DOI:
10.1039/D1SC01611C
(Edge Article)
Chem. Sci., 2021,
12, 8746-8754
Helix-mediated over 1 nm-range chirality recognition by ligand-to-ligand interactions of dinuclear helicates†
Received
20th March 2021
, Accepted 13th May 2021
First published on 19th May 2021
Abstract
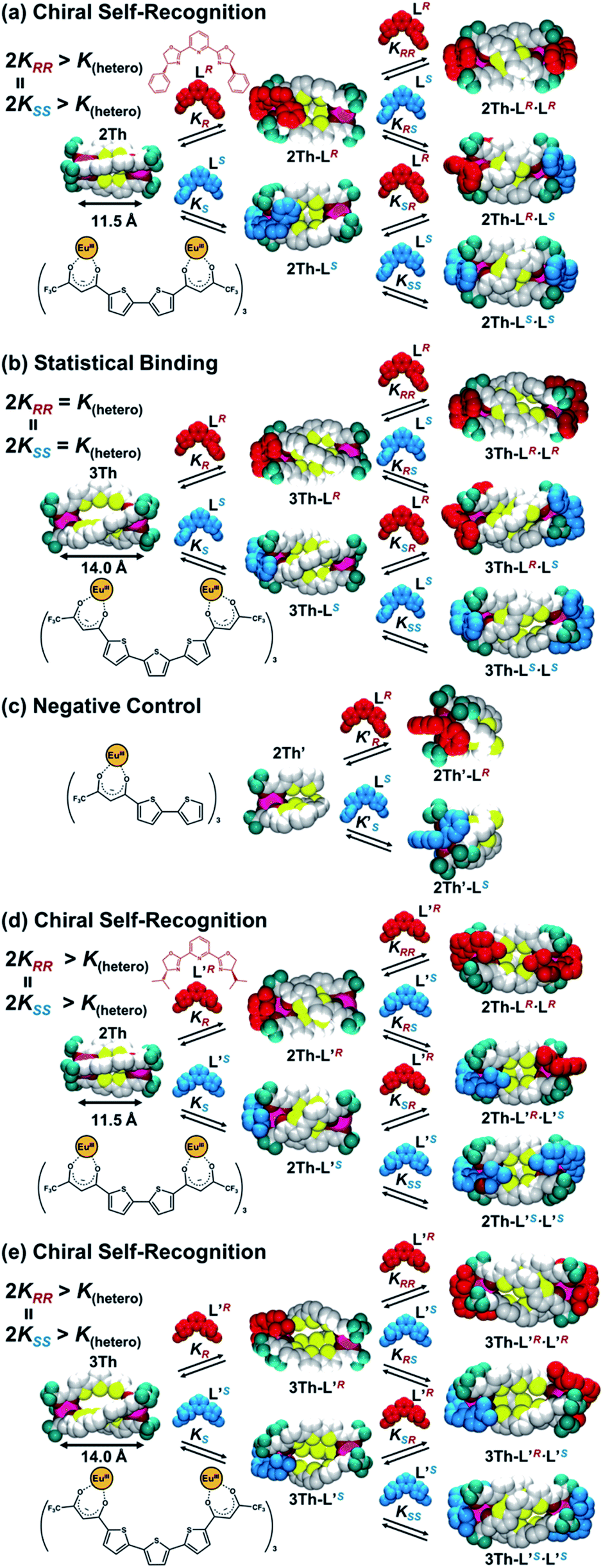
Long-range chirality recognition between the two chiral guest ligands can be tuned based on the helix distances (dLn–Ln = 11.5 and 14.0 Å) of bis-diketonate bridged dinuclear lanthanide complexes (2Th and 3Th, respectively) used as mediators. Both 2Th and 3Th form one-dimensional (1D) helical structures upon terminal binding of two chiral guest co-ligands (LR or LS). Long-range chiral self-recognition is achieved in self-assembly of 2Th with LR and LS to preferentially form homochiral assemblies, 2Th-LR·LR and 2Th-LS·LS, whereas there is no direct molecular interaction between the two guest ligands at the terminal edges. X-ray crystal structure analysis and density functional theory studies reveal that long-range chiral recognition is achieved by terminal ligand-to-ligand interactions between the bis-diketonate ligands and chiral guest co-ligands. Conversely, in self-assembly of 3Th with a longer helix length, statistical binding of LR and LS occurs, forming heterochiral (3Th-LR·LS) and homochiral (3Th-LR·LR and 3Th-LS·LS) assemblies in an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. When phenyl side arms of the chiral guest co-ligands are replaced by isopropyl groups (L′R and L′S), chiral self-recognition is also achieved in the self-assembly process of 3Th with the longer helix length to generate homochiral (3Th-L′R·L′R and 3Th-L′S·L′S) assemblies as the favored products. Thus, subtle modification of the chiral guests is capable of achieving over 1.4 nm-range chirality recognition.
:1 ratio. When phenyl side arms of the chiral guest co-ligands are replaced by isopropyl groups (L′R and L′S), chiral self-recognition is also achieved in the self-assembly process of 3Th with the longer helix length to generate homochiral (3Th-L′R·L′R and 3Th-L′S·L′S) assemblies as the favored products. Thus, subtle modification of the chiral guests is capable of achieving over 1.4 nm-range chirality recognition.
Introduction
Molecular recognition has an important role in chemistry. High-fidelity molecular recognition is typically achieved by short-range direct intermolecular contact that enables recognition of one molecule from another through noncovalent interactions.1 Conversely, long-range molecular recognition is inherently important for understanding how molecular information is recognized and transferred across long distances in supramolecular systems, even without direct intermolecular interactions.2 In this context, long-range molecular recognition is particularly challenging for enantiomers that have the same size and shape, with their only distinguishing feature being the spatial orientations of certain groups.3–9 Such interactions have a pivotal role in various chemical and biological phenomena.
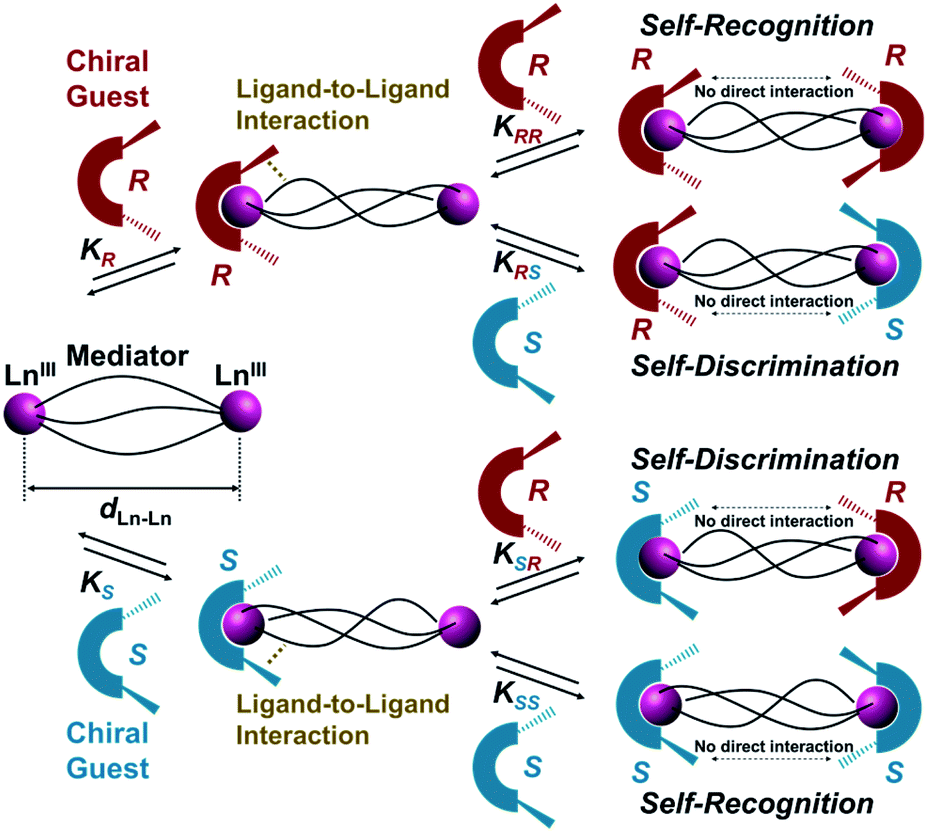
Herein, we report helix-length dependent over 1 nm-range chiral recognition by terminal binding of two chiral guest ligands across a one-dimensional (1D) helix. A helix is intrinsically chiral with either P- or M-helicity, where the preferred helicity is induced by local chirality at the terminal edges. Hence, a helix is a prototypical motif that can mediate terminal chiral information across long distances.10 In particular, a dinuclear helicate is a smart platform for investigating long-range chiral recognition because the helix distance can be tuned by adjusting the number of spacer units immobilized in bridging ligands.11–17 However, most transition metal-based dinuclear helicates (such as M2L3) are coordinatively saturated, therefore guest ligands are not expected to have any access. Conversely, lanthanide-based dinuclear helicates (such as Ln2L3) are often coordinatively unsaturated because of the wide range of coordination numbers (n ≥ 8) of lanthanide(III) ions,12,13 which enable sequential interactions with chiral co-ligands. In this context, we have extensively developed ternary lanthanide self-assembly systems comprising β-diketonate ligands and chiral co-ligands, which revealed that the attractive ligand-to-ligand interactions between β-diketonate ligands and chiral co-ligands give rise to overall chirality in the final self-assembled molecule.18 Such ligand-to-ligand interactions would be suitable for transfer of molecular chirality information across long distances. Scheme 1 illustrates helix-mediated long-range chirality recognition using the ligand-to-ligand interaction approach, where there is no direct molecular interaction between two chiral ligands at the terminal edges. Herein, an initial chiral ligand binding induces P- or M-helicity of a helicate (mediator) through ligand-to-ligand interactions at the terminal edge. When the resulting initial helicate interacts preferably with the second ligand of the same chirality (2KRR = 2KSS > K(hetero)), chiral self-recognition takes place, forming homochiral assemblies. Here, K(hetero) = KRS + KSR = 2KRS = 2KSR (since KRS = KSR). Conversely, chiral self-discrimination occurs when the second ligand binding prefers the opposite chirality (2KRR = 2KSS < K(hetero)) to form heterochiral assemblies. Alternatively, when the initial helicates have no sufficient energy bias to distinguish the same and opposite chirality in the second binding event (2KRR = 2KSS = K(hetero)), statistical binding of both enantiomers takes place. In this study, we show that chirality recognition of two chiral guest co-ligands (L(R or S)) is highly sensitive to the helix length of the mediator, where a shorter helix (dLn–Ln = 11.5 Å, 2Th) causes chiral self-recognition and a longer helix (dLn–Ln = 14.0 Å, 3Th) results in statistical binding. Conversely, subtle modification of the chiral guest ligands (L(R or S) → L′(R or S)) enables even the longer helix (dLn–Ln = 14.0 Å, 3Th) to demonstrate chiral self-recognition. Thus, the present findings provide valuable insights for unravelling the principles of molecular recognition and transfer across long distances in supramolecular systems.
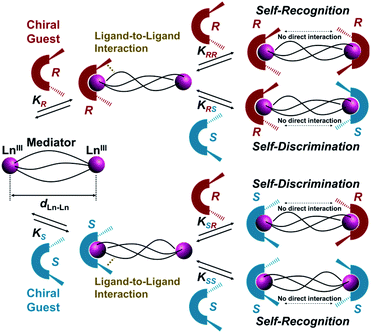 |
| | Scheme 1 Helix-mediated long-range chirality recognition: chiral self-recognition is favored when 2KRR = 2KSS > K(hetero). Conversely, chiral self-discrimination will be predominant when 2KRR = 2KSS < K(hetero). Alternatively, statistical binding will occur when 2KRR = 2KSS = K(hetero). Here, K(hetero) = KRS + KSR = 2KRS = 2KSR. | |
Results and discussion
X-ray structure of dinuclear helicates
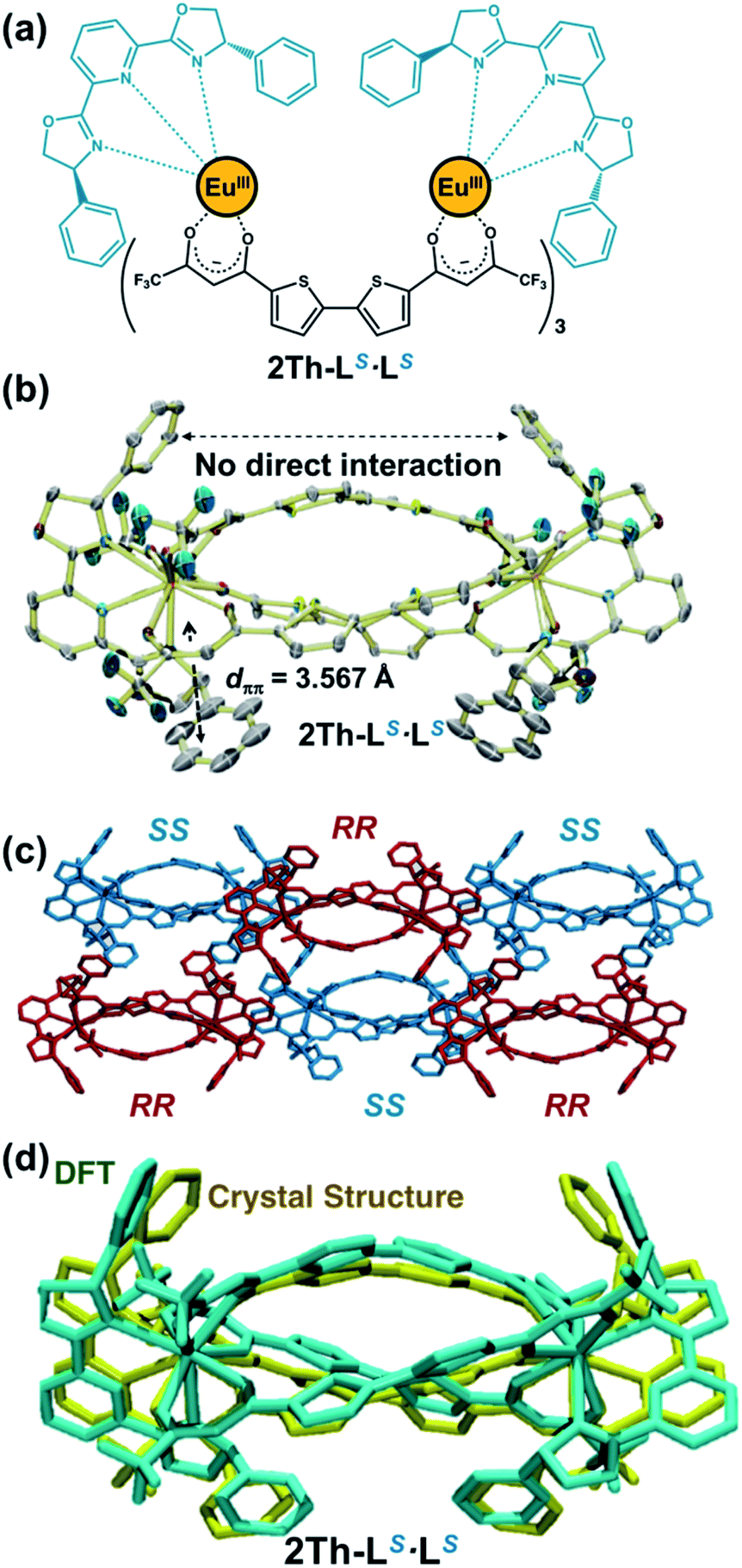
For the chirality mediator, we used selected dinuclear lanthanide complexes (2Th and 3Th) having two and three thiophene spacer units, respectively, which were synthesized by reacting the corresponding bis-diketone ligands (L2 and L3, respectively) and europium acetate in a 3:2 stoichiometry (Fig. S1–S3†).13b In this context, Yan et al. successfully revealed the helix structure of dinuclear lanthanide complexes with the bis-diketone ligands.13b Both 2Th and 3Th formed 1D helical assemblies upon terminal binding of two chiral guest co-ligands [L(R or S) or L′(R or S)] (vide infra).19,20 Among the series of helicate assemblies, we successfully grew crystals suitable for X-ray analysis by slow evaporation of an acetone solution containing 2Th with the presence of 2 equiv. of a racemic mixture of the chiral guest co-ligands (LR and LS) having phenyl side arms. The X-ray structure analysis (Fig. 1) revealed that the both terminal EuIII ions of 2Th were occupied by chiral guest co-ligands of the same chirality to afford homochiral assemblies, 2Th-LR·LR and 2Th-LS·LS, which arranged alternatively in the crystal packing diagram (Fig. 1c). These results indicate that 2Th with chiral guest co-ligands favored ligand chiral self-recognition over chiral self-discrimination and statistical binding. Helical structures were found in the thiophene spacer units, i.e., P-helicity for 2Th-LR·LR and M-helicity for 2Th-LS·LS (Fig. 1c), whose helicity was induced by ligand-to-ligand interactions between the chiral guest co-ligands and the diketonate ligand units.18 While no direct interaction between the two chiral ligands at the terminal edges was detected, intra-complex π–π stacking (dπ–π = 3.567 Å) was indicated between the phenyl rings of LS and the β-diketonate plane (Fig. 1b). Notably, the optimized structure of 2Th-LS·LS calculated by density functional theory (DFT) [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)] reproduced the crystal structure well (Fig. 1d and Table S1†).21 Therefore, rational structural modelling of the other helicates, such as 3Th with a flexible longer thiophene chain length that is not suitable for crystal growth, would be also possible with appropriate use of DFT calculations (vide infra).
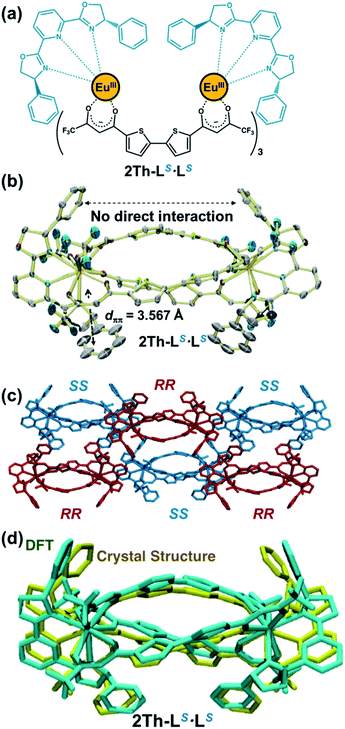 |
| | Fig. 1 (a) Chemical structure, (b) X-ray crystal structure (ORTEP view, 50% probability) of 2Th-LS·LS (CCDC 2036830†). (c) Crystal packing diagrams of the crystal structures of 2Th-LR·LR (red) and 2Th-LS·LS (blue). (d) Overlapping image of X-ray crystal structure (yellow) of 2Th-LS·LS and optimized structure [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)] of 2Th-LS·LS, where the Eu atoms were replaced by La atoms. Hydrogen atoms are omitted for clarity. One of the disordered structures was shown for clarity (Fig. S5†). | |
Allosteric effects on formation of dinuclear helicates
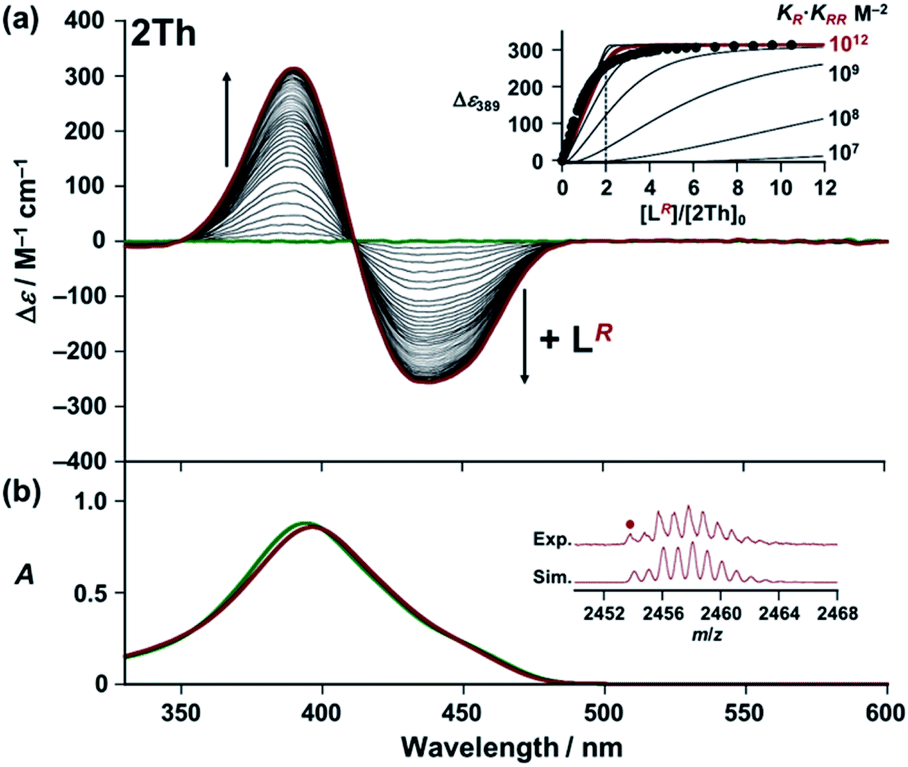
Next, we used circular dichroism (CD) spectroscopy to investigate the helicate formation of 2Th and 3Th through the terminal binding of chiral guest co-ligands [L(R or S) and L′(R or S)] in solution.22–24 Upon addition of the chiral guest co-ligand, 2Th and 3Th began to show biphasic (splitting) CD spectra with intense CD signals, whereas neither 2Th nor 3Th themselves exhibited CD (Fig. 2a and S5–S8†). Conversely, no notable UV-vis absorption spectral changes were observed because of the lack of absorption by the chiral guest co-ligand (LR) in this region (Fig. 2b and ESI Fig. S5–S8†). A titration plot was obtained by plotting Δε at 389 nm against the molar ratio of 2Th and the chiral guest co-ligand ([LR]/[2Th]0), which exhibited saturation at approximately [LR]/[2Th]0 = 2.0 (Fig. 2a inset). Subtle difference between the experimental titration curve and the theoretical one around [LR]/[2Th]0 = 2.0 (as well as less defined saturation point) should be ascribed to a minor contribution of the mono-coordinated species (2Th-LR). This result indicates a 1:2 binding mode between 2Th and LR (Scheme 2a and Fig. S5–S8†) as confirmed by the X-ray structure (vide supra, Fig. 1b). The 2Th-LR·LR assembly was also analyzed by ESI mass spectrometry of 2Th with LR in acetone solution (Fig. 2b inset).25
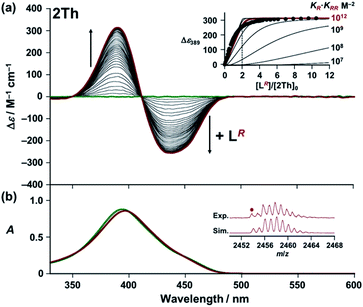 |
| | Fig. 2 (a) CD and (b) UV-vis absorption spectra of 2Th (6.9 × 10−6 M) in the presence of LR [0 (green line) – 7.2 × 10−5 M (red line)] in acetone at 298 K. (Insets) (a) Plot of Δε at 389 nm versus [LR]/[2Th]0. Simulated curves were obtained based on the consecutive binding model (KRR ≫ KR): [LR] = (α/(KR·KRR(1 − α)))0.5 + 2α[2Th]0, using KR·KRR = 107–1014 M−2. Here, [2Th]0 denotes the initial concentration of 2Th, and α = [2Th-LR·LR]/[2Th]0. (b) Negative ESI mass spectrum of 2Th in the presence of LR in acetone with the calculated isotopic distributions for {(Eu)2(L2-2H)3(LR)2 + CH3COCH3 + 2H2O}. For ESI mass spectrum in the whole region (Fig. S9†). | |
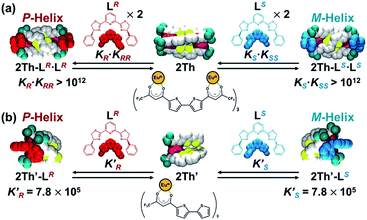 |
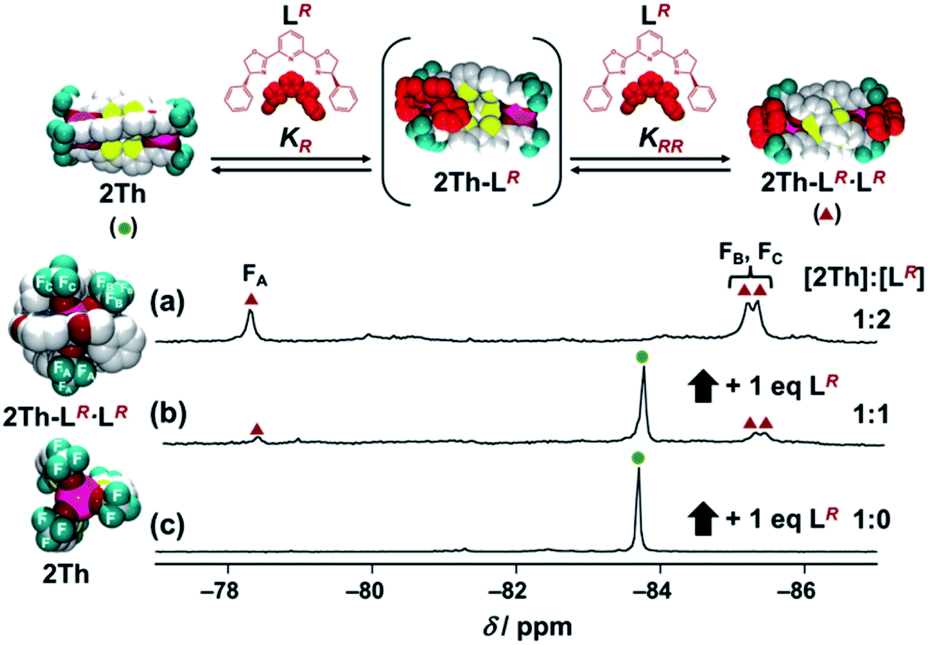
| | Scheme 2 Self-assembly formation of (a) 2Th and (b) 2Th′ with LR (left) and that with LS (right). Here, KR denotes the binding constant between 2Th and LR to form 2Th-LR, and KRR means the binding constant between 2Th-LR and LR to generate the final assembly 2Th-LR·LR. Conversely, K′R denotes the binding constant between 2Th′ and LR to form 2Th′-LR. | |
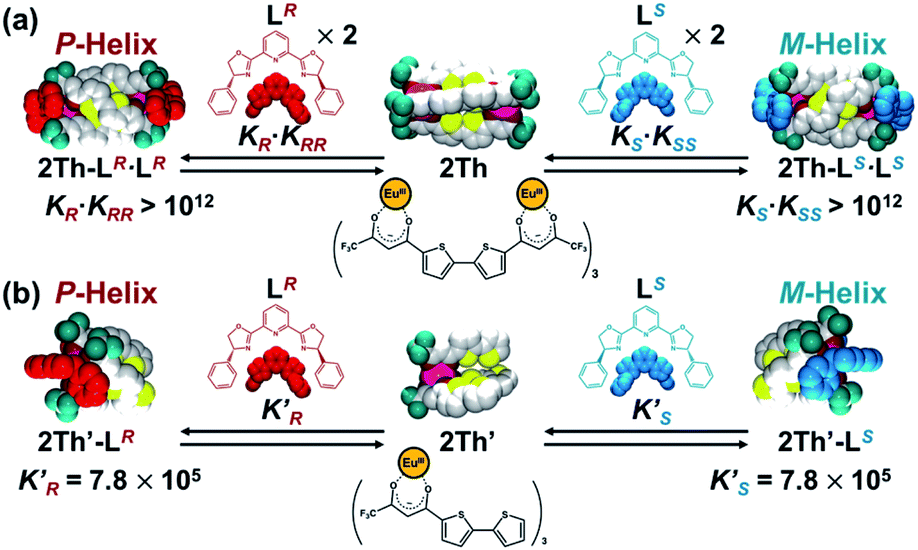
Next, by using 19F NMR spectroscopy, we investigated whether the two chiral guest co-ligands (LR) associate with 2Th sequentially or continuously in terms of allosteric effects.26,27 If the second binding constant is larger than the initial binding constant (KRR ≥ KR), continuous association of the two guest co-ligands (LR) to 2Th will occur to yield the final assembly of 2Th-LR·LR in a formally single step. Conversely when the second binding constant is comparable with the initial binding constant (KRR ≤ KR), association of LR to 2Th will occur sequentially through the mono-coordinated species (2Th-LR). Upon addition of 2 equiv. of LR to 2Th, a single 19F NMR signal at −83.7 ppm assigned to 2Th (itself) split into three signals at −78.3, −85.2, and −85.4 ppm (Fig. 3c to a). Since the original 2Th has a threefold axis, the terminal CF3 groups are in the same chemical environment (Fig. 3c left).18a However, the terminal binding of the chiral guest co-ligands removes this threefold axis and places the three CF3 groups in different chemical environments (Fig. 3a left). Thus, the observed 19F NMR signal split, clearly suggested the formation of a helical structure of 2Th-LR·LR based on terminal binding of the two guest co-ligands (LR) to 2Th (Fig. S5–S8†). Conversely when addition of 1 eq. of LR into 2Th in acetone-d6, 19F NMR signals corresponding to 2Th (green circle) and 2Th-LR·LR (red triangles) were observed, while that corresponding to the mono-coordinated species (2Th-LR) was not detected (Fig. 3b). Further addition of LR (2 eq.) resulted in development of 19F NMR signals due to 2Th-LR·LR (red triangles), where the signals corresponding to 2Th (green circle) were completely disappeared (vide supra, Fig. 3a). Such observation suggested the continuous binding model.
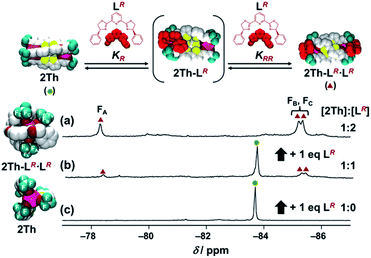 |
| | Fig. 3
19F NMR spectra of 2Th (c) in the absence and presence of (b) 1 eq. and (a) 2 eq. of LR in acetone-d6 at 298 K. Left figures show the optimized structures of 2Th-LR·LR and 2Th [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)]. | |
With these results in hand, we rationalized the above CD titration plot (Fig. 2a inset) based on the theoretical titration curve based on the continuous binding model. Since binding between 2Th and LR is almost stoichiometric judging from the titration curve (Fig. 2a inset), the binding constant (KR·KRR) could not be determined accurately. Conversely, the theoretical titration curve suggested that the observed stoichiometric binding agreed with the binding constant at least larger than 1012 M−2 (KR·KRR > 1012 M−2, red line in Fig. 2a inset). For further verification on the allosteric effects, we prepared a mononuclear complex 2Th′ and a 1:1 binding constant (K′R) with LR was determined to be (7.2 ± 0.7) × 105 M−1 from a CD titration experiment of 2Th′ with LR (Scheme 2b and Fig. S8†). Since the twice value of the resulting 1:1 binding constant (K′R) between 2Th′ and LR is probably nearly equal to the initial binding constant (KR) between 2Th and LR (2K′R ≈ KR), the second binding constant (KRR) should be larger than 6.9 × 105 M−1 (KRR > 6.9 × 105 M−1). Although it could not be concluded whether binding between 2Th and LR follows the continuous binding model based on the estimated binding constants, the determined 4KRR/KR value (4KRR/KR > 1.9) suggested the allosteric effect in the present system. The observed allosteric effect could result from the initial binding event, which causes a conformational change of the helicate suitable for the second binding of the chiral co-ligand with the same chirality. The initial and the second binding energy (E(1)R and E(2)RR, respectively) were estimated from the optimized structures of LR, 2Th, 2Th-LR, and 2Th-LR·LR [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)], where the second binding energy (E(2)RR) is 7.66 kcal mol−1 larger than that of the initial binding energy (E(1)R) (Table 1), which agreed with the experimentally observed allosteric effect.
Table 1 Initial and second binding constants (KR and KRR), binding energy (E(1)R and E(2)RR) in self-assembly formation of 2Th with LR
|
|
K
R
(M−1) |
K
RR
(M−1) |
E
(1)
R
(kcal mol−1) |
E
(2)
RR
(kcal mol−1) |
|
Determined from the titration experiments of 2Th′ with chiral guest co-ligands.
Determined from the titration experiments of 2Th′ and 2Th with the chiral guest co-ligands.
Determined from the optimized structures of LR, 2Th, 2Th-LR, and 2Th-LR·LR [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)].
|
|
|
1.4 × 106 |
>6.9 × 105 |
36.54 |
44.20 |
A DFT prediction on long-range chirality recognition
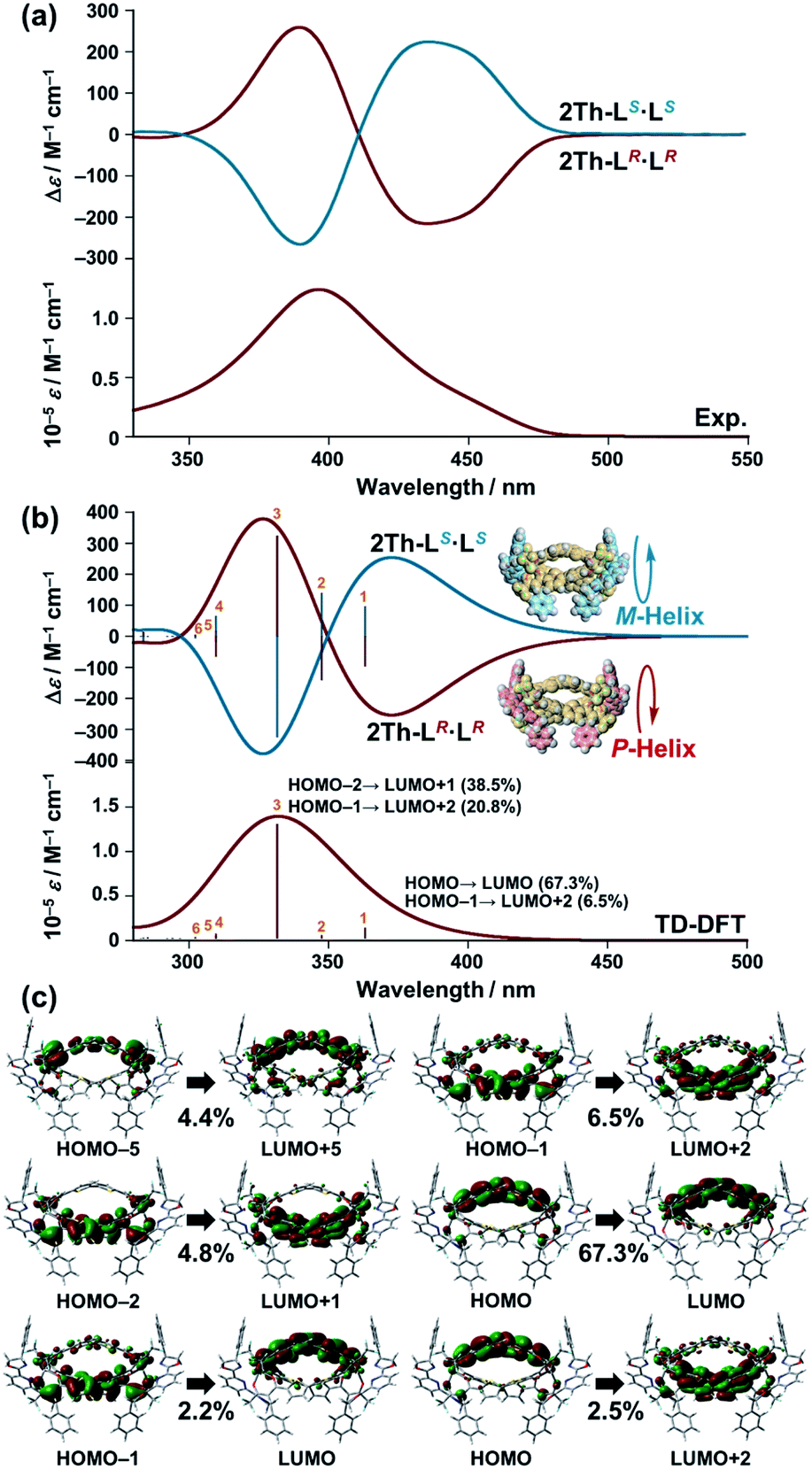
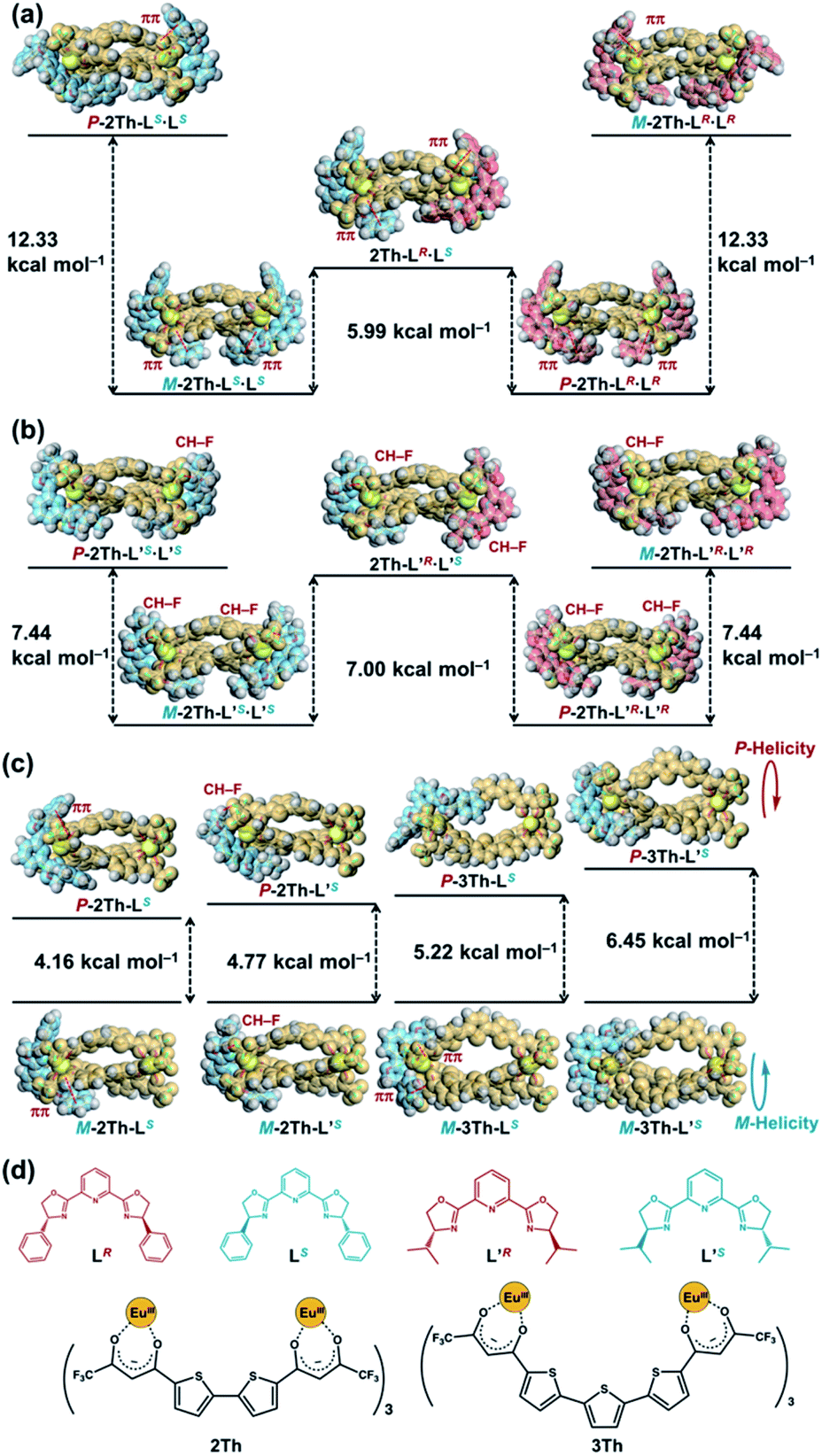
The biphasic CD spectra of 2Th-LR·LR and 2Th-LS·LS (Fig. 4a, red and blue lines, respectively) were successfully reproduced in calculated CD spectra obtained with the use of time-dependent (TD) DFT (Fig. 4b, red and blue lines, respectively), from the structures of 2Th-LR·LR optimized with P-helicity and 2Th-LS·LS optimized with M-helicity (Fig. 4b, inset). The TD DFT calculation suggested that electronic transition corresponding to the first cotton band (excited state 1) arises from molecular orbitals along with the three bridged bis-diketone ligands (Fig. 4c), indicating that the origin of the biphasic CD spectra is excitonic splitting inside the helicates.18 The helix sense of the other helicates was determined in the same manner (Fig. S10–S12†), to reveal that both 2Th and 3Th favor P-helicity with R guest co-ligands (LR and L′R), and M-helicity with S guest co-ligands (LS and L′S). The DFT calculations also indicated that M-2Th-LS·LS and P-2Th-LR·LR are 12.33 kcal mol−1 lower in energy than that of P-2Th-LS·LS and M-2Th-LR·LR (diastereomers), respectively (Fig. 5a). The M-2Th-LS·LS and P-2Th-LR·LR have two ligand-to-ligand interactions (π–π interactions) with the guest co-ligands, while P-2Th-LS·LS and M-2Th-LR·LR (diastereomers) contain only one ligand-to-ligand interaction inside the assembly (Fig. 5a). In the same manner, the optimized structures of M-2Th-L′S·L′S and P-2Th-L′R·L′R are 7.44 kcal mol−1 lower in energy than that of P-2Th-L′S·L′S and M-2Th-L′R·L′R (diastereomers), respectively (Fig. 5b). These DFT results can explain the fact that 2Th favored P-helicity with the R-guest co-ligands (LR and L′R), and M-helicity with the S-guest co-ligands (LS and L′S). More importantly, the DFT calculations predict that homochiral assemblies (2Th-LR·LR, 2Th-LS·LS, 2Th-L′R·L′R, and 2Th-L′S·L′S) are 5.99–7.00 kcal mol−1 lower in energy than that of heterochiral assemblies (2Th-LR·LS and 2Th-L′R·L′S) [Fig. 5a and b].28 Additionally, the DFT calculations provide an important prediction concerning on the performed complexes (1:1 complexes): even the initial terminal binding of the chiral guest co-ligands will induce the helical conformation of 2Th and 3Th, where the M-helix structures (1:1 complexes) with the S-guest co-ligands (LS and L′S) are 4.16–6.45 kcal mol−1 lower in energy than that of the P-helix structures (Fig. 5c) as in the case of the final assemblies (1:2 complexes).
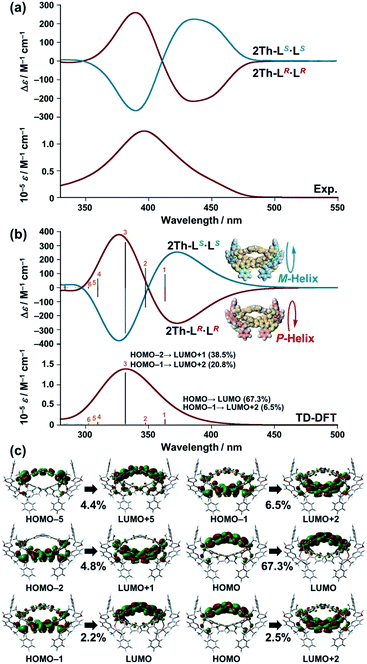 |
| | Fig. 4 (a) Experimentally obtained UV-vis and CD spectra of 2Th-LR·LR (red line) and 2Th-LS·LS (blue line) in acetone. (b) Theoretical CD spectrum [time dependent-DFT/CAM-B3LYP-6-31G(d) [C H N O S F]/LANL2DZ (Sc)] of the optimized structure [DFT/CAM-B3LYP-6-31G(d) [C H N O S F]/LANL2DZ (Sc)] of P-2Th-LR·LR and M-2Th-LS·LS, where Eu atoms are replaced by Sc atoms to reduce the calculation complexity (Fig. S10–S12†). (c) Summary of excited state 1. | |
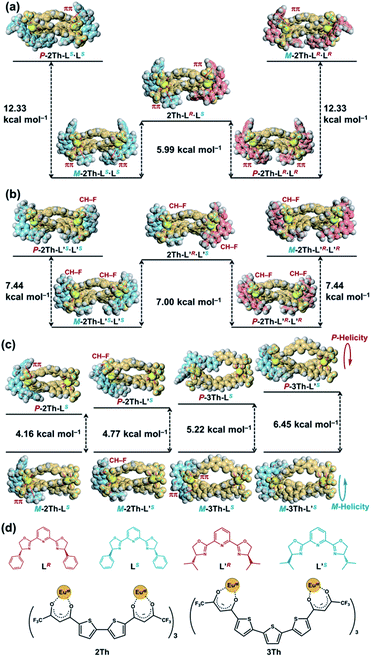 |
| | Fig. 5 Relative energy difference between the optimized structures of (a) P-2Th-LS·LS, M-2Th-LS·LS, 2Th-LR·LS, P-2Th-LR·LR, and M-2Th-LR·LR, (b) P-2Th-L′S·L′S, M-2Th-L′S·L′S, 2Th-L′R·L′S, P-2Th-L′R·L′R, and M-2Th-L′R·L′R, (c) M-2Th-LS and P-2Th-LS, M-2Th-L′S and P-2Th-L′S, M-3Th-LS and P-3Th-LS, M-3Th-L′S and P-3Th-L′S [DFT/CAM-B3LYP/def2SVP (C H N O S F)/def2TZVPP (La)], where the Eu atoms were replaced by La atoms. (d) Chemical structures of LR, LS, L′R, L′S, 2Th, and 3Th. | |
Helix length-dependent over 1 nm-range chirality recognition
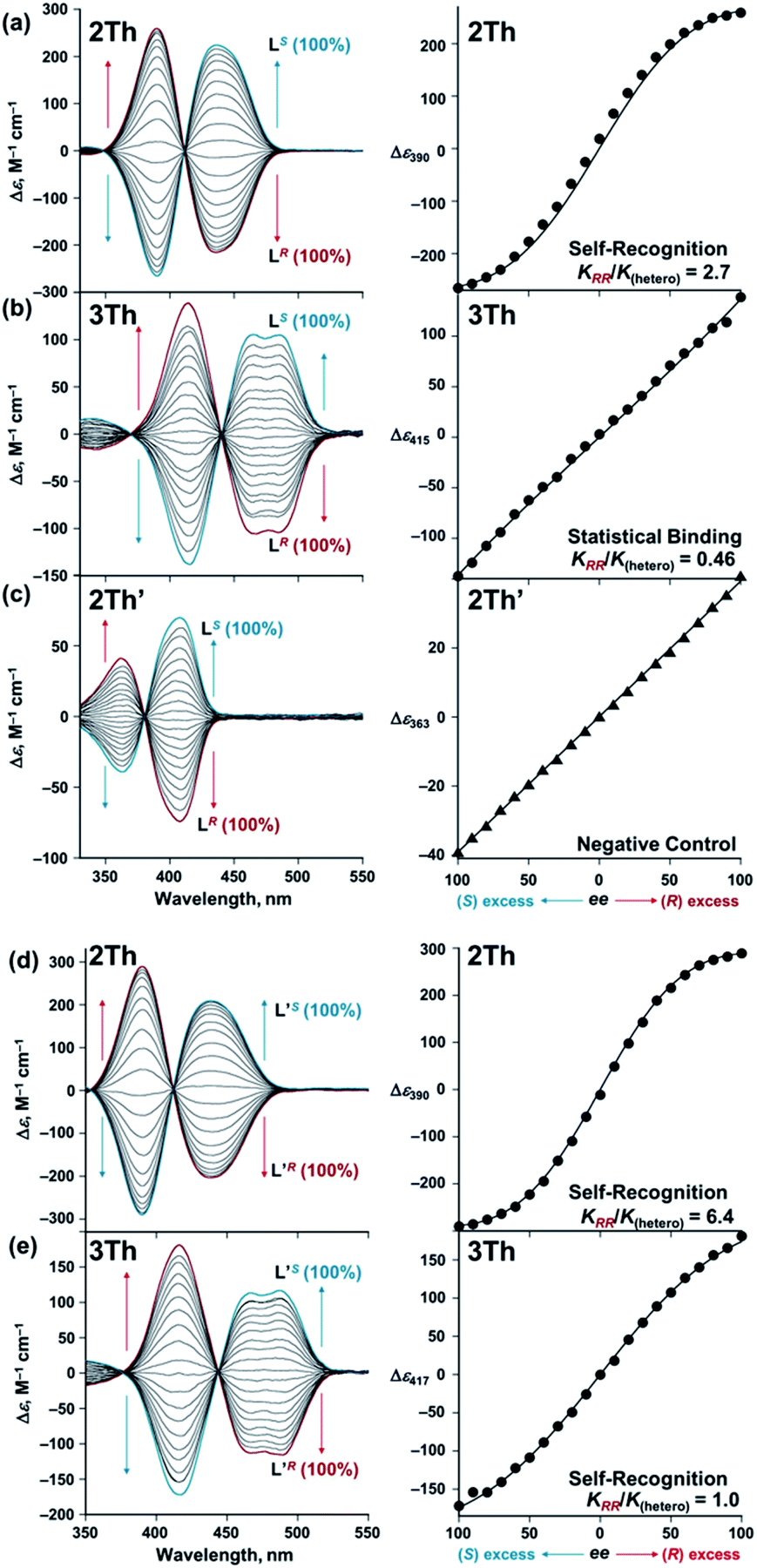
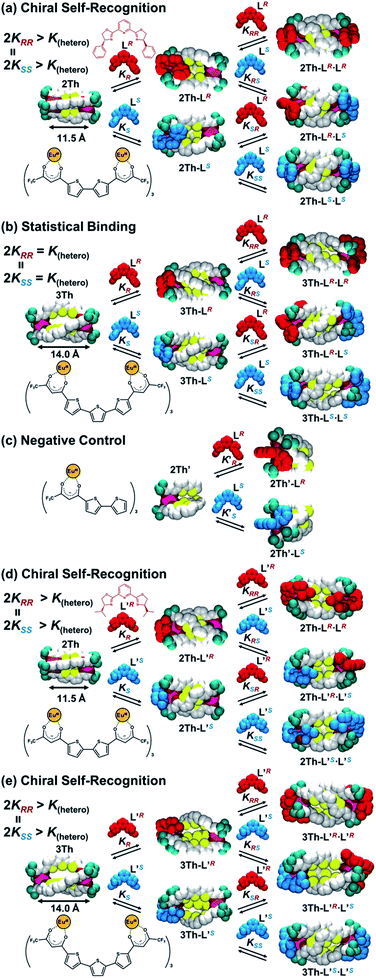
On the basis of these results, we investigated the effects of the helix length on the long-range chirality recognition by monitoring the induced CD signals of 2Th and 3Th (dLn–Ln = 11.5 and 14.0 Å, respectively) on changing the enantiomeric excess (ee) of the chiral guest co-ligands in the range from ee = ([R] − [S])/([R] + [S]) = 0 to +1 and to −1 (Fig. 6 left, red and blue lines, respectively). Here, the total concentration of chiral ligands ([R] + [S]) remained constant at each molar ratio, ca. a 90-fold excess against 2Th and 3Th. Such excess conditions are required for a clear distinguishment on whether the plot of Δε versus ee (Fig. 6 right) follows the positive nonlinear (sigmoidal) or linear relationship (vide infra). A positive nonlinear relationship was clearly observed for the combination of 2Th with LR and LS (Fig. 6a, right). The experimentally obtained nonlinear relationship is explained by eqn (1) derived from Scheme 3a (Appendix 1†):| | | Δε/Δε∞ = 4ee(KRR/K(hetero))/(2(1 + ee2)(KRR/K(hetero)) + (1 − ee2)) | (1) |
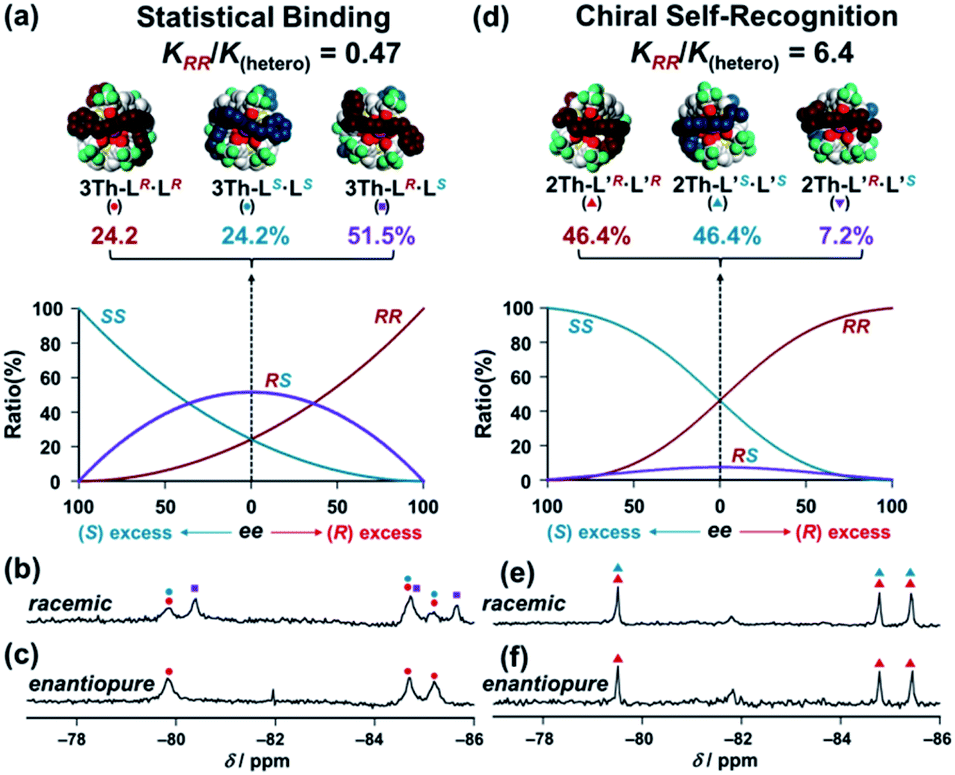
where Δε∞ denotes the Δε value of the homochiral assemblies at ee = +1. A positive nonlinear relationship is expected when the system follows chiral-self recognition because KRR/K(hetero) > 0.50, and the degree of sigmoidal character increases together with the KRR/K(hetero) value (here, K(hetero) = KRS + KSR = 2KRS = 2KSR).29,30 The observed positive nonlinear relationship (Fig. 6a, right) agreed well with the calculated curve with KRR/K(hetero) = 2.7, with the use of eqn (1). Therefore, chiral self-recognition occurred in the self-assembly process of 2Th with LR and LS (Scheme 3a). The larger KRR value than that of KRS suggested a higher thermodynamic stability of the homochiral 2Th-LR·LR over the heterochiral 2Th-LR·LS, which is consistent with the above energy estimation from their DFT-optimized structures (ΔE = 5.99 kcal mol−1, Fig. 5a). Notably, the degree of chiral self-recognition is highly temperature dependent. Whereas a high KRR/K(hetero) value was obtained at lower temperature, KRR/K(hetero) = 2.4 at 323 K, 2.7 at 298 K, and 7.0 at 273 K (Table 2 and Fig. S15†). This trend implies a major role of the enthalpy term in the chiral-self-recognition nature, which likely arises from ligand-to-ligand interactions between 2Th and LR or LS (vide supra, Fig. 1b). Conversely, 3Th with a longer helix length (dLn–Ln = 14.0 Å) had an almost completely linear relationship between Δε and ee in their self-assembly process with LR and LS (Fig. 6b, right). The linear relationship between Δε and ee can be described by eqn (1) with KRR/K(hetero) = 0.46, suggesting statistical binding of LR and LS to 3Th (Scheme 3b). The value of KRR/K(hetero) = 0.46 indicates that homochiral (3Th-LR·LR and 3Th-LS·LS) and heterochiral assemblies (3Th-LR·LS) have almost the same thermodynamic stability, which is likely attributed to the longer thiophene spacer units of 3Th that create flexibility in the helix chain and prohibit effective chirality information transfer.28 Hence, the value of KRR/K(hetero) in self-assembly of 3Th with LR and LS was almost temperature independent (Table 2 and Fig. S16†). Conversely, a complete linear relationship was success-fully obtained between Δε and ee during the self-assembly of mononuclear complex 2Th′ with LR and LS (Fig. 6c, right), where 2Th′ exhibited induced biphasic CD signals (Fig. 6c, left) similar to the case of the dinuclear helicates (2Th and 3Th). Therefore, the negative control experiments revealed that even the initial terminal binding of LR or LS to 2Th induced helicate structures prior to the final assembly (Scheme 3), which is also in good agreement with the DFT prediction (Fig. 5c, vide supra). Conversely, when the phenyl groups of LR and LS were replaced by isopropyl groups (L′R and L′S, respectively), a higher degree of chiral self-recognition was achieved by a combination of 2Th with L′R and L′S in terms of a higher value of KRR/K(hetero) (2.7 → 6.4, Fig. 6d and Table 1), where the chiral self-recognition feature was maximized at 273 K (KRR/K(hetero) > 10.0, Fig. S15† and Table 2). The above DFT calculations indicate that homochiral 2Th-L′S·L′S is 7.00 kcal mol−1 lower in energy than that of heterochiral 2Th-L′R·L′S (Fig. 5b), which was larger than that between 2Th-LS·LS and 2Th-LR·LS (5.99 kcal mol−1, Fig. 5a). Notably, a plot of Δε versus ee obtained in combination of 3Th with L′R and L′S suggested KRR/K(hetero) = 1.0 (Fig. 6e), indicating that chiral self-recognition can be achieved even for 3Th with longer helix lengths (Scheme 3e and Fig. S17–S19†).
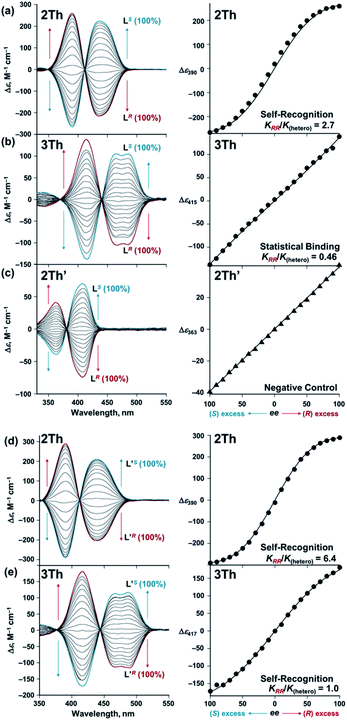 |
| | Fig. 6 Left: CD spectra of (a) and (d) 2Th (7.2 × 10−6 M), (b) and (e) 3Th (5.3 × 10−6 M), and (c) 2Th′ (9.4 × 10−6 M) in the presence of different enantiomeric excess (ee) of L(R or S) [total concentrations: (a) 6.6 × 10−4 M, (b) 4.9 × 10−4 M, (c) 8.7 × 10−4 M], L′ (R or S) [total concentrations: (d) 7.5 × 10−4 M and (e) 5.1 × 10−4 M] in acetone at 298 K. Right: plots of Δε versus ee of (a)–(c) L(R or S), (d) and (e) L′ (R or S). | |
 |
| | Scheme 3 Summary of self-sorting for LR and LS with (a) 2Th, (b) 3Th, (c) 2Th′, and those for L′R and L′S with (d) 2Th and (e) 3Th, where K(hetero) = KRS + KSR = 2KRS = 2KSR. | |
Table 2 Ratios of binding constants (KRR/K(hetero)) in self-assembly formation of 2Th and 3Th with chiral guest co-ligands, where K(hetero) = KRS + KSR = 2KRS = 2KSR
|
|
2Th
K
RR
/K(hetero) |
3Th
K
RR
/K(hetero) |
| 323 K |
298 K |
273 K |
323 K |
298 K |
273 K |
| R = Ph (LR, LS) |
2.4 |
2.7 |
7.0 |
0.70 |
0.46 |
0.56 |
| R = iPr (L′R, L′S) |
4.6 |
6.4 |
>10.0 |
0.90 |
1.00 |
1.05 |
Finally, conclusive evidence for the chiral self-recognition versus statistical binding mechanism was obtained by 19F NMR measured under enantiopure and racemic conditions of the chiral guest co-ligands (Fig. 7). Under the racemic conditions ([3Th]:[LR]:[LS] = 1:1:1), the 19F NMR spectrum of 3Th (Fig. 7b) had new signals (purple squares) other than those corresponding to the homochiral assemblies (3Th-LR·LR and 3Th-LS·LS, red and blue circles) observed under the enantiopure conditions ([3Th]:[LR]:[LS] = 1:2:0, Fig. 7c). Hence, the new signals (Fig. 7b, purple squares) are responsible for the heterochiral assembly(3Th-LR·LS) formed under the racemic conditions. The ratio between the homochiral and heterochiral assemblies was determined to be [3Th-LR·LR + 3Th-LS·LS]:[3Th-LR·LS] = 1.0:1.2 from the integrated ratio of the 19F NMR signals, which agreed well with the theoretical product ratio at ee = 0 calculated from KRR/K(hetero) = 0.46 (Fig. 7a). This assignment is consistent with the statistical binding mechanism for a self-assembly process of 3Th with LR and LS (Scheme 3b). Conversely, the 19F NMR spectrum of 2Th measured under racemic conditions ([2Th]:[L′R]:[L′S] = 1:1:1, Fig. 7e) was identical to that obtained under the enantiopure conditions ([2Th]:[L′R]:[L′S] = 1:2:0, Fig. 7f). The theoretical product ratio between homochiral and heterochiral assemblies is [2Th-L′R·L′R + 2Th-L′S·L′S]:[2Th-L′R·L′S] = 92.8:7.2 (at ee = 0) calculated from KRR/K(hetero) = 6.4 (Fig. 4d),31 which also underlines the strong chiral self-recognition behavior for self-assembly of 2Th with L′R and L′S (Scheme 3d).
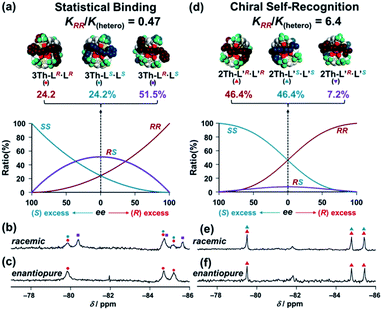 |
| | Fig. 7 Theoretical product ratio at ee = 0 between (a) 3Th-LR·LR (red) and 3Th-LS·LS (blue) and 3Th-LR·LS (purple) calculated based on eqn (1) with KRR/K(hetero) = 0.47, (d) 2Th-L′R·L′R (red) and 2Th-L′S·L′S (blue) and 2Th-L′R·L′S (purple) calculated based on eqn (1) with KRR/K(hetero) = 6.4, where K(hetero) = KRS + KSR = 2KRS = 2KSR. 19F NMR spectra of 3Th under the (b) racemic conditions [3Th]:[LR]:[LS] = 1:1:1, (c) enantiopure conditions [3Th]:[LR]:[LS] = 1:2:0, those of 2Th under the (e) racemic conditions [2Th]:[LR]:[LS] = 1:1:1, (f) enantiopure conditions [2Th]:[LR]:[LS] = 1:2:0, in acetone-d6 at 298 K. Symbols correspond to those for the structures of (a) and (d), red circles: 3Th-LR·LR, blue circles: 3Th-LS·LS, purple squares: 3Th-LR·LS, red triangles: 2Th-L′R·L′R, blue triangles: 2Th-L′S·L′S. | |
Conclusions
In conclusion, we successfully demonstrated that helix chain (mediator) length can control when long-range chiral recognition occurs through chiral self-recognition or in statistical manner. Self-assembly of the shorter helicate 2Th (dLn–Ln = 11.5 Å) with chiral guest co-ligands (LR and LS) resulted in chiral self-recognition (Scheme 3a), forming mainly homochiral assemblies (2Th-LR·LR and 2Th-LS·LS) rather than heterochiral assemblies (2Th-LR·LS). Conversely, self-assembly of the longer helicate 3Th (dLn–Ln = 14.0 Å) with LR and LS occurred in a statistical binding manner (Scheme 3b), resulting in formation of the heterochiral (3Th-LR·LS) and homochiral assemblies (3Th-LR·LR and 3Th-LS·LS) in an almost 1:1 ratio. Subtle modification on the side arms of the chiral guest co-ligands (L(R or S) → L′(R or S)) enables even the longer helicate 3Th to achieve over 1.4 nm-range chirality recognition (Scheme 3e). Thus, the present findings open up opportunities for rational planning of molecular communication through the use of chirality as simple chemical input across long distances.
Author contributions
N. S., H. I., and N. O. performed the synthesis and characterization of materials, and also contributed the titration experiments. Y. O. contributed the analysis of the X-ray crystal structure. J. Y. designed the study, analysed the experimental data and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Numbers JP19H02693, JP19K22207, and JP19H04596 in Scientific Research on Innovative Areas “Coordination Asymmetry”, Grant-in-Aid for the ASAHI Glass Foundation. We thank the Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Notes and references
- G. Cooke and V. M. Rotello, Chem. Soc. Rev., 2002, 31, 275 RSC.
- L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555 RSC.
-
(a) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed;
(b) H. Jędrzejewska and A. Szumna, Chem. Rev., 2017, 117, 4863 CrossRef PubMed;
(c) R. L. Greenaway, V. Santolini, A. Pulido, M. A. Little, B. M. Alston, M. E. Briggs, G. M. Day, A. I. Cooper and K. E. Jelfs, Angew. Chem., Int. Ed., 2019, 58, 16275 CrossRef CAS PubMed;
(d) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed.
-
(a) J. Zhong, L. Zhang, D. P. August, G. F. S. Whitehead and D. A. Leigh, J. Am. Chem. Soc., 2019, 141, 14249 CrossRef CAS PubMed;
(b) S. Kai, T. Kojima, F. L. Thorp-Greenwood, M. J. Hardiea and S. Hiraoka, Chem. Sci., 2018, 9, 4104 RSC;
(c) E. Chinnaraja, R. Arunachalam, E. Suresh, S. K. Sen, R. Natarajan and P. S. Subramanian, Inorg. Chem., 2019, 58, 4465 CrossRef CAS PubMed;
(d) V. E. Pritchard, D. Rota Martir, S. Oldknow, S. Kai, S. Hiraoka, N. J. Cookson, E. Zysman-Colman and M. J. Hardie, Chem.–Eur. J., 2017, 23, 6290 CrossRef CAS PubMed;
(e) S. A. Boer and D. R. Turner, Chem. Commun., 2015, 51, 17375 RSC;
(f) O. Gidron, M. Jirásek, N. Trapp, M. O. Ebert, X. Zhang and F. Diederich, J. Am. Chem. Soc., 2015, 137, 12502 CrossRef CAS PubMed;
(g) Y. Tsunoda, M. Takatsuka, R. Sekiya and T. Haino, Angew. Chem., Int. Ed., 2017, 56, 2613 CrossRef CAS PubMed;
(h) W. Naito, K. Urakawa, R. Sato, Y. Shigeta, N. Yasuda and H. Maeda, Org. Biomol. Chem., 2019, 17, 1163 RSC.
-
(a) C. Kaes, M. W. Hosseini, C. E. F. Rickard, B. W. Skelton and A. H. White, Angew. Chem., Int. Ed., 1998, 37, 920 CrossRef CAS;
(b) M. A. Masood, E. J. Enemark and T. D. P. Stack, Angew. Chem., Int. Ed., 1998, 37, 928 CrossRef;
(c) S. G. Telfer, T. Sato, R. Kuroda, J. Lefebvre and D. B. Leznoff, Inorg. Chem., 2004, 43, 421 CrossRef CAS PubMed;
(d) T. W. Kim, M. S. Lah and J.-I. Hong, Chem. Commun., 2001, 743 RSC;
(e) J. M. Rowland, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2002, 41, 1545 CrossRef CAS PubMed;
(f) G. Zhang, G. Gil-Ramírez, A. Markevicius, C. Browne, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2015, 137, 10437 CrossRef CAS PubMed.
-
(a) L. Volbach, N. Struch, F. Bohle, F. Topić, G. Schnakenburg, A. Schneider, K. Rissanen, S. Grimme and A. Lützen, Chem.–Eur. J., 2020, 26, 3335 CrossRef CAS PubMed;
(b) J. Anhäuser, R. Puttreddy, L. Glanz, A. Schneider, M. Engeser, K. Rissanen and A. Lützen, Chem.–Eur. J., 2019, 25, 12294 CrossRef PubMed;
(c) T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562 CrossRef CAS PubMed.
-
(a) D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244 CrossRef CAS PubMed;
(b) L. Y. Yao, T. K. M. Lee and V. W. W. Yam, J. Am. Chem. Soc., 2016, 138, 7260 CrossRef CAS PubMed;
(c) C. G. Claessens and T. Torres, J. Am. Chem. Soc., 2002, 124, 14522 CrossRef CAS PubMed.
-
(a) S. Iseki, K. Nonomura, S. Kishida, D. Ogata and J. Yuasa, J. Am. Chem. Soc., 2020, 142, 15842 CrossRef CAS PubMed;
(b) D. Ogata and J. Yuasa, Chem. Commun., 2020, 56, 8679 RSC;
(c) K. Nonomura and J. Yuasa, Inorg. Chem., 2019, 58, 6474 CrossRef CAS PubMed;
(d) Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., Int. Ed., 2018, 57, 8973 CrossRef CAS PubMed;
(e) Y. Imai and J. Yuasa, Chem. Sci., 2019, 10, 4236 RSC;
(f) Y. Imai and J. Yuasa, Chem. Commun., 2019, 55, 4095 RSC;
(g) D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424 CrossRef CAS PubMed.
-
(a) L. L. Yan, C. H. Tan, G. L. Zhang, L. P. Zhou, J. C. Bünzli and Q. F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed;
(b) Y. Zhou, H. Li, T. Zhu, T. Gao and P. Yan, J. Am. Chem. Soc., 2019, 141, 19634 CrossRef CAS PubMed;
(c) C.-T. Yeung, K.-H. Yim, H.-Y. Wong, R. Pal, W.-S. Lo, S.-C. Yan, M. Yee-Man Wong, D. Yufit, D. E. Smiles, L. J. McCormick, S. J. Teat, D. K. Shuh, W.-T. Wong and G.-L. Law, Nat. Commun., 2017, 8, 1128 CrossRef PubMed;
(d) H.-Y. Wong, W.-S. Lo, K.-H. Yim and G.-L. Law, Chem, 2019, 5, 3058 CrossRef CAS;
(e) J. Lisowski, Inorg. Chem., 2011, 50, 5567 CrossRef CAS PubMed;
(f) M. Iwamura, M. Fujii, A. Yamada, H. Koike and K. Nozaki, Chem.–Asian J., 2019, 14, 561 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
-
(a) M. Albrecht, Chem. Rev., 2001, 101, 3457 CrossRef CAS PubMed;
(b) M. Boiocchi and L. Fabbrizzi, Chem. Soc. Rev., 2014, 43, 1835 RSC.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244 RSC.
-
(a) J. Shi, Y. Hou, W. Chu, X. Shi, H. Gu, B. Wang and Z. Sun, Inorg. Chem., 2013, 52, 5013 CrossRef CAS PubMed;
(b) B. Li, H. Li, P. Chen, W. Sun, C. Wang, T. Gao and P. Yan, Phys. Chem. Chem. Phys., 2015, 17, 30510 RSC;
(c) B. Li, H. Li, P. Chen, W. Sun, C. Wang, T. Gao and P. Yan, Dalton Trans., 2016, 45, 11459 RSC.
- B. El Aroussi, S. Zebret, C. Besnard, P. Perrottet and J. Hamacek, J. Am. Chem. Soc., 2011, 133, 10764 CrossRef CAS PubMed.
-
(a) A. M. Johnson, M. C. Young, X. Zhang, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2013, 135, 17723 CrossRef CAS PubMed;
(b) A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2015, 54, 5641 CrossRef CAS PubMed.
-
(a) T. Lathion, L. Guénée, C. Besnard, A. Bousseksou and C. Piguet, Chem.–Eur. J., 2018, 24, 16873 CrossRef CAS PubMed;
(b) D. Zare, Y. Suffren, H. Nozary, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2017, 56, 14612 CrossRef CAS PubMed.
-
(a) Y. Zhou, Y. Yao, Z. Cheng, T. Gao, H. Li and P. Yan, Inorg. Chem., 2020, 59, 12850 CrossRef CAS PubMed;
(b) D. Liu, Y. Zhou, Y. Zhang, H. Li, P. Chen, W. Sun, T. Gao and P. Yan, Inorg. Chem., 2018, 57, 8332 CrossRef CAS PubMed;
(c) W. Chen, X. Tang, W. Dou, B. Wang, L. Guo, Z. Ju and W. Liu, Chem.–Eur. J., 2017, 23, 9804 CrossRef CAS PubMed;
(d) C.-T. Yeung, W. T. K. Chan, S.-C. Yan, K.-L. Yu, K.-H. Yim, W.-T. Wong and G.-L. Law, Chem. Commun., 2015, 51, 592 RSC.
-
(a) T. Y. Bing, T. Kawai and J. Yuasa, J. Am. Chem. Soc., 2018, 140, 3683 CrossRef CAS PubMed;
(b) J. Yuasa, T. Ohno, K. Miyata, H. Tsumatori, Y. Hasegawa and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9892 CrossRef CAS PubMed;
(c) Y. Okayasu and J. Yuasa, Mol. Syst. Des. Eng., 2018, 3, 66 RSC;
(d) Y. B. Tan, M. Yamada, S. Katao, Y. Nishikawa, F. Asanoma, J. Yuasa and T. Kawai, Inorg. Chem., 2020, 59, 12867 CrossRef CAS PubMed;
(e) Y. B. Tan, Y. Okayasu, S. Katao, Y. Nishikawa, F. Asanoma, M. Yamada, J. Yuasa and T. Kawai, J. Am. Chem. Soc., 2020, 142, 17653 CrossRef CAS PubMed.
- G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119 CrossRef CAS PubMed.
- A. de Bettencourt-Dias, P. S. Barber and S. Bauer, J. Am. Chem. Soc., 2012, 134, 6987 CrossRef CAS PubMed.
- M. Leonzio, A. Melchior, G. Faura, M. Tolazzi, F. Zinna, L. Di Bari and F. Piccinelli, Inorg. Chem., 2017, 56, 4413 CrossRef CAS PubMed.
-
(a) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673 RSC;
(b) G. Muller, Dalton Trans., 2009, 9692–9707 RSC;
(c) D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977 CrossRef CAS PubMed;
(d) C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS;
(e) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC;
(f) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1 CrossRef CAS.
- J. M. Dragna, G. Pescitelli, L. Tran, V. M. Lynch, E. V. Anslyn and L. Di Bari, J. Am. Chem. Soc., 2012, 134, 4398 CrossRef CAS PubMed.
- S. P. Morcillo, D. Miguel, L. Á. de Cienfuegos, J. Justicia, S. Abbate, E. Castiglioni, C. Bour, M. Ribagorda, D. J. Cárdenas, J. M. Paredes, L. Crovetto, D. Choquesillo-Lazarte, A. J. Mota, M. C. Carreño, G. Longhi and J. M. Cuerva, Chem. Sci., 2016, 7, 5663 RSC.
- The negative charge may come from reduction of EuIII or the bis-diketone ligand having thiophene spacers during the ionization process of mass.
-
(a) L. Babel, T. N. Y. Hoang, L. Guénée, C. Besnard, T. A. Wesolowski, M. Humbert-Droz and C. Piguet, Chem.–Eur. J., 2016, 22, 8113 CrossRef CAS PubMed;
(b) A. Zaïm, H. Nozary, L. Guénée, C. Besnard, J.-F. Lemonnier, S. Petoud and C. Piguet, Chem.–Eur. J., 2012, 18, 7155 CrossRef PubMed.
- The small 19F signals should be ascribed to a minor complex (e.g., coordination isomer) or the minor contribution of 2Th-LR.
- Number of atoms in the 3Th-assemblies is too large to perform frequency calculation required for the zero point energy correction, and therefore the energy estimation of the assemblies of the 3Th-assemblies could not be accomplished in this study (Fig. S13 and S14†).
-
(a) D. Guillaneux, S.-H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430 CrossRef CAS;
(b) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef CAS.
- Self-recognition and self-discrimination was distinguished by whether the KRR (or KSS) values are the half of the K(hetero) value.
-
19F NMR signals due to the heterochiral assembly (2Th-L′R·L′S) should be less than detection level.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and additional data. CCDC 2036830. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01611c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*