, 5bp). The substrate bearing an ortho-ester group (4f) was also compatible.
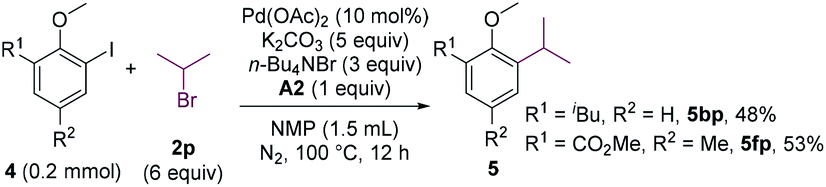 | ||
| Scheme 4 Coupling of 2-iodoanisoles with 2-bromopropane. | ||
The methoxy group of ortho-iodoanisoles acts as a mediator to enable the cross-electrophile coupling and C–H alkylation reaction. We envisioned that other alkoxy groups could also facilitate such a cascade reaction. Gratefully, the benzyloxy group was found to be an effective mediator for the dialkylation reaction (Scheme 5, 7aa).19 Substituted benzyl groups also enabled the alkylation (7ba–7da), and functionalized iodobenzenes and alkyl chlorides could undergo the cascade reaction smoothly (7ea, 7fa, 7ad, and 7af). It should be noted that benzyl groups could be cleaved readily, which allows for the further manipulation of the resulted phenol products.
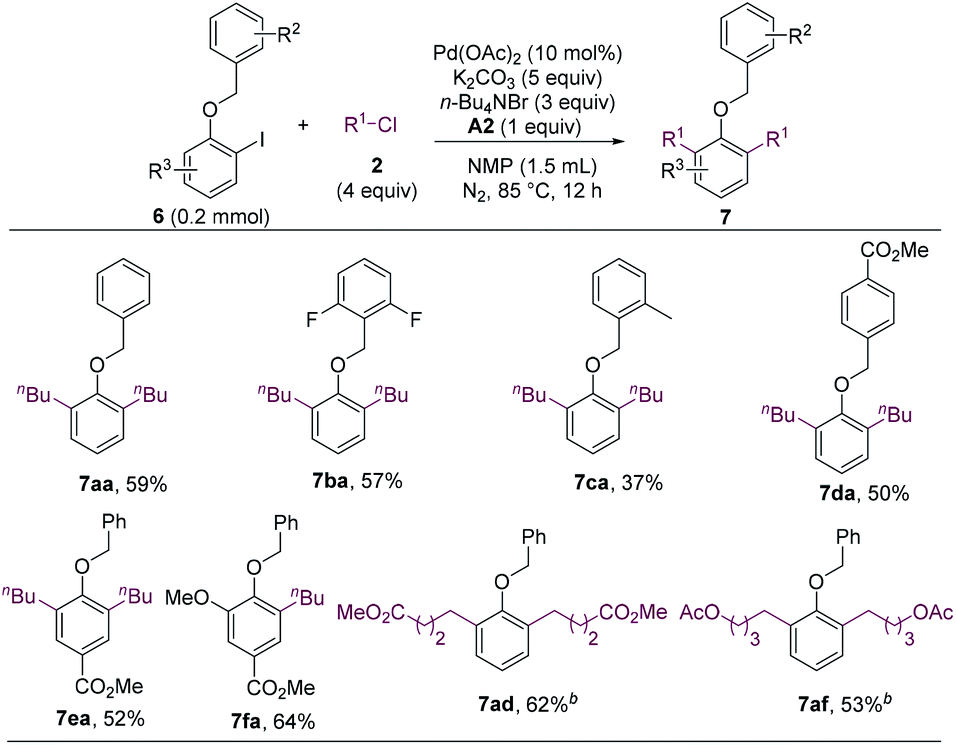 | ||
| Scheme 5 Coupling reaction utilizing a benzyloxy group as a mediator. a Isolated yields. b 2.5 equiv. of alkyl chloride. | ||
The dialkylation reaction provides a straightforward method for the synthesis of ortho-dialkylated phenol derivatives. The current synthetic methods for ortho-dialkylated phenols primarily include: (1) Claisen rearrangement of phenoxy allyl ether;20 (2) Coupling of 2,6-dihalophenol derivatives with alkyl organometallic reagents.21 The first method requires multiple-step synthesis, and the Claisen rearrangement was usually carried out under harsh conditions. The second method employs alkylmetallic reagents, and the use of dihalogenated substrates is not atom-economic. Our dialkylation reaction features readily available reagents, comparatively mild conditions, and high atom-economy.
It should be noted that dialkylated phenol derivatives are essential structural motifs widely found in bioactive molecules.22 Furthermore, they are also key intermediates in the synthesis of biologically active compounds and organic functional materials. For example, compound 3aq, which was synthesized by the dialkylation of 1a with 2q under the standard conditions, could be hydrolyzed to 3aq-A (Fig. 2a). 3aq-A was the synthetic intermediate for a novel H-shaped chromophore.20a And 7er-A, which could be readily obtained from compound 7er, was the intermediate in the synthesis of a liver X receptor b-selective agonist.20b For the original method, 7er-A was synthesized in eight steps under harsh conditions. The monoalkylated anisoles were also intermediates in the synthesis of bioactive compounds. For instance, compound 5ea was the synthetic intermediate for an antibacterial agent.23 Furthermore, monoalkylated product 5di could afford 5di-A, which was used to synthesize an anti-cancer agent.24 It should be noted that all the 2-iodoanisole substrates in our alkylation reactions were prepared in one or two steps by using low-cost reagents.
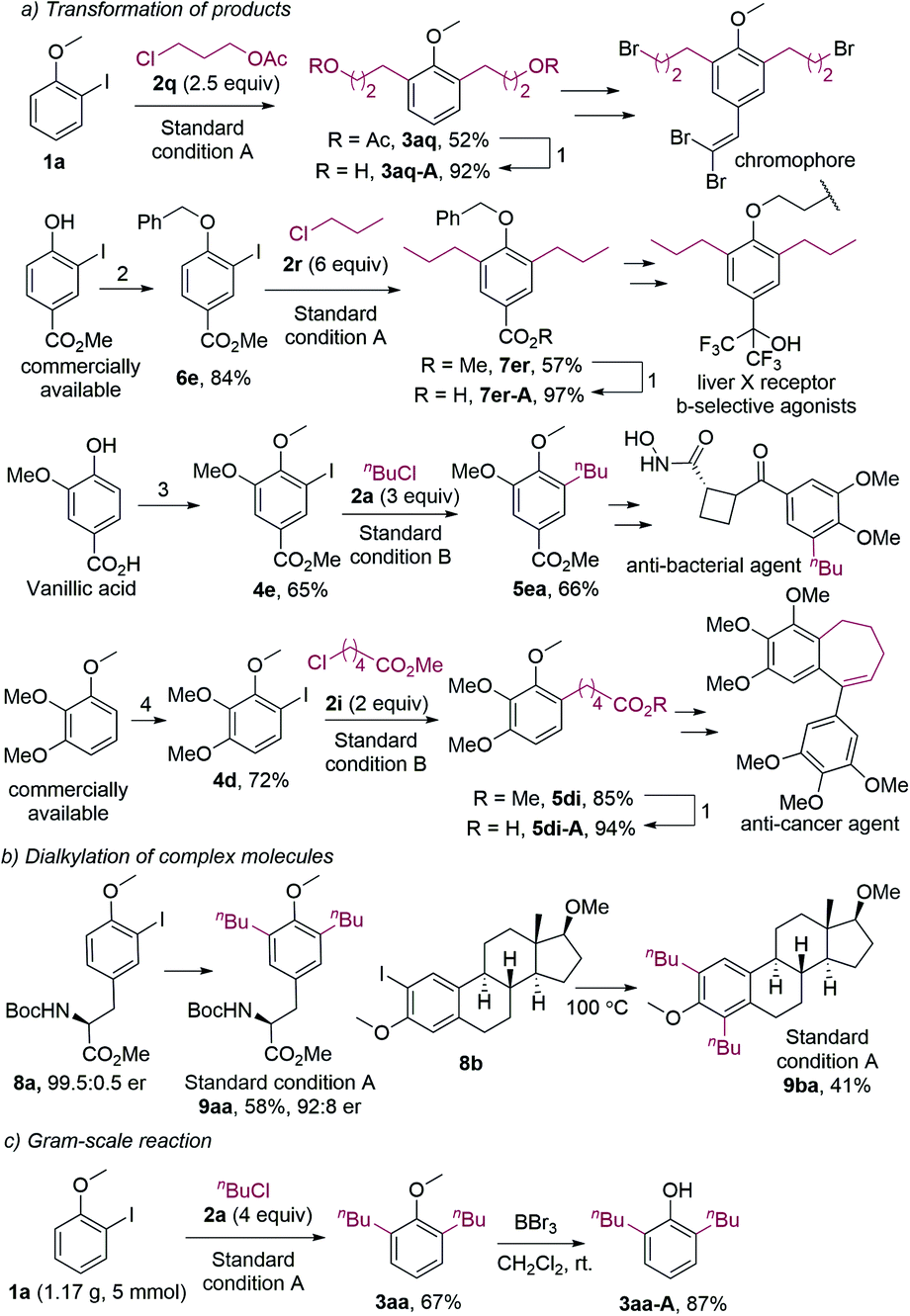 | ||
| Fig. 2 Practical applications. Standard condition A: 10 mol% of Pd(OAc)2, 5 equiv. of K2CO3, 3 equiv. of n-Bu4NBr, 1 equiv. of A2, NMP, 85 °C, 12 h; standard condition B: 10 mol% of Pd(OAc)2, 4 equiv. of K2CO3, 2 equiv. of n-Bu4NBr, 1 equiv. of A2, NMP, 85 °C, 12 h. Other conditions: (1) NaOH, MeOH/H2O, 60 °C; (2) BnCl, K2CO3, DMF, rt; (3) a. NaI, NaOH, NaClO aq., MeOH, 0 °C; b. MeI, K2CO3, DMF, rt; (4) CF3CO2H, NIS, MeCN, rt. | ||
The dialkylation reaction is also applicable to the dialkylation of complex molecules. For example, the estradiol- and tyrosine-derived iodides could be dialkylated under the standard conditions (Fig. 2b). It should be mentioned that the product 9aa was slightly racemized, which could be caused by the base and the high temperature. Furthermore, the dialkylation reaction was scalable, and the methyl group of dialkylated anisoles could be removed (Fig. 2c). The resulting hydroxy group allows for further functionalization.
Preliminary mechanistic studies were conducted (Fig. 3). When 3-iodoanisole 10a (or 4-iodoanisole 10b) and 1-chlorobutane were subjected to the standard conditions, the alkylated products were not detected, and only the homocoupling product 11a (or 11b) was obtained (Fig. 3a). The outcomes indicated that the presence of the ortho-methoxy group is crucial for the alkylation reaction. Deuterium-labeling experiments were also carried out (Fig. 3b). When the deuterated analogue 1a-D3 was subjected to the standard conditions, 3aa-D2 was obtained as the major product. The proportion of 3aa-D2 was further enhanced to 95% when 20 equivalents of water was added. Moreover, when the alkylation reaction of 1a was carried out using CD3OD as the reductant, deuteration occurred at the methoxy group in 40% yield. These experiments implied that the activation of the methoxyl C(sp3)–H bond occurred and the alcohol was one of the major reductants.25 Intermolecular competition experiments between 1a and 1a-D3 were conducted, and the KIE value was 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 3c). The kinetic isotope effect supported the involvement of the methoxy group in the reaction via C–H bond activation, which might be involved in the rate-determining step.
:1 (Fig. 3c). The kinetic isotope effect supported the involvement of the methoxy group in the reaction via C–H bond activation, which might be involved in the rate-determining step.
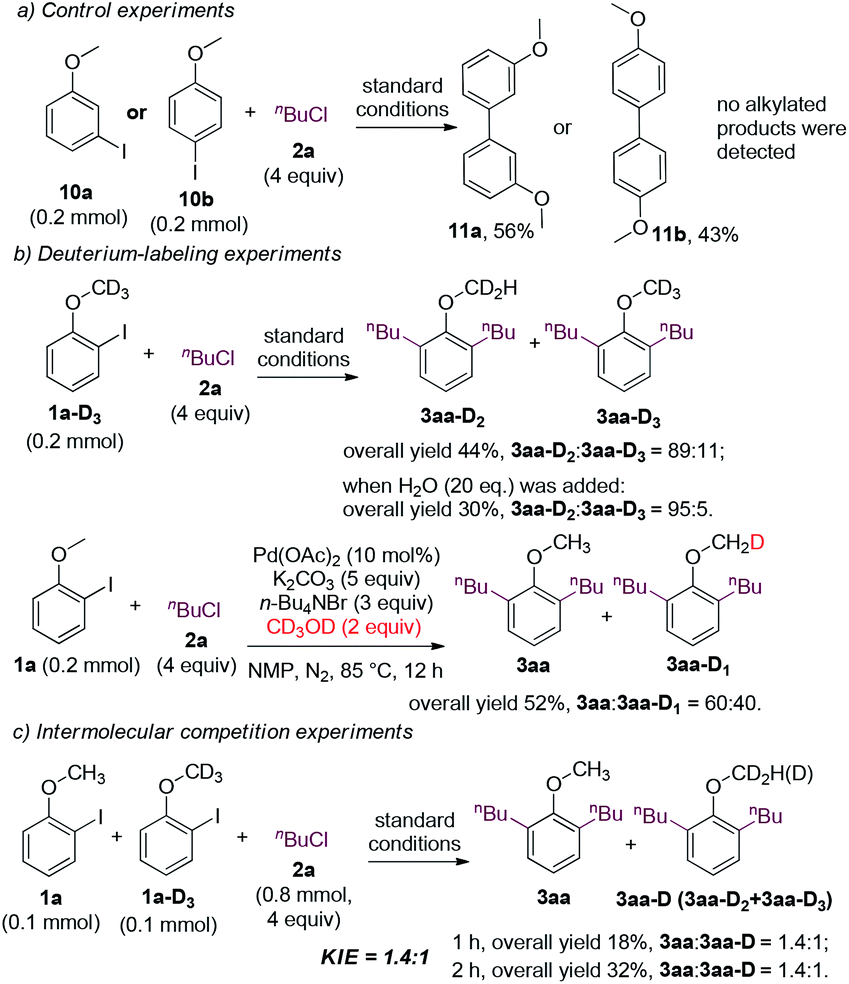 | ||
| Fig. 3 Preliminary mechanistic studies. | ||
On the basis of the mechanistic studies and the previous reports,14,15,26 a possible mechanism was proposed in Fig. 4 for the dialkylation reaction. The substrate undergoes oxidative addition and C(sp3)–H activation to afford palladacycle B. Palladacycle B has high reactivity towards alkyl halides, and undergoes the oxidative addition to generate Pd(IV) species C.27 The reductive elimination of C gives D. Then, the aryl C–H is cleaved, affording a second palladacycle E. E undergoes the same process as that for the formation of D to introduce a second alkyl group. Eventually, alkylpalladium G is reduced, yielding the product and releasing Pd0.
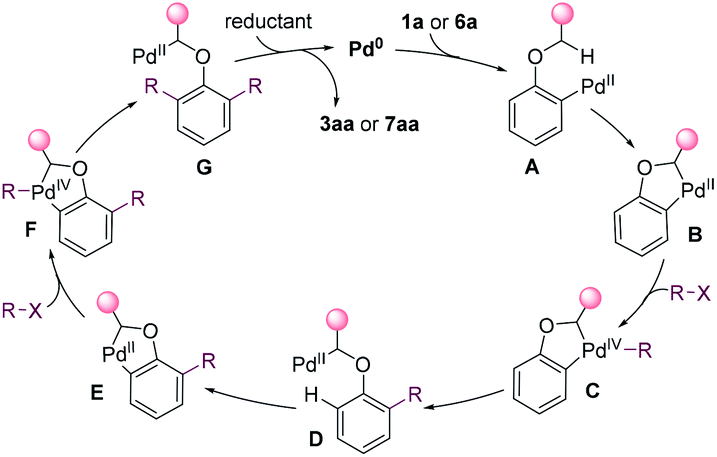 | ||
| Fig. 4 Plausible mechanism. | ||
Conclusions
In conclusion, we have developed cascade Pd-catalyzed cross-electrophile coupling and C–H alkylation reaction of 2-iodo-alkoxylarenes with alkyl chlorides. The ortho-methoxy or benzyloxy group acted as a mediator via primary or secondary C(sp3)–H activation. It is very rare and intriguing that the cross-electrophile coupling is achieved by the assistance of an alkoxyl group, which can be readily removed. The methoxy and benzyloxy groups also served as non-heteroatom-chelating directing groups for C–H alkylation reaction. The reaction provides an efficient and innovative method for the synthesis of alkylated, especially 2,6-dialkylated, phenol derivatives, which are ubiquitous in bioactive compounds and organic functional materials.Author contributions
Z. Wu and H. Jiang performed the experiments and analysed the data. Y. Zhang conceived the project and analysed experimental data. The manuscript was written by Y. Zhang and Z. Wu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 21971196).Notes and references
- (a) G. Manolikakes, Coupling Reactions Between sp3 and sp2 Carbon Centers, in Comprehensive Organic Synthesis, ed. P. Knochel and G. Molander, Elsevier, Oxford, 2nd edn, vol. 3, 2014, pp. 392–464 Search PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed.
- (a) J. Liu, Y. Ye, J. L. Sessler and H. Gong, Acc. Chem. Res., 2020, 53, 1833–1845 CrossRef CAS PubMed; (b) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed; (c) C. E. I. Knappke, S. Grupe, D. Gaertner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (d) W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- (a) M. Durandetti, C. Gosmini and J. Perichon, Tetrahedron, 2007, 63, 1146–1153 CrossRef CAS; (b) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed; (c) S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352–3355 CrossRef CAS PubMed; (d) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed; (e) J. Sheng, H.-Q. Ni, H.-R. Zhang, K.-F. Zhang, Y.-N. Wang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 7634–7639 CrossRef CAS PubMed; (f) S. Kim, M. J. Goldfogel, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 9902–9907 CrossRef CAS PubMed.
- (a) P. Gomes, C. Gosmini and J. Perichon, Org. Lett., 2003, 5, 1043–1045 CrossRef CAS PubMed; (b) M. Amatore and C. Gosmini, Chem.–Eur. J., 2010, 16, 5848–5852 CrossRef CAS PubMed; (c) S. Pal, S. Chowdhury, E. Rozwadowski, A. Auffrant and C. Gosmini, Adv. Synth. Catal., 2016, 358, 2431–2435 CrossRef CAS.
- The use of organic reductants, photoredox catalysts, and electrochemical methods have been alternative strategies to avoid the usage of metallic reductants: (a) L. L. Anka-Lufford, K. M. M. Huihui, N. J. Gower, L. K. G. Ackerman and D. J. Weix, Chem.–Eur. J., 2016, 22, 11564–11567 CrossRef CAS PubMed; (b) A. Garcia-Dominguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef CAS PubMed; (c) Z. Duan, W. Li and A. Lei, Org. Lett., 2016, 18, 4012–4015 CrossRef CAS PubMed; (d) P. Zhang, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (e) K.-J. Jiao, D. Liu, H.-X. Ma, H. Qiu, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 6520–6524 CrossRef CAS PubMed.
- For reviews, see: (a) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7202–7236 CrossRef CAS PubMed; (b) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7558–7598 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC; (d) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC.
- For selected examples, see: (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875–1877 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279–9282 CrossRef CAS PubMed; (d) N. Kimura, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2017, 139, 14849–14852 CrossRef CAS PubMed.
- For selected examples, see: (a) P. S. Thuy-Boun, G. Villa, D. Dang, P. Richardson, S. Su and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 17508–17513 CrossRef CAS PubMed; (b) H. Wang, N. Schrçder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386–5389 CrossRef CAS PubMed; (c) R.-Y. Zhu, J. He, X.-C. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194–13197 CrossRef CAS PubMed; (d) B. M. Monks, E. R. Fruchey and S. P. Cook, Angew. Chem., Int. Ed., 2014, 53, 11065–11069 CrossRef CAS PubMed.
- For selected examples, see: (a) S. J. Tremont and H. U. Rahman, J. Am. Chem. Soc., 1984, 106, 5759–5760 CrossRef CAS; (b) S. R. Neufeldt, C. K. Seigerman and M. S. Sanford, Org. Lett., 2013, 15, 2302–2305 CrossRef CAS PubMed; (c) Z. Ruan, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 3153–3157 CrossRef CAS PubMed; (d) X. Zhou, J. Xia, G. Zheng, L. Kong and X. Li, Angew. Chem., Int. Ed., 2018, 57, 6681–6685 CrossRef CAS PubMed; (e) Z. Shen, H. Huang, C. Zhu, S. Warratz and L. Ackermann, Org. Lett., 2019, 21, 571–574 CrossRef CAS PubMed; (f) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789–1792 CrossRef CAS PubMed.
- For selected examples, see: (a) B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109–1113 CrossRef CAS PubMed; (b) J. M. Wiest, A. Pothig and T. Bach, Org. Lett., 2016, 18, 852–855 CrossRef CAS PubMed; (c) X. Wang, X. Ji, C. Shao, Y. Zhang and Y. Zhang, Org. Biomol. Chem., 2017, 15, 5616–5624 RSC; (d) K. Korvorapun, M. Moselage, J. Struwe, T. Rogge, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 18795–18803 CrossRef CAS PubMed.
- For a review on phenol and its derivative-directed C–H functionalization, see: (a) Z. Huang and J.-P. Lumb, ACS Catal., 2019, 9, 521–555 CrossRef CAS; For phenol and derivative-directed C–H alkylation, see: (b) R. Dorta and A. Togni, Chem. Commun., 2003, 760–761 RSC; (c) Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914–9915 CrossRef CAS PubMed; (d) J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828–12832 CrossRef CAS PubMed; (e) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS PubMed; (f) S. S. Bera and M. S. Maji, Org. Lett., 2020, 22, 2615–2620 CrossRef CAS PubMed.
- Although Pd-catalyzed cross-coupling between aryl and alkyl halides has been reported, the actual reactive species were in situ formed alkylzincs. (a) A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592–15593 CrossRef CAS PubMed; (b) C. Duplais, A. Krasovskiy and B. H. Lipshutz, Organometallics, 2011, 30, 6090–6097 CrossRef CAS PubMed.
- Single electron transfer (SET) processes involving Pd(I) and Pd(III) intermediates are not common. For reviews, see: (a) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS; (b) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed.
- Although the alkylation of palladacycles formed via C–H activation with alkyl halides has been reported, the works involve C(sp2),C(sp2)- and tert-butyl-derived C(sp3),C(sp2)-palladacycles: (a) D. Chen, G. Shi, H. Jiang, Y. Zhang and Y. Zhang, Org. Lett., 2016, 18, 2130–2133 CrossRef CAS PubMed; (b) Z. Wu, D. Ma, B. Zhou, X. Ji, X. Ma, X. Wang and Y. Zhang, Angew. Chem., Int. Ed., 2017, 56, 12288–12291 CrossRef CAS PubMed.
- G. Dyker, Angew. Chem., Int. Ed., 1992, 31, 1023–1025 CrossRef.
- For the dimethylation of 2-iodoanisoles reported very recently, see: Z. Wu, F. Wei, B. Wan and Y. Zhang, J. Am. Chem. Soc., 2021, 143, 4524–4530 CrossRef CAS PubMed.
- M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC.
- For a review, see: (a) Z. Qureshi, C. Toker and M. Lautens, Synthesis, 2017, 49, 1–16 CAS. For selected Pd-catalyzed C–H alkylation with secondary alkyl halides, see: (b) Q. Wang, S. An, Z. Deng, W. Zhu, Z. Huang, G. He and G. Chen, Nat. Catal., 2019, 2, 793–800 CrossRef CAS; (c) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531–539 CrossRef CAS PubMed; (d) Y.-C. Luo, C. Yang, S.-Q. Qiu, Q.-J. Liang, Y.-H. Xu and T.-P. Loh, ACS Catal., 2019, 9, 4271–4276 CrossRef CAS.
- (a) D. Dailler, R. Rocaboy and O. Baudoin, Angew. Chem., Int. Ed., 2017, 56, 7218–7222 CrossRef CAS PubMed; (b) J. Pedroni, M. Boghi, T. Saget and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 9064–9067 CrossRef CAS PubMed; (c) D. Katayev, M. Nakanishi, T. Bürgi and E. P. Kündig, Chem. Sci., 2012, 3, 1422–1425 RSC.
- (a) J.-P. Shi, D.-L. Wu, Y. Ding, D.-H. Wu, H.-W. Hu and G.-Y. Lu, Tetrahedron, 2012, 68, 2770–2777 CrossRef CAS; (b) M. Koura, T. Matsuda, A. Okuda, Y. Watanabe, Y. Yamaguchi, S. Kurobuchi, Y. Matsumoto and K. Shibuya, Bioorg. Med. Chem. Lett., 2015, 25, 2668–2674 CrossRef CAS PubMed.
- A. V. Predeus, V. Gopalsamuthiram, R. J. Staples and W. D. Wulff, Angew. Chem., Int. Ed., 2013, 52, 911–915 CrossRef CAS.
- (a) K. J. Lafferty and J. A. Panetta, EP 500337 A1, 1992; (b) D. S. Dhanoa, K. J. Fitch, D. F. Veber, T. F. Walsh and D. L. Williams Jr, GB 2276383 A, 1994; (c) M. M. Butler, WO 2004094370 A2, 2004; (d) G. Q. Shi, J. F. Dropinski, B. M. McKeever, S. Xu, J. W. Becker, J. P. Berger, K. L. MacNaul, A. Elbrecht, G. Zhou, T. W. Doebber, P. Wang, Y.-S. Chao, M. Forrest, J. V. Heck, D. E. Moller and A. B. Jones, J. Med. Chem., 2005, 48, 4457–4468 CrossRef CAS.
- B. G. Raju, H. Odowd, H. Gao, D. V. Patel and J. Trias, WO 2004007444 A2, 2004.
- R. P. Tanpure, C. S. George, T. E. Strecker, L. Devkota, J. K. Tidmore, C.-M. Lin, C. A. Herdman, M. T. MacDonough, M. Sriram, D. J. Chaplin, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem., 2013, 21, 8019–8032 CrossRef CAS PubMed.
- An example where alkyl halide was used as a source of reductant or hydrogen transfer reagent has been reported: A. Martins and M. Lautens, Org. Lett., 2008, 10, 5095–5097 CrossRef CAS PubMed.
- D. J. Cárdenas, C. Mateo and A. M. Echavarren, Angew. Chem., Int. Ed., 1995, 33, 2445–2447 CrossRef.
- The palladacycle intermediates in Catellani reaction also show great reactivity towards alkyl halides. For a review, see: N. D. Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01731d |
| This journal is © The Royal Society of Chemistry 2021 |