
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding polyoxometalates as water oxidation catalysts through iron vs. cobalt reactivity†
Khalid
Azmani
ab,
Maria
Besora
 *b,
Joaquín
Soriano-López
*c,
Meriem
Landolsi‡
b,
Anne-Lucie
Teillout
d,
Pedro
de Oliveira
d,
Israël-Martyr
Mbomekallé
d,
Josep M.
Poblet
*b and
José-Ramón
Galán-Mascarós
ae
*b,
Joaquín
Soriano-López
*c,
Meriem
Landolsi‡
b,
Anne-Lucie
Teillout
d,
Pedro
de Oliveira
d,
Israël-Martyr
Mbomekallé
d,
Josep M.
Poblet
*b and
José-Ramón
Galán-Mascarós
ae
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans, 16, Tarragona, E-43007, Spain
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo 1, E-43007 Tarragona, Spain. E-mail: maria.besora@urv.cat; josepmaria.poblet@urv.cat
cSchool of Chemistry & AMBER Center, Trinity College, University of Dublin, Dublin, D02 PN40, Ireland. E-mail: sorianoj@tcd.ie
dEquipe d'Electrochimie et de Photo-électrochimie, Institut de Chimie Physique, UMR 8000, CNRS, Université Paris Saclay, Orsay, F-91405, France
eICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 19th May 2021
Abstract
Cobalt polyoxometalates (Co-POMs) have emerged as promising water oxidation catalysts (WOCs), with the added advantage of their molecular nature despite being metal oxide fragments. In comparison with metal oxides, that do not offer well-defined active surfaces, POMs have a controlled, discrete structure that allows for precise correlations between experiment and computational analyses. Thus, beyond highly active WOCs, POMs are also model systems to gain deeper mechanistic understanding on the oxygen evolution reaction (OER). The tetracobalt Weakley sandwich [CoII4(H2O)2(B-α-PW9O34)2]10− (Co4-WS) has been one of the most extensively studied. We have compared its activity with that of the iron analog [FeIII4(H2O)2(B-α-PW9O34)2]6− (Fe4-WS) looking for the electronic effects determining their activity. Furthermore, the effect of POM nuclearity was also investigated by comparison with the iron- and cobalt-monosubstituted Keggin clusters. Electrocatalytic experiments employing solid state electrodes containing the POMs and the corresponding computational calculations demonstrate that CoII-POMs display better WOC activity than the FeIII derivatives. Moreover, the activity of POMs is less influenced by their nuclearity, thus Weakley sandwich moieties show slightly improved WOC characteristics than Keggin clusters. In good agreement with the experimental data, computational methods, including pKa values, confirm that the resting state for Fe-POMs in neutral media corresponds to the S1 (FeIII–OH) species. Overall, the proposed reaction mechanism for Fe4-WS is analogous to that found for Co4-WS, despite their electronic differences. The potential limiting step is a proton-coupled electron transfer event yielding the active S2 (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) species, which receives a water nucleophilic attack to form the O–O bond. The latter has activation energies slightly higher than those computed for the Co-POMs, in good agreement with experimental observations. These results provide new insights for the accurate understanding of the structure–reactivity relationships of polyoxometalates in particular, and or metal oxides in general, which are of utmost importance for the development of new bottom-up synthetic approaches to design efficient, robust and non-expensive earth-abundant water oxidation catalysts.
O) species, which receives a water nucleophilic attack to form the O–O bond. The latter has activation energies slightly higher than those computed for the Co-POMs, in good agreement with experimental observations. These results provide new insights for the accurate understanding of the structure–reactivity relationships of polyoxometalates in particular, and or metal oxides in general, which are of utmost importance for the development of new bottom-up synthetic approaches to design efficient, robust and non-expensive earth-abundant water oxidation catalysts.
Introduction
The development of sustainable, carbon-free energy concepts has become of paramount importance to meet the growing global energy demands while diminishing anthropogenic CO2 emissions.1,2 Solar energy is considered the most powerful source of clean and renewable energy.3–5 However, due to its intermittency, solar energy needs to be harvested and stored, preferably in the form of chemical bonds such as H2, to be supplied on demand. In the hydrogen economy, water is used as a clean fuel owing to its ubiquity, providing the required reducing equivalents to produce H2 through the water splitting reaction, thus mimicking natural photosynthesis.6,7 Notwithstanding, the water oxidation half-reaction to produce O2 is both thermodynamically and kinetically highly energy demanding, typically illustrated by high overpotentials (η) and sluggish kinetics during the O–O bond formation.8–10 Hence, the water oxidation half-reaction is considered a bottleneck within the scheme, hampering the development and deployment of this technology.The use of water oxidation catalysts (WOCs) is imperative in order to alleviate the high energy demands of the reaction. In this regard, noble metal-based WOCs have shown promising catalytic capabilities.11,12 However, their implementation in large-scale applications is limited due to their scarcity and prohibitive cost. Intensive efforts have been made during the last years in WOCs research with earth abundant elements.13–15 Interestingly, first row transition metal oxides provide excellent catalytic activity in alkaline media, but their stability rapidly decays at lower pH, where only cobalt oxide analogues sustain good WOC performance in neutral media with the aid of phosphate electrolytes.16,17
To overcome the hydrolysis of transition metal oxides in neutral and acidic media, catalytically active metal ions have been incorporated into polyoxometalate (POM) frameworks, thus providing good stability over a large range of pH values.18,19 Moreover, as redox-active materials, POMs can afford different oxidation states without suffering major structural changes, exhibiting high stability under harsh working conditions.20 For instance, in 2008 a Ru-containing POM, [Ru4O4(OH)2(H2O)4(γ-SiW10O36)2]10−, was reported as the first POM showing efficient water oxidation catalytic activity.21,22 Regarding POMs based on earth abundant elements, Co-containing POMs (Co-POMs) are the most studied because of their good activity and remarkable stability under controlled working conditions. In 2010, the Co-POM with the formula [Co4(H2O)2(B-α-PW9O34)2]10− (Co4-WS) was reported as the first WOC of its kind.23 This POM is composed of a tetracobalt-oxo core stabilized by two trilacunary Keggin moieties (B-α-PW9O349−) forming the also-known Weakley sandwich (Fig. 1).24 In 2012, a high nuclearity Co-POM, [Co9(H2O)6(OH)3(HPO4)2(PW9O34)3]16−, was reported to render efficient WOC activity, evolving O2 for weeks without sign of fatigue or decomposition.25 Later on, it was demonstrated that this Co-POM maintains its water oxidation capabilities with outstanding stability in the solid-state over a wide range of pH, surpassing the catalytic activity shown by benchmarking IrO2 in strong acidic conditions.26,27
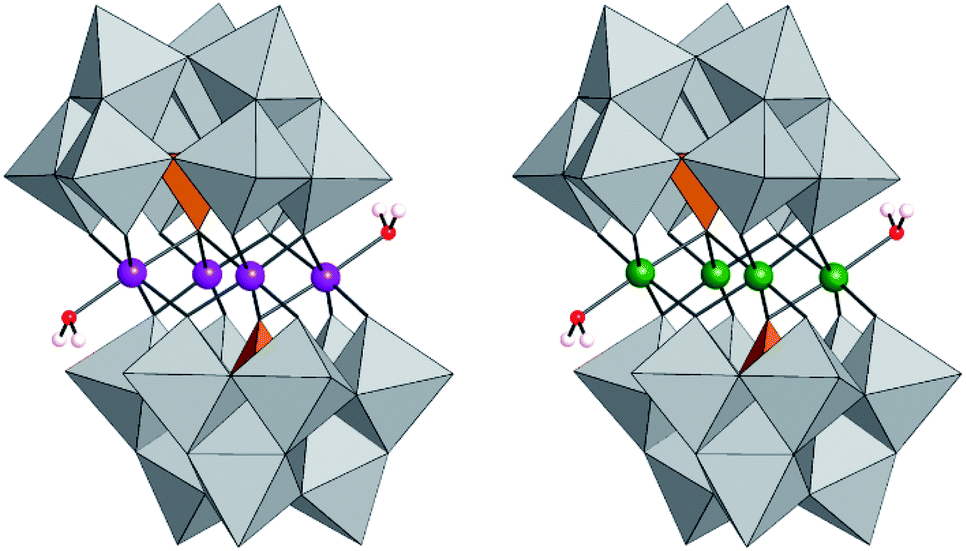 | ||
| Fig. 1 Polyhedral representation of [(CoIIOH2)2CoII2(PW9O34)2]10− (Co4-WS), [(FeIIIOH2)2FeIII2(PW9O34)2]6− (Fe4-WS). | ||
Iron, as one of the most abundant and less toxic transition metals, is a promising element to design cost-effective and durable catalysts.28,29 The redox properties of iron make it essential in biological systems for O2 involving processes, such as the nonheme iron complex that acts as an electron-transmitter in Photosystem-II.30 Many iron-based catalysts have been reported with interesting and promising performance and good stability towards water oxidation catalysis.31 Hydrated oxides (FeOOH),32 FeOx/CNT composites (FeP@CNT),33 iron-phosphate (FePO4)34 and a trimetallic FeCoW oxide35 are some examples of Fe-based WOCs used in basic (1 M KOH) media performing an overpotential of ca. 200 mV at 10 mA cm−2. Moreover, incorporation of traces of iron into the nickel oxyhydroxide structure is known to dramatically enhance the catalytic activity.36 Nevertheless, the studies of Fe-containing POMs as WOCs are scarce37,38 and in some cases the POM acts as a catalyst precursor and not as the true active species.39
In addition, the reaction mechanism of the water oxidation catalysed by Co4-WS at close-to-neutral pH was recently studied by computational means.40 There, the active species and the O–O bond formation steps were explored using a simple model system. Within this proposed mechanism the potential-determining step corresponds to the O–H bond cleavage to facilitate the formation of the active species, a radical Co-oxyl species, through a proton-coupled electron transfer step. Thereafter the O–O bond formation occurs when this electrophilic, active species is attacked by a water molecule, working as a nucleophile. Taking advantage of this theoretical investigation and the extensive experimental studies carried out on the Co4-WS, other POMs with similar structural features may be studied to gain a deeper mechanistic understanding. This knowledge will allow to define a strategy for the design of novel efficient and cost-effective POMs working as WOCs. Hence, a clear case study is the Fe-containing POM [(FeIIIOH2)2FeIII2(PW9O34)2]6− firstly described by Hill and co-workers.41 The electronic structure and redox properties of Fe-based anions have been reported previously.42–45
Here we report the water oxidation catalytic activity of [(FeIIIOH2)2FeIII2(B-α-PW9O34)2]6− (Fe4-WS) and compare it with that of the Co4-WS. We have carried out our catalytic study at neutral pH and in the solid-state using the POM water-insoluble barium salt to assure structural stability. Moreover, we have investigated the reaction mechanism employing computational methods to determine the key aspects in comparison with those of the Co4-WS. In addition, in order to attain more insight on the structure–reactivity relationship of POMs, we also investigated the OER catalytic properties of the Fe- and Co-monosubstituted Keggin moieties.
Experimental and theoretical details
Materials
All chemicals and solvents were used as purchased without further purification. Milli-Q water (ca. 18.2 MΩ cm resistivity) was employed to prepare all aqueous solutions and to clean and rinse the electrodes. Carbon paste (CP) was purchased from ALS (CPO Carbon Paste Oil: uniform-sized graphite powder and paraffin oil).Electrode preparation
The CP blends were prepared by mixing the barium salts of the POMs with CP in the desired weight ratios using an agate mortar. Then, the blend was introduced into the CP electrode pocket. A small piece of staple cotton was used to cover the electrode surface for long-term electrocatalytic experiments to avoid the expulsion of the blend out of the electrode pocket (pictures available, Fig. S1†).Electrochemical methods
All electrochemical experiments were performed using a Biologic SP150 potentiostat. Ohmic drop was compensated using the positive feedback compensation implemented in the instrument. All experiments were performed with a three-electrode configuration using the CP electrode as a working electrode, whereas a Saturated Calomel Electrode (SCE) and a Pt mesh were used as a reference and counter electrodes, respectively. Linear sweep voltammetry experiments (LSV) were performed using an ALS RRDE-3A set-up, with a CP Rotating Disk Electrode (RDE, surface area = 0.07 cm2) at 1600 rpm and at scan rate of 1 mV s−1. Bulk water electrolysis was performed at a constant current density of 1 mA cm−2 for 20 h. All current densities were calculated based on the geometrical surface area of the electrodes (0.07 cm2).The experiments were carried out in an aqueous potassium phosphate (50 mM) buffer solution containing KNO3 (1 M) as the electrolyte at pH = 6.9. The thermodynamic potential for the water oxidation was corrected by the pH value using the Nernst equation:
| EO2|H2O = 1.229 − (0.059 × pH) (V) vs. NHE at 25 °C |
All potentials were converted to the NHE reference scale using ENHE = ESCE + 0.241 (V). The water oxidation overpotentials (η) were calculated by subtracting the thermodynamic water oxidation potential to the applied potential:
| η = Eapp − EO2|H2O |
Oxygen evolution
The oxygen evolved was measured using an Ocean Optics NeoFOX oxygen sensing system equipped with a FOXY probe. The FOXY probe was calibrated employing a two-point calibration, taking (a) 0% of O2 determined under a N2 flow and (b) 20.9% of O2 measured in the air. The experiments were carried out in an H-cell, with the anode and cathode compartments separated by a porous frit. The FOXY probe was inserted into the gas space of the anodic compartment (VGas space ≈ 21 mL). The solution was completely deaerated by purging with N2 for at least 1 h. The N2 flow was then removed and a base line of 30 min was recorded before starting the chronopotentiometry at a constant current density of 1 mA cm−2. The O2 generated during the electrochemical experiment was expressed as the partial pressure of O2.The number of moles of oxygen produced were calculated using the Ideal Gas Law as:
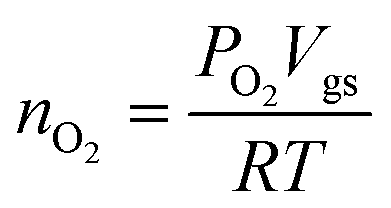
The theoretical amount of O2 produced was calculated from the number of charges passed through the working electrode during the chronopotentiometry experiment as:
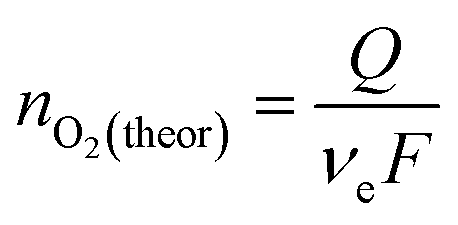
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).
Recovery of the catalyst
After the electrochemical experiments, the Ba[POM]/CP blend (ca. 40 mg) was suspended in acetone (30 mL) and sonicated for 30 minutes. The supernatant liquid (containing carbon black and the organic oil binder) was decanted to retain the POM catalyst in the beaker. This procedure was repeated 10 times to get a clean catalyst sample, ready for post-catalytic characterization.Characterization methods
All the POMs were characterized by thermogravimetric analysis (TGA) (Fig. S2 and S3†), infrared spectroscopy (Fig. S4 and S5†), Raman spectroscopy (Fig. S6 and S7†) and energy dispersive X-ray spectroscopy (EDX) (Table S1†).Thermogravimetric analysis (TGA) was performed under N2 flow with powder samples using a TGA/SDTA851 Mettler Toledo with a MT1 microbalance. The experiments were carried out by weighing about 3 mg of sample in an Al-crucible of 40 μL and with a temperature scan range from 30 °C to 600 °C at a heating rate of 10 °C min−1. IR spectra were collected with a FT-IR Bruker spectrometer model Alpha equipped with an ATR accessory. The spectra were acquired in the range 400–4000 cm−1 with 32 scans. Raman measurements were acquired using a Renishaw inVia Reflex Raman confocal microscope (Gloucester-Shire, UK), equipped with a diode laser emitting at 785 nm at a nominal power of 300 mW, and a Peltier-cooled CCD detector (−70 °C) coupled to a Leica DM2500 microscope. Calibration was carried out daily by recording the Raman spectrum of an internal Si standard. Rayleigh scattered light was appropriately rejected by using edge-type filters. The spectra were acquired in the range 100–1800 cm−1. Laser power was used at nominal 1% to avoid sample damage. Spectra were recorded with the accumulation of at least 3 scans with a 30 s scan time. Cation content was determined by energy-dispersive X-ray spectroscopy (EDX), which was collected with a JEOL-JMS6400 environmental scanning electron microscope equipped with an Oxford Instruments X-ray elemental analyser. The sample was adhered on a carbon tape using Al-stubs as a support. Before measurements, the sample was cleaned with an airflow to remove non-attached compound. The measurements were performed under low-vacuum conditions with a Large-Field Detector at 20 kV using a probe current of 17 pA. Quantification of metal leached into the buffer solution was carried out with an Inductively Coupled Plasma Quadrupolar Mass Spectrometer (ICP-MS) (Thermo, iCAP Qc, Xt interphase and PFA micronebulizer) at the University of Valencia. Elemental analysis was carried out with an Agilent 725-ES inductively coupled plasma optical emission spectrometer (ICP-OES) at the University of Valladolid, taking the following standards: Ba (455.403 nm), Cs (697.327 nm), W (207.912 nm), Co (238.892 nm), P (213.618 nm) and Si (250.690 nm). The digestion of the POM was carried out by dissolving 10 mg of POM in 10 mL of acidic solution (concentrated HNO3, concentrated HCl).
Computational methods
The calculations were performed using the Gaussian-16 package.46 The B3LYP functional47–49 was chosen based on the successful results obtained for similar systems.40 We used two types of basis set combinations, herein BS1 and BS2. BS1 is a combination of small basis sets firstly used to explore in detail the potential energy surface of the computed POMs. In BS1, we used the 6-31G(d,p) basis set for all the H and O atoms directly bonded to either the iron or the cobalt atoms and the 6-31G basis set for the rest of the oxygens.50–52 In the BS2 combination, the 6-31G(d,p) basis set was used for all the oxygens to re-optimize all the relevant structures. In both methods, the LANL2DZ effective core potential (ECP) and associated basis sets were used for the P, W, Co and Fe atoms.53 All the structures were optimized using the IEF-PCM approach to model implicitly the solvent effects of water (ε = 78.36 and UFF radii).54 We tested Grimme's dispersion corrections GD2,55 GD356 and GD3BJ57 to our calculations. However, these calculations lead to a poor reproduction of the experimentally observed pKa's, while the combination of the B3LYP functional with BS2 yielded results in close agreement with the experimental values. The nature of all the stationary points was verified by computation of the vibrational frequencies. All the energies reported correspond to the computed free Gibbs energies in solution; electrochemical steps are reported either in V or eV, and chemical steps in eV and kcal mol−1. All the experimental and computed potentials and overpotentials given in the present work are referred to the NHE reference scale. A collection data set of all computational data is accessible in the ioChem-BD repository and may be accessed viahttps://doi.org/10.19061/iochem-bd-2-48.58
Results and discussion
Electrocatalytic oxygen evolution reaction (OER)
We have studied the WOC performance of Fe4-WS at neutral pH and compared it with that of the cobalt analogue Co4-WS,24 with the aim of elucidating the effect of the metal nature of the tetra-oxo core belt on the catalytic activity. Furthermore, we have also examined the WOC activity of the iron and cobalt derivatives of the monosubstituted Keggin species, [FeIII(H2O)PW11O39]4− (Fe–K) and [CoII(H2O)PW11O39]5− (Co–K), to gain a deeper understanding on the structure–reactivity relationship of POMs.The electrocatalytic water oxidation experiments were carried out in the solid-state using the water-insoluble barium salts of the POMs to prepare modified carbon paste (CP) electrodes. The choice of the countercation was made based on our previous works, where we showed its superior WOC performance compared with the caesium salts.27,59 The catalyst content in the carbon blend was limited to 20% to avoid mechanical sensitivity due to the formation of oxygen bubbles.
The water-insoluble barium salts were obtained by metathesis from POM aqueous solutions. The catalytic performance was studied by running multiple (≥5) linear sweep voltammetry (LSV) scans using a three-electrode configuration with a Pt mesh as the counter-electrode and a Saturated Calomel Electrode (SCE) as the reference electrode. Three independent electrodes were tested in order to evaluate the reproducibility of the measurements.
LSV with 20% Ba[Fe4-WS]/CP electrodes show a significant catalytic activity with increasing current density rapidly deviating from that obtained with catalyst-free CP electrodes (Fig. 2a). Repetitive LSV demonstrates chemical stability during turnover conditions (see ESI, Fig. S8†). The experimental applied potential values required to reach 1 mA cm−2 for 20% Ba[Fe4-WS]/CP and Ba[Co4-WS]/CP are 1.560 V (η = 732 mV) and 1.512 V (η = 684 mV), respectively (see Fig. 2a and Table 1), which represents an overpotential difference of 48 mV. Ba[Fe–K] and Ba[Co–K] show an apparent better WOC activity than the corresponding Weakley sandwich POMs, with a decrease in the overpotential values of ca. 70 mV to reach 1 mA cm−2, with applied potentials of 1.495 V and 1.442 V, respectively. Again, the influence of the nature of the catalytically active metal center on the activity is noticeable with the cobalt analogue requiring a 53 mV lower potential than Ba[Fe–K] to achieve a current density of 1 mA cm−2. This lower overpotential at the same total weight derives from the lower molecular weight of the POM-K salts. When we normalized the current density per total number of moles of catalyst, both POM structures show similar activity, with a slightly better performance in the case of the Weakley sandwich POMs, and a significantly better performance for the Co-POMs (Fig. 2b and Table 1). This comparison suggests that the nature of the metal center seems to play a more important role in the WOC performance than the POM structure.
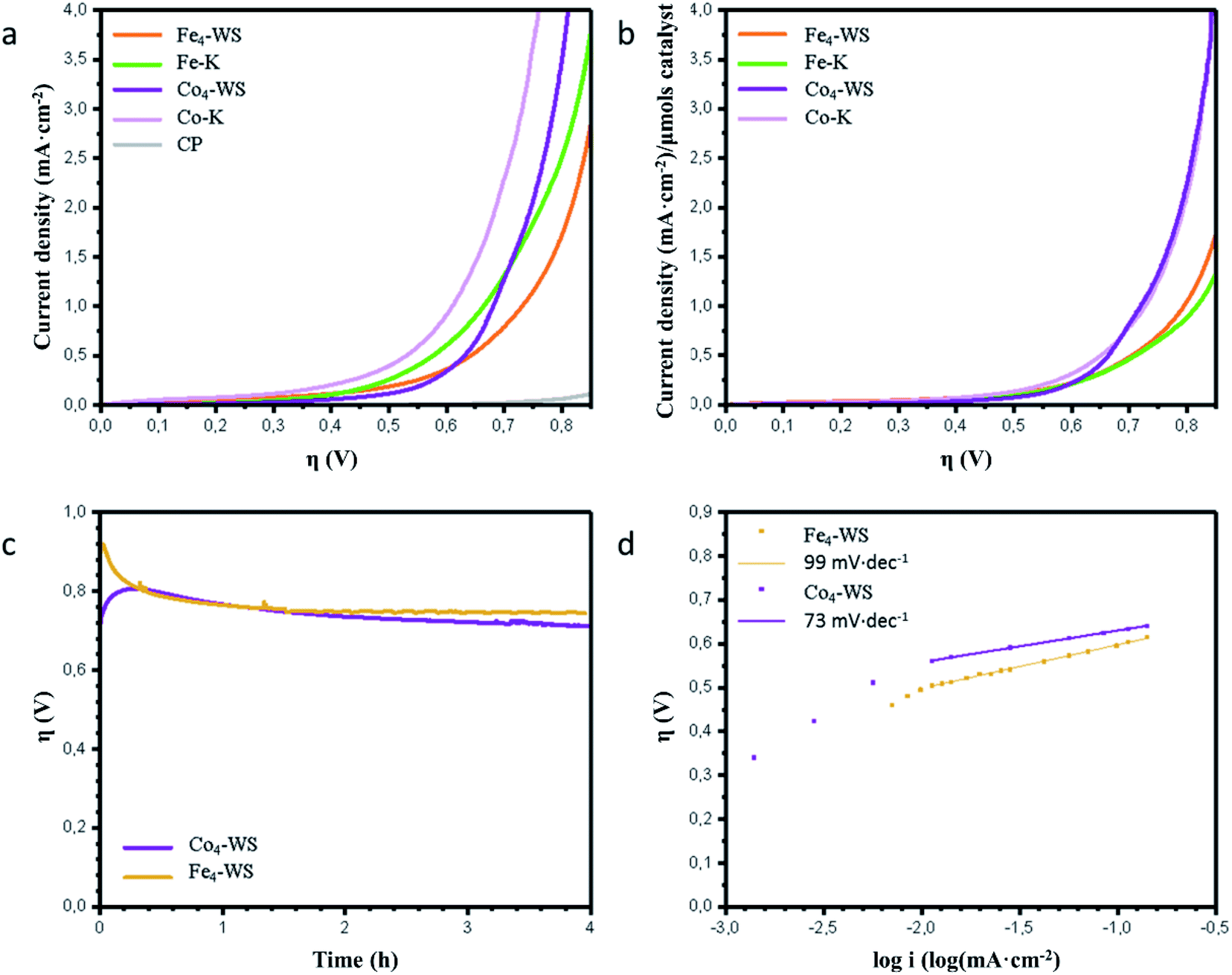 | ||
| Fig. 2 (a) Linear sweep voltammetry of the 20% Ba[POM]/CP working electrodes, (b) linear sweep voltammetry normalized by the total number of moles of POM used in each blend, (c) chronopotentiometry at a constant current density of 1 mA cm−2 using 20% Ba[Fe4-WS]/CP and Ba[Co4-WS]/CP electrodes, (d) Tafel behaviour of 20% Ba[Fe4-WS]/CP and Ba[Co4-WS]/CP electrodes obtained from steady-state chronoamperometric experiments. All the measurements where done in an aqueous potassium phosphate (50 mM) buffer solution at pH = 6.9 with KNO3 (1 M) as the electrolyte. | ||
| Potential/overpotential | Set | Co4-WS | Co–K | Fe4-WS | Fe–K |
|---|---|---|---|---|---|
| a Two sets of potentials and overpotentials were determined from measured current densities (Fig. 2a) and from current densities normalized per μmol of catalyst (Fig. 2b). The experimental errors for the potentials were estimated as a standard deviation of three independent measurements. b Computed potentials and overpotentials were determined using a smaller (BS1) and a larger (BS2) basis set (see Computational section for more details). Computed overpotentials values may be compared with experimental overpotentials at 1 mA cm−2.66 c An Eapp of 1.52 V was reported for Co4-WS using a Nafion ink on a GC electrode.67 d Value from ref. 40 corrected at pH 7. | |||||
| E app | Measured @1 mA cm−2 | 1.512 ± 0.004 | 1.442 ± 0.010 | 1.560 ± 0.013 | 1.495 ± 0.008 |
| η | 684 ± 4 | 614 ± 10 | 732 ± 13 | 666 ± 8 | |

|
Normalized @1 mA cm−2 per μmol of catalyst | 1.553 ± 0.006 | 1.555 ± 0.009 | 1.610 ± 0.010 | 1.642 ± 0.012 |
| η′ | 724 ± 6 | 727 ± 9 | 793 ± 10 | 813 ± 12 | |
| E DFT | BS1 theory level | 1.54d | 1.69 | 1.59 | 1.67 |
| η DFT | 723 | 870 | 773 | 853 | |
| E DFT | BS2 theory level | 1.54 | 1.58 | 1.54 | 1.64 |
| η DFT | 723 | 763 | 723 | 823 | |
The long-term stability of Ba[Fe4-WS] was confirmed by chronopotentiometric measurements at a constant current density of 1 mA cm−2 for 4 hours (Fig. 2c) and 20 hours (Fig. S9†). All POMs show good stability. Moreover, the oxygen evolved employing Ba[Fe4-WS] during a chronopotentiometric experiment at 1 mA cm−2 for 30 minutes was measured employing a fluorescence probe. This measurement confirms a faradaic oxygen production (>91%) compared with the theoretical amount of oxygen expected from a stoichiometric 4e− reaction (Fig. S10†).
Post-catalytic characterization of the recovered POMs after bulk water electrolysis confirm the structural stability of the compounds. No signs suggesting the evolution of the catalysts towards a new species, metal leaching or POM decomposition were detected. Raman spectroscopy is a surface-sensitive characterization technique that allows the identification of traces of new species formed on the surface of the catalyst.59,60 The Raman and IR spectra of the recovered Ba[Fe4-WS] matches that of the freshly made compound (Fig. S11†). Moreover, the electrolyte solution was analysed by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) to identify metal leaching (Table S2†). The concentration of all the elements were below the detection limit of the technique, suggesting a good chemical stability under the working conditions.
In order to understand better the origin of the different WOC activities shown by Ba[Fe4-WS] and Ba[Co4-WS], we calculated the double-layer capacitance (Cdl) of the modified CP electrodes, which is expected to be proportional to the total electrocatalytic active surface area. Ba[Fe4-WS] electrodes show lower Cdl values than Ba[Co4-WS] ones, which indicates that the cobalt analogue possesses an intrinsically higher electrocatalytic surface area than the iron derivative (Fig. S12†). This difference can be somehow related to the total charge of the POM, which determines the number of cations needed to counterbalance the high anionic charge of the POMs. Hence, a higher number of countercations may lead to a higher number of accessible active sites on the insoluble POM particles.
We also studied the Tafel behaviour of these POMs through steady-state analysis (Fig. 2d). The Tafel slope depends exclusively on the rate-determining step of the catalytic reaction, being independent on the total number of active sites.61 We found Tafel slopes of 99 mV dec−1 and 73 mV dec−1 for Ba[Fe4-WS] and Ba[Co4-WS], respectively. These values indicate a competition between a chemical and an electron-transfer limiting step for both catalysts, and confirm the faster kinetics for the Co-POM.27
Determination of the pKa values
The protonation–deprotonation equilibrium is key to understand and predict the mechanistic behaviour of POMs as WOCs.40 Luckily, we have been able to determine experimentally a pKa value of 6.0 for Fe4-WS (Fig. S13†) by acid–base titration. In our previous work we computationally estimated a pKa value for Co4-WS above 10,40 where the POM becomes unstable hindering the experimental determination.20,62,63 The different pKa values between Fe4-WS and Co4-WS are attributed to a stronger metal center–Owater interaction in the case of iron with respect to the cobalt analogue, due to the higher oxidation state of the metal (FeIIIvs. CoII).We have also studied the protonation–deprotonation equilibrium by computational means. The computed pKa value for Fe4-WS is 6.1, which is in very good agreement with experiments. It is worth noting that the computational M–Ow distances in the aqua system are 2.151 Å and 2.220 Å for Fe4-WS and Co4-WS, respectively, supporting that Fe4-WS has a stronger M–Ow interaction than the cobalt derivative.
In order to get more insight into the deprotonation of these type of POMs, we have computed and determined experimentally the pKa value of the Fe–monosubstituted Keggin POM (Fe–K) to be 4.4 and 4.3, respectively. The value is slightly lower than that of Fe4-WS, probably due to the difference in the overall charge of these two POMs (−6 for the sandwich Fe4-WS and −4 for the monosubstituted Fe–K). We have used the Bond Valence Sum model64 to estimate the oxidation state of the Fe in both systems, obtaining values of 2.7 for the sandwich Fe4-WS and 2.9 in the case of the monosubstituted Fe–K. This also agrees with the Fe–O distances that are a slightly larger for the sandwich Fe4-WS.
Fe4-WS possesses a C2h symmetry with two equivalent aqua ligands. Consequently, this POM shows two different pKa values derived from the removal of a proton from each H2O ligand. The first deprotonation changes the overall charge of the POM to −7, thus rendering the second deprotonation higher in energy. According to our DFT calculations, the value of pKa2 is 9.0. Unfortunately, we could not establish the pKa2 value during the acid–base titration experiments up to pH = 10, after which Fe4-WS becomes unstable. Nevertheless, we can assume that the second deprotonation would occur at higher pH values than the catalytic working conditions we are considering in this work and, hence, it will not be involved in our proposed reaction mechanism. We can conclude that, at neutral pH, both Fe-POMs are present as POM-FeIII–OH species at the resting state (vide infra).
Reaction mechanism
The Co4-WS, analogue to Fe4-WS, was mechanistically investigated in our previous work.40 The postulated mechanism for Co4-WS was described in two stages: first the electrochemical formation of the active species via an electron-then-proton transfer event followed by a proton coupled electron transfer (PCET) step. In the second stage of the mechanism, the active species leads to the O–O bond via a water nucleophilic attack (WNA). Additionally, the electronic structure of Fe4-WS was computationally investigated by some of us back in 2007.65 In the present work, we followed a similar strategy defining as starting system the four iron centers to be FeIII with a high-spin d5 electron configuration. The magnetic coupling between the iron centers was not considered. We investigated the influence of the metallic center on the key steps leading to the formation of the active species and the O2 bond formation process. Experimentally, water oxidation catalysis is performed electrochemically, where an applied potential drives the catalytic reaction. As found in the cobalt analogue, we expect to differentiate two governing factors: (i) the oxidative character of the active center, i.e. the ability to lose electrons and reach high oxidation states and (ii) the rate of the non-electrochemical steps, i.e. the WNA, which is driven by the electrophilicity of the oxidized POM, and the proton transfer events that are dictated by the acid/base properties. In addition, the chemically controlled equilibrium can determine, as we will see below, the resting state species and the kinetics of the global process. Thus, we have analysed each step of the Fe4-WS catalysed water oxidation cycle at pH 7 starting from the S1 FeIII–OH species. The different steps involved in the catalytic cycle are summarized in Fig. 3 and they will be described below.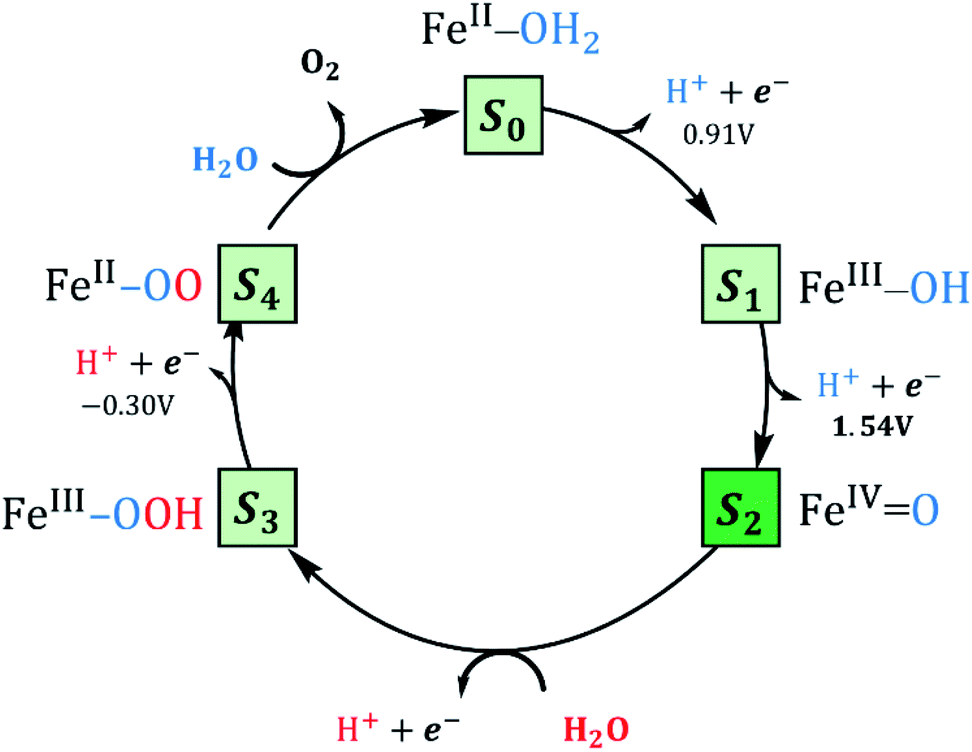 | ||
| Fig. 3 Water oxidation mechanism using Fe-POMs considered as single-site catalysts. Potentials and energies correspond to the catalytic cycle computed for the Fe4-WS electrocatalyst. The dark green square indicates the active species. | ||
Formation of the active species
As discussed above, Fe4-WS (S1′, the (FeIII–OH2) species) has an experimental pKa value of 6.0, which is in very good agreement with our computed value of 6.1. This means that the speciation of Fe4-WS depends on the pH. Above pH 6, there is a high deprotonation degree of Fe4-WS, leading to the formation of the hydroxyl species S1 (FeIII–OH), whereas at pH values below 6 the aqua species (S1′) is favoured. As the present work is carried at pH 7 (one pH unit higher than the pKa value), S1 is the predominant form of the catalyst, where [S1]/[S1′] ≈ 8 (according to Henderson–Hasselbalch equation). Furthermore, any oxidation process from the iron-aqua species (S1′) would be highly energy demanding because the computed potentials are 2.34 and 2.16 V involving either an electron transfer (ET) or a proton-coupled electron transfer (PCET), respectively (see Fig. 4).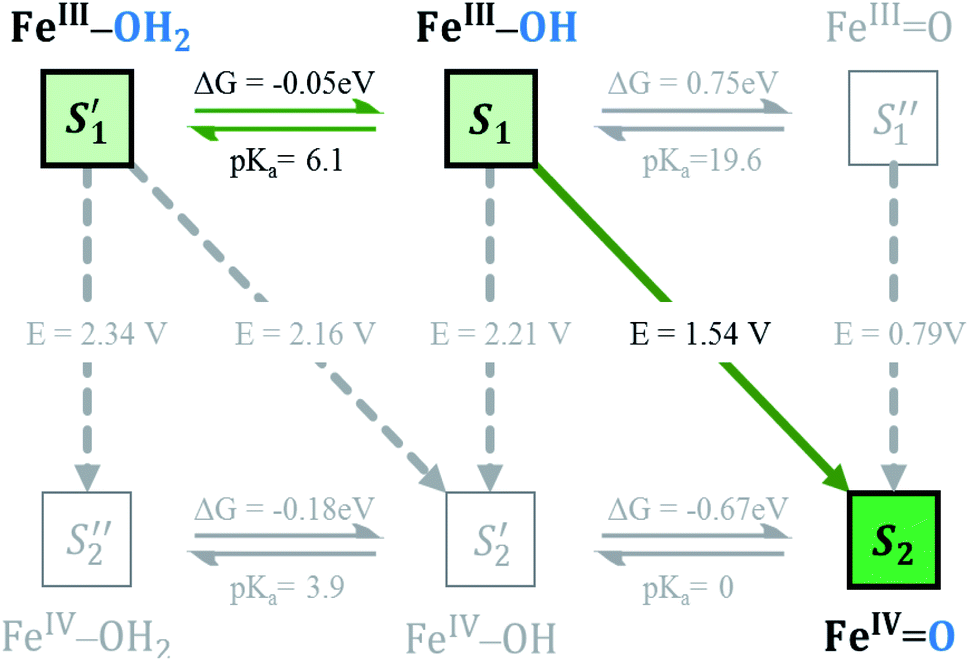 | ||
| Fig. 4 Square diagram representation of the PCET, PT and ET events for Fe4-WS acting as WOC, starting from the resting state species at pH = 7 to reach the active species. Potentials are given in V vs. NHE for the electrochemical processes (PCET and ET), while the chemical processes related to acid–base equilibria (PT) are expressed as Gibbs free energy (in eV) with the corresponding pKa value. | ||
The first step of the reaction starting from the resting state S1 (FeIII–OH) species clearly consists of a PCET event leading to the iron-oxo species S2 (FeIVO). The sequential electron-then-proton transfer pathways are more energy demanding, as can be seen in the square diagram in Fig. 4. In this figure, vertical processes correspond to oxidation reactions (ET), horizontal ones to proton cleavage (O–H, only proton transfer (PT) acid–base reaction) and diagonal arrows represent PCET steps with one electron and one proton loss. Therefore, the first step S1 (FeIII–OH) → S2 (FeIVO) takes place, according to calculations, at 1.54 V. An ET would require a very high potential of 2.21 V. This is because the hydroxyl ligand does not stabilize high oxidation states such as FeIV. Moreover, the deprotonation of S1 is thermodynamically unfavourable, and it would require strong basic conditions, where the Fe4-WS is unstable. The S1 (FeIII–OH) species is expected to have five unpaired electrons in each iron. However, DFT calculations tend to somewhat over-delocalize the electron spin density leading to values of 4.1e on all four irons and 0.4e on the hydroxyl oxygen (see Table 2). S2 (FeIVO), shows an electron spin density on the reactive iron of 3.1e and 0.6e on the terminal oxo moiety. Hence, this species can be regarded as an average of the two Lewis structures FeIVO and FeIII–O˙. The computed spin density is more consistent with a FeIVO species, since a complete electron has been removed from the iron center upon one-electron oxidation of the FeIII–OH species. The computed Fe–O bond length of 1.609 Å also suggests that S2 must be seen preferably as an FeIVO species (Fig. 5). However, the high electron spin density localized at the terminal oxygen in S2, gives a significant reactivity to this oxygen. At this point, the reaction mechanism can proceed through two different pathways, either the S2 (FeIVO) species is the electrophilic reactive center (WNA takes place at this point) or the S2 is one-electron oxidized to give a FeVO species.
| S0 FeIIOH2 | S1′ FeIIIOH2 | S1 FeIIIOH | S2 FeIVO | TS2 [FeIIO⋯OH2 + H2O] | Int FeII–O–OH2 | S3 FeIII–O–OH | S4 FeII–O–O | |
|---|---|---|---|---|---|---|---|---|
| a Bonds lengths are in Å and Mulliken spin densities in e. All data correspond to BS2 calculations without explicit water molecules except for TS2 and Int, whose values correspond to the calculations performed with an extra water molecule. b Int: intermediate species generated from TS2. | ||||||||
| Charge | −7 | −6 | −7 | −7 | −7 | −7 | −7 | −7 |
| ρ(Fe) | 3.778 | 4.137 | 4.100 | 3.125 | 3.769 | 3.744 | 3.912 | 3.741 |
| ρ(O) | 0.027 | 0.070 | 0.414 | 0.563 | 0.139 | 0.094 | 0.552 | 1.007 |
| ρ(OWNA) | — | — | — | — | −0.126 | −0.003 | 0.198 | 1.005 |
| d(Fe–O) | 2.227 | 2.151 | 1.836 | 1.609 | 1.818 | 2.001 | 1.964 | 3.924 |
| d(O–OWNA) | — | — | — | 1.700 | 1.501 | 1.375 | 1.214 | |
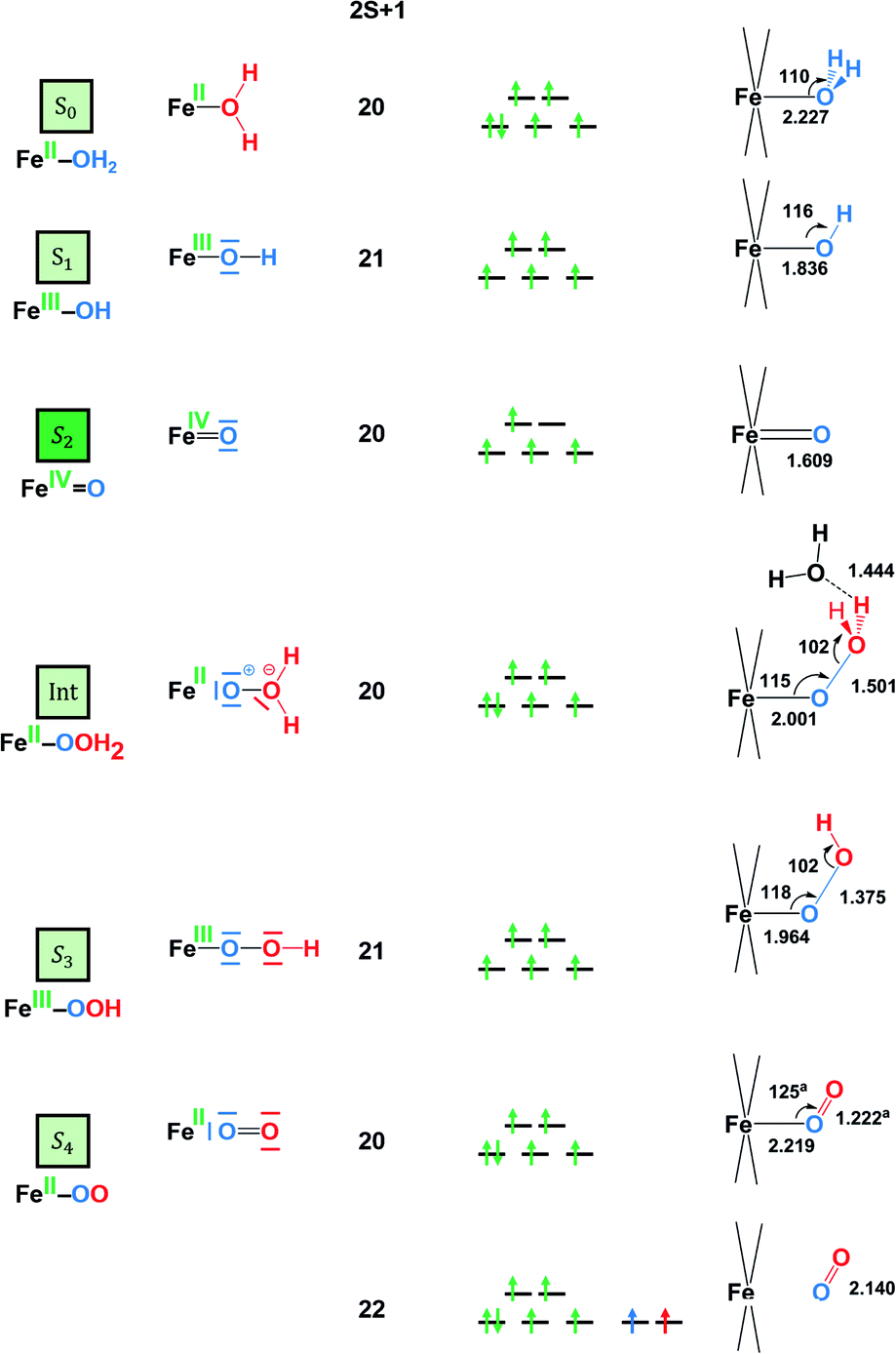 | ||
| Fig. 5 Electronic and structural data for the S0 to S4 species. The angles are given in degrees and the bond lengths in Angstroms. a The geometrical parameters of this structure were obtained using BS1. | ||
The latter requires a high applied potential (2.11 V), similar to that reported for the cobalt analogue anion.39 Given that the LUMO of S2 (Fig. 6a) has an important p(oxo) contribution, it is expected that S2 may act as an electrophile and be the active species that oxidizes water (see below).
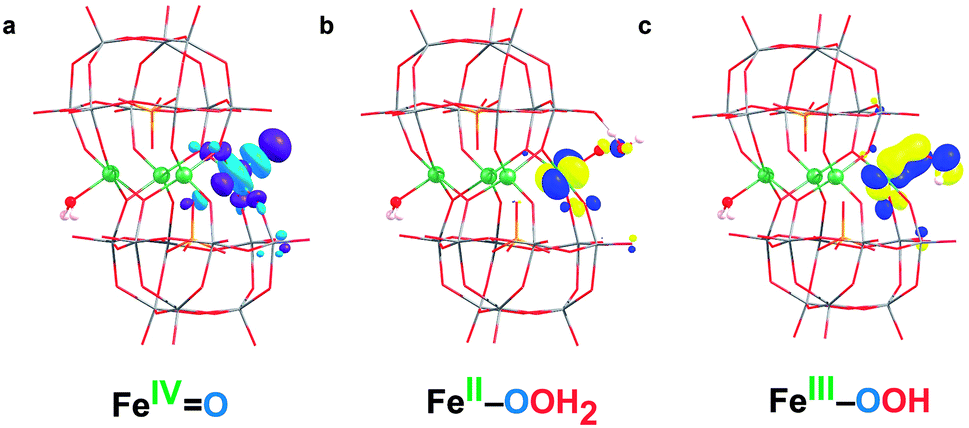 | ||
| Fig. 6 (a) LUMO of FeIVO (S2), (b) SOMO (β) of FeII–OOH2 (Int) and (c) SOMO (β) of FeIII–OOH (S3) species. | ||
O–O bond formation
After reaching the active species S2 (FeIVO), the O–O bond formation should take place via a water nucleophilic attack, leading to the formation of an intermediate species Int (FeII–OOH2). As shown in Fig. 5, the attack of a water molecule induces a significant electronic reorganization at the iron centre, see ESI† for a discussion on the Water Nucleophilic Attack. The formation of a relatively strong O–O bond of 1.501 Å is combined with an elongation of the Fe–O bond from 1.609 Å in S2 to 2.001 Å in Int. In turn, the iron centre is formally reduced from IV to II. We computed the corresponding transition state (TS) exploring different mechanistic possibilities, i.e. different hydrogen bond modes between the attacking water molecule and the oxygen atoms of the POM cluster, and different number of water molecules. Using the basis set BS2, two different TSs were located when computing just one water molecule (see ESI†). Both TSs showed relatively high activation energies (25.6 and 25.7 kcal mol−1). In the first TS, namely TS1B, the attacking water molecule is stabilized by the formation of a hydrogen bond with one of the bridging oxygens of the POM (Hwat–OPOM distance of 1.482 Å). In this case, the O–O and Fe–O distances are 1.715 Å and 1.846 Å. In the second TS, TS1T, the hydrogen bond occurs between the attacking molecule of water and a terminal oxygen atom of the POM, with a larger Hwat–OPOM distance (1.508 Å) than in the previous case. The weaker hydrogen bond interaction yields a shorter O–O distance of 1.664 Å, and a longer Fe–O distance of 1.860 Å, compared to TS1B. Remarkably, both TSs have almost identical activation energies, even though the oxygen atoms placed at the Fe–O–W bridging positions have been shown to be the most basic atoms in the POM structure.68–71 However, the computed structure in TS1B is more constrained than that in TS1T, where the OPOM–Owat–OFe angle is 81.1° for TS1B and 94.0° for TS1T. Arguably, both factors may compensate each other, resulting in almost isoenergetic structures (see Fig. S14†).
When a second molecule of water is included in the calculations (TS2), the activation barrier significantly decreases to 19.8 kcal mol−1 (0.86 eV). In this case, we could only find a transition state that exhibits hydrogen bonds with terminal oxygens (Fig. 7). We have repeated these series of calculations using the smaller basis set BS1. In general, the results are very similar but with lower activation energy barriers.
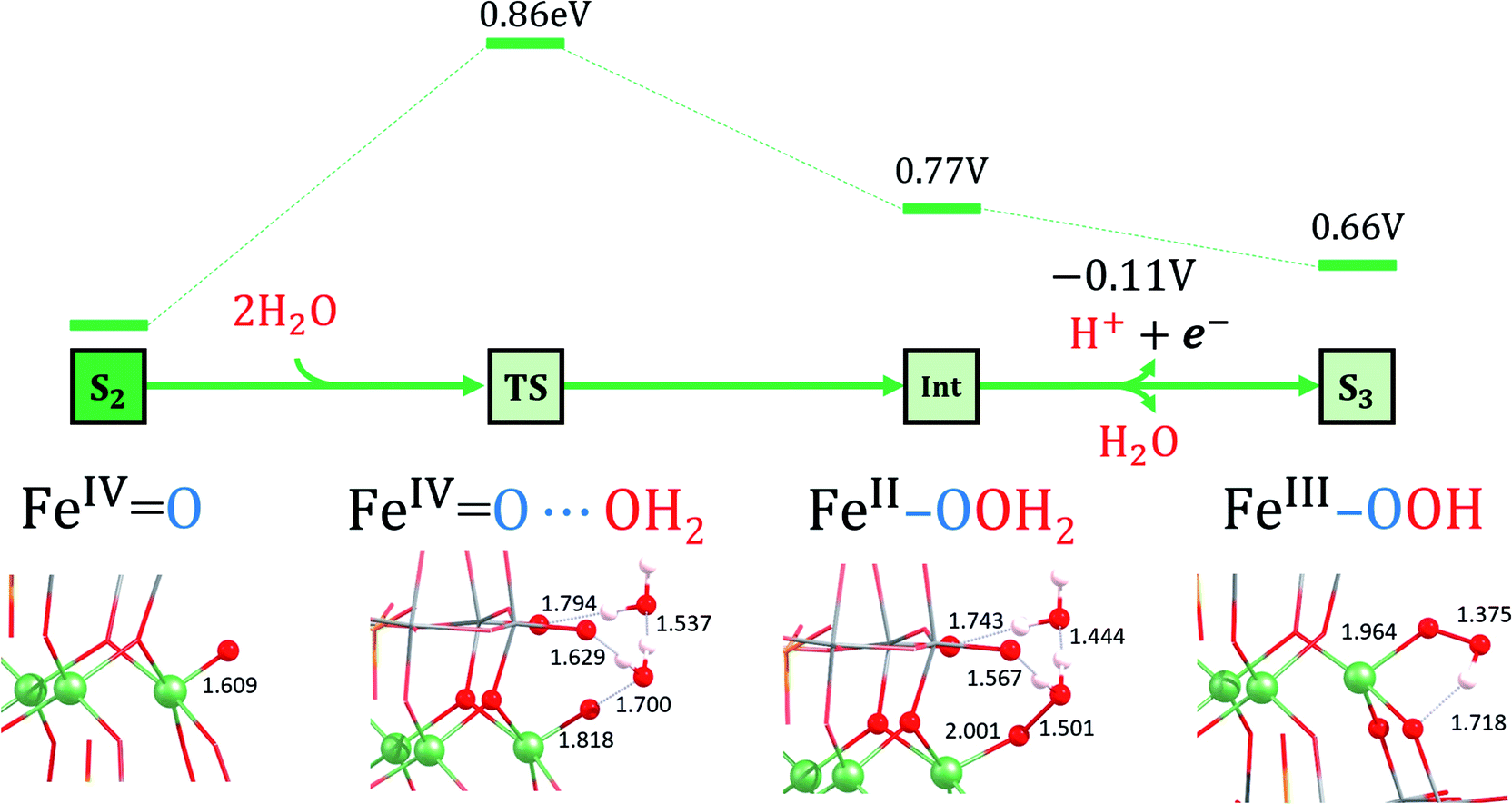 | ||
| Fig. 7 Ball and stick representations of the S2, TS2, Int and S3 species. For a description see footnote in Table 2. Selected distances are in Å. Energies are given in eV. The S2 to Int transformation is controlled thermally, whereas the Int to S3 one is controlled electrochemically. | ||
According to these results, TS2 is the preferred pathway for the O–O bond formation. This step should be rather fast, since the computed activation energy is easily surmountable at room temperature. Moreover, bulk water and buffering molecules may assist in speeding up the process. The transition state TS2 leads to the Int (FeII–OOH2) species, where the hydrogen atoms of the inserted water molecule form hydrogen bonds with both a terminal oxygen atom of the POM and the oxygen atom of the assisting water molecule. Notably, the WNA process involves the reorganization of the electrons in the active metal center, which is the result of an electron-donation effect from the attacking water molecule to the iron center (Fig. 5). This is reflected in a reduction of the oxidation state of the active iron center from FeIV to FeII. Indeed, Int (FeII–OOH2) species presents a spin density of 3.7e on the reactive iron, whereas a negligible spin density is localized on the nearby oxygens (Table 2). The FeII nature of this atom is also confirmed by orbital inspection. Fig. 6b shows the SOMO beta localized in the reactive iron atom indicating that it cannot have a higher oxidation state. In addition, a bond distance of 1.501 Å indicates the formation of a relatively strong O–O single bond, which is combined with an elongation of the Fe–O bond length from 1.609 Å in S2 to 2.001 Å in Int. The influence of the POM structure was analysed by computing the corresponding TSs for the Fe–K anion. The results are very similar to those obtained for Fe4-WS, even though all the free activation energies are slightly higher (Table 3). One possible explanation for this is that all hydrogen bonds occur with bridging oxygen sites (Fig. S15†). The computed energies agree well with the experimental normalized current densities (Fig. 2b), where Fe4-WS shows slightly better OER activity than Fe–K.
| POM | Basis set | Transition states | ||
|---|---|---|---|---|
| a All energies are in kcal mol−1. b TS1B and TS1T represent the two different transition states computed with only one molecule of water, which forms a hydrogen bond interaction with either a bridging oxygen or a terminal oxygen, respectively. c TS2 results from the computation of two molecules of water that form a hydrogen bond with a terminal oxygen. d The Keggin anion only presents hydrogen bond interactions with a bridging oxygen. Two TSs (TS1B1 and TS1B2) were located using one molecule of water. | ||||
| TS1B | TS1T | TS2 | ||
| Fe4-WS | BS2 | 25.6 | 25.7 | 19.8 |
| BS1 | 23.2 | 20.0 | 14.2 | |
| TS1B1 | TS1B2 | TS2 | ||
| Fe–K | BS2 | 27.3 | 27.3 | 24.2 |
| BS1 | 25.5 | 25.1 | 21.2 | |
We also compared the activation barriers between the Fe and Co based POMs using the small basis set BS1. The reported Gibbs free energy for Co4-WS in TS1B was 22.9 kcal mol−1,40 which corresponds to a 0.3 kcal mol−1 lower energy than the corresponding value for Fe4-WS. Additionally, we have obtained a systematic series of results for the Co–K catalyst using BS1 and BS2. In this case, the activation barriers were found to be ca. 2 kcal mol−1 lower in energy than the Fe analogue (Table S3†). These results are consistent with the greater catalytic activity shown by the cobalt compounds (Fig. 2).
O2 evolution
Once the Int (FeII–OOH2) species is reached, the following steps in the reaction mechanism are two PCET events that lead to the oxygen evolution from the reaction site. Finally, the POM is regenerated by adding a water molecule followed by a last PCET event, leading to the resting sate S1 (FeIII–OH) species. Int (FeII–OOH2) is not a very stable intermediate, which easily transforms into S3 (FeIII–OOH) by removing one electron from the SOMO (β) localized at the Fe atom (Fig. 6b). A direct consequence is that the electron spin density slightly increases at the Fe center, together with a much larger increase at the reactive O atom (Table 2). This suggests that S3 would probably be better represented by a combination of the two resonance forms
The next PCET event involves the oxidation and deprotonation of the hydroperoxo ligand of the S3 (FeIII–OOH) species due to the significant contribution of the p(O) orbital in its SOMO (Fig. 6c). This step leads to the formation of the S4 (FeII–OO) species in a very favoured process with an associated potential of −0.30 V. It is worth mentioning that an electron-donation effect is again observed from ligand to metal, which implies the formal reduction of the iron center from FeIII to FeII. It must be noted that all attempts to allocate S4 (FeII–OO) as “end-on” (η1) or “side-on” (η2) led to an adduct with a long Fe–O2 distance, where O2 can be considered uncoordinated to the metal center. A structure with coordinated O2 similar to the one found for the cobalt analogue could be located by using BS1. However, this structure is found 0.8 kcal mol−1 above the uncoordinated equivalent (2S + 1 = 20) and at 17.0 kcal mol−1 above the equivalent structure with uncoordinated triplet oxygen (2S + 1 = 22). This indeed suggests that O2 is a very labile ligand in this POM. Once the oxygen molecule is released, a new water molecule will favourably coordinate to iron (ΔG = −8.6 kcal mol−1), to form the S0 (FeII–OH2) species. The last step consists in the regeneration of the catalyst to its resting state from S0 (FeII–OH2). This step can occur via a PCET step that leads directly to S1 (FeIII–OH), or through a sequential electron-then-proton transfer event at pH 7. Both paths are expected to occur applying a potential below +1 V.
The overall mechanism can be described in two equivalent ways: (i) taking the initial species S1 (FeIII–OH) as the initial stage (PCET + WNA + 2× PCET + O2 evolution + PCET) or (ii) taking the S0 (FeII–OH2) species as the initial stage (2× PCET + WNA + 2× PCET + O2 evolution). The latter mechanism is analogous to other postulated ones for water oxidation, in which usually two protons and two electrons are released (coupled or not) before the water nucleophilic attack.72 The major difference here is that the S0 (FeII–OH2) species naturally suffers a PCET event in solution under aerobic conditions, yielding the S1 (FeIII–OH) species in the crystalline solid. Once it enters the WOC reaction, the complete cycle is needed to move the reaction further. Either way, the potential-limiting step in the reaction mechanism is the S1 (FeIII–OH) → S2 (FeIVO) PCET event with a required applied potential of 1.54 V, whereas a chemical barrier of 19.8 kcal mol−1 (0.86 eV) must be overcome to form the O–O bond through a WNA step.
Conclusions
We have studied the electrocatalytic OER activity in neutral media of the [(FeIIIOH2)2FeIII2(B-α-PW9O34)2]6− (Fe4-WS) and [(FeIIIOH2)PW11O39]4− (Fe–K) polyanions. The insoluble barium salts of the POMs were used to modify carbon paste electrodes and their activities were compared with that of the isostructural cobalt derivatives. Experiments show that both Fe-POMs display similar OER activity, and slightly slower kinetics than the Co-POM counterparts.The speciation of molecular WOCs at the working conditions is key to understand the reaction mechanisms. Acid–base titration experiments and DFT calculations allowed us to calculate a pKa1 value of 6.0 in the case of Fe4-WS and 4.3 for Fe–K. This indicates that under our experimental conditions of pH 7, both Fe-POM catalysts are found as POM-FeIII–OH species (S1) in the solid. Thus, a detailed DFT study of the OER mechanism promoted by Fe4-WS shows two key steps in the catalytic reaction, (i) the potential-limiting step, S1 (FeIII–OH) → S2 (FeIVO), proceeds via a PCET event and requires an applied potential of 1.54 V; (ii) the chemical-limiting step corresponds to the O–O bond formation during the WNA and has an associated activation barrier of 19.8 kcal mol−1 (0.86 eV). Moreover, we have seen that the presence of hydrogen bonds during the WNA helps in decreasing the activation barrier for the O–O bond formation. These hydrogen bonds are more effective in the sandwich species compared to the monosubstituted Keggin structures, as illustrated by their calculated lower activation energies.
Despite the different initial states in the solid for Fe4-WS (S1) and Co4-WS (S0) at neutral pH, the computed potential-limiting steps FeIII–OH → FeIVO and CoIII–OH → CoIII–O˙ involve analogous associated energies. These results are in good agreement with experimental observations, since Fe4-WS requires just a higher applied potential than Co4-WS. Moreover, the lower Tafel slope displayed by Co4-WS indicates faster kinetics than Fe4-WS, which correlates well with the lower activation barriers found for the Co-POM.
It is very illustrative to see that even though POM-FeIIIOH and POM-CoIIOH2 anions show significant differences in their electronic structures, the observed overpotentials are rather similar and trends are well reproduced by our computational studies. This is very relevant for at least two reasons: (1) CoII-POMs have shown excellent OER activity under strong acidic conditions; (2) given the questioned stability of POMs under electrocatalytic conditions and the complexity of the reaction mechanisms, the agreement between experimental and theoretical observations reinforces both experimental characterizations and theoretical studies. The current results strongly suggest that FeIII-POMs are stable under heterogeneous electrocatalytic conditions and active as OER catalysts. With these promising results, our groups are currently working to show that FeIII-POMs can be active and stable in acidic conditions.
Author contributions
J.-R. G.-M. and J. M. P. conceived the work. A.-L. T., I.-M. M. and P. d. O. synthesized the compounds, K. A., J. S.-L. and J.-R. G.-M. performed and analysed electro-catalytic experiments, M. L., M. B. and J. M. P. performed and analysed theoretical calculations. K. A., M. B. and J. S.-L. wrote the paper with the input of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministerio de Ciencia e Innovación through Severo Ochoa Excellence Accreditation 2020–2023 (CEX2019-000925-S, MIC/AEI); the FEDER/Ministerio de Ciencia, Innovación y Universidades – Agencia Estatal de Investigación (projects RTI2018-095618-B-100 and CTQ2017-87269-P), the Generalitat de Catalunya (2017-SGR-797, 2017-SGR-629) and the CERCA program. J.M.P. also thanks the ICREA foundation for an ICREA ACADEMIA award and the URV for support. K.A. thanks MINECO for a predoctoral F.P.U. fellowship. M.L. thanks the Tunisian Ministry of Science for a grant. J.S.-L. acknowledges the funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 713567. A.-L.T., I.-M.M. and P.d.O. thank the Université Paris-Saclay and the CNRS for financial support.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- W. Steffen, K. Richardson, J. Rockstrom, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sorlin, Science, 2015, 347(6223), 1259855 CrossRef PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- J. R. Galán-Mascarós, Catal. Sci. Technol., 2020, 10, 1967–1974 RSC.
- S. A. Sherif, F. Barbir and T. N. Veziroglu, Sol. Energy, 2005, 78, 647–660 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, Springer International Publishing, Cham, 2016, pp. 41–104 Search PubMed.
- J. Soriano-López, W. Schmitt and M. García-Melchor, Curr. Opin. Electrochem., 2018, 7, 22–30 CrossRef.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- J. R. Galán-Mascarós, ChemElectroChem, 2015, 2, 37–50 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- A. J. Esswein, Y. Surendranath, S. Y. Reece and D. G. Nocera, Energy Environ. Sci., 2011, 4, 499–504 RSC.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- D. Gao, I. Trentin, L. Schwiedrzik, L. González and C. Streb, Molecules, 2020, 25, 1–20 Search PubMed.
- S. Goberna-Ferrón, J. Soriano-López, J. R. Galán-Mascarós and M. Nyman, Eur. J. Inorg. Chem., 2015, 2015, 2833–2840 CrossRef.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- Q. Yin, J. M. Tan, C. Besson, Y. V Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- T. J. R. Weakley, H. T. Evans, J. S. Showell, G. F. Tourné and C. M. Tourné, J. Chem. Soc., Chem. Commun., 1973, 139, 140 Search PubMed.
- S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707–11715 CrossRef PubMed.
- J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753–4755 CrossRef PubMed.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2017, 10, 24 CrossRef PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- C. Casadevall, A. Bucci, M. Costas and J. Lloret-Fillol, in Water Oxidation Catalysts, ed. R. van Eldik and C. D. Hubbard, Academic Press, 2019, vol. 74, pp. 151–196 Search PubMed.
- F. Müh and A. Zouni, Photosynth. Res., 2013, 116, 295–314 CrossRef.
- H. Bandal, K. K. Reddy, A. Chaugule and H. Kim, J. Power Sources, 2018, 395, 106–127 CrossRef CAS.
- P. T. Babar, B. S. Pawar, A. C. Lokhande, M. G. Gang, J. S. Jang, M. P. Suryawanshi, S. M. Pawar and J. H. Kim, J. Energy Chem., 2017, 26, 757–761 CrossRef.
- Y. Yan, B. Zhao, S. C. Yi and X. Wang, J. Mater. Chem. A, 2016, 4, 13005–13010 RSC.
- D. Zhong, L. Liu, D. Li, C. Wei, Q. Wang, G. Hao, Q. Zhao and J. Li, J. Mater. Chem. A, 2017, 5, 18627–18633 RSC.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. Garcia de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- X. Du, Y. Ding, F. Song, B. Ma, J. Zhao and J. Song, Chem. Commun., 2015, 51, 13925–13928 RSC.
- M. Zheng, Y. Ding, X. Cao, T. Tian and J. Lin, Appl. Catal., B, 2018, 237, 1091–1100 CrossRef CAS.
- M. Zheng, X. Cao, Y. Ding, T. Tian and J. Lin, J. Catal., 2018, 363, 109–116 CrossRef CAS.
- J. Soriano-López, D. G. Musaev, C. L. Hill, J. R. Galán-Mascarós, J. J. Carbó and J. M. Poblet, J. Catal., 2017, 350, 56–63 CrossRef.
- X. Zhang, Q. Chen, D. C. Duncan, R. J. Lachicotte and C. L. Hill, Inorg. Chem., 1997, 36, 4381–4386 CrossRef CAS PubMed.
- N. Vilà, P. A. Aparicio, F. Sécheresse, J. M. Poblet, X. López and I. M. Mbomekallé, Inorg. Chem., 2012, 51, 6129–6138 CrossRef PubMed.
- I. M. Mbomekalle, B. Keita, L. Nadjo, P. Berthet, K. I. Hardcastle, C. L. Hill and T. M. Anderson, Inorg. Chem., 2003, 42, 1163–1169 CrossRef CAS PubMed.
- C. S. Ayingone Mezui, P. de Oliveira, A.-L. Teillout, J. Marrot, P. Berthet, M. Lebrini and I. M. Mbomekallé, Inorg. Chem., 2017, 56, 1999–2012 CrossRef CAS PubMed.
- S. Romo, J. A. Fernández, J. M. Maestre, B. Keita, L. Nadjo, C. de Graaf and J. M. Poblet, Inorg. Chem., 2007, 46, 4022–4027 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. G. FoxW. C. Inc., 2016.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
- J. T. Arens, M. Blasco-ahicart, K. Azmani, J. Soriano-lópez, A. García-eguizábal and J. M. Poblet, J. Catal., 2020, 389, 345–351 CrossRef CAS.
- A. G. MacDiarmid, Inorg. Synth., 1977, vol. 17, ISBN: 978-0-470-13283-8 Search PubMed.
- M. N. Kushner-Lenhoff, J. D. Blakemore, N. D. Schley, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2013, 42, 3617–3622 RSC.
- C. A. Ohlin, S. J. Harley, J. G. McAlpin, R. K. Hocking, B. Q. Mercado, R. L. Johnson, E. M. Villa, M. K. Fidler, M. M. Olmstead, L. Spiccia, R. D. Britt and W. H. Casey, Chem.–Eur. J., 2011, 17, 4408–4417 CrossRef CAS PubMed.
- D. Lieb, A. Zahl, E. F. Wilson, C. Streb, L. C. Nye, K. Meyer and I. Ivanović-Burmazović, Inorg. Chem., 2011, 50, 9053–9058 CrossRef CAS PubMed.
- S. M. Kanowitz and G. J. Palenik, Inorg. Chem., 1998, 37, 2086–2088 CrossRef CAS.
- S. Romo, J. A. Fernández, J. M. Maestre, B. Keita, L. Nadjo, C. de Graaf and J. M. Poblet, Inorg. Chem., 2007, 46, 4022–4027 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355(6321), eaad4998 CrossRef PubMed.
- M. Martin-sabi, J. Soriano-lópez, R. S. Winter, J. Chen, L. Vilà-nadal, D. Long, J. R. Galán-mascarós and L. Cronin, Nat. Catal., 2018, 1, 4–9 Search PubMed.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- H. Lei, A. Han, F. Li, M. Zhang, Y. Han, P. Du, W. Lai and R. Cao, Phys. Chem. Chem. Phys., 2014, 16, 1883–1893 RSC.
- L. Vigara, M. Z. Ertem, N. Planas, F. Bozoglian, N. Leidel, H. Dau, M. Haumann, L. Gagliardi, C. J. Cramer and A. Llobet, Chem. Sci., 2012, 3, 2576–2586 RSC.
- W. Lai, R. Cao, G. Dong, S. Shaik, J. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef CAS PubMed.
- J. Lloret-Fillol and M. Costas, in Advances in Organometallic Chemistry, ed. C. Pérez, Academic Press, 2019, vol. 71, pp. 1–52 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01016f |
| ‡ Permanent address: Laboratory of Chemical Materials (LR13ES08), Faculty of Sciences of Bizerte, Carthage University, 7021 Zarzouna, Tunisia. |
| This journal is © The Royal Society of Chemistry 2021 |