
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
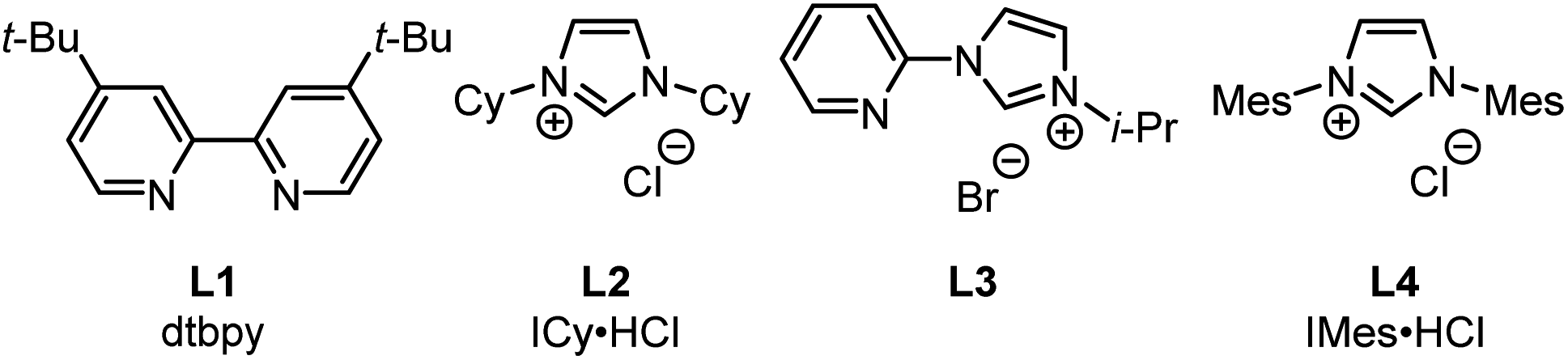
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hydrosilylboronates via the monoborylation of a dihydrosilane Si–H bond and their application for the generation of dialkylhydrosilyl anions†
Takumi
Takeuchi
a,
Ryosuke
Shishido
a,
Koji
Kubota
*ab and
Hajime
Ito
 *ab
*ab
aDivision of Applied Chemistry and Frontier Chemistry Center, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido, Japan. E-mail: kbt@eng.hokudai.ac.jp; hajito@eng.hokudai.ac.jp
bInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo, Hokkaido, Japan
First published on 3rd August 2021
Abstract
The synthesis of a series of hydrosilylboronates via the selective iridium- or nickel-catalyzed monoborylation of dihydrosilane Si–H bonds is described. The synthesized silylboronates, which bear a single Si–H bond, can be used as novel silicon nucleophiles in the presence of transition-metal catalysts or bases. The first 29Si{1H} NMR spectroscopic evidence for the formation of (t-Bu)2HSiLi, generated by the reaction of (t-Bu)2HSi–B(pin) with MeLi, is reported as the first example of a dialkylhydorosilyl lithium species.
Introduction
Silicon-based compounds have many applications in catalyst design, drug discovery, materials science, and polymer chemistry.1 The development of new reagents for the synthesis of organosilicon compounds is therefore of considerable interest in a broad range of scientific fields.1 Since the pioneering study of Suginome and Ito in 1996, silylboronates have become indispensable silylation reagents on account of their reactivity and the ease with which they can be handled.1–3 In the presence of a transition-metal catalyst or a base, silylboronates can be activated and used as silicon nucleophiles for reactions with a wide variety of electrophiles.1–3 Given their high synthetic utility, the development of efficient methods for the preparation of new silicon-based boron nucleophiles would expand the range of silicon-based compounds that are synthetically accessible.The typical synthetic route to silylboronates involves a stoichiometric reaction between a silyl anion and a boron electrophile.2 As silyl anions are in most cases produced by the reduction of chlorosilanes with alkali metals, the variety of substituents tolerated on the silicon atom is quite limited; moreover, at least one aromatic group is required at the silicon center in order to reduce chlorosilanes and disilane intermediates.4,5 Additionally, due to the harsh reduction conditions, this method suffers from low functional-group compatibility. Therefore, only a limited range of silylboronates can be prepared using this approach.
Instead, the direct borylation of Si–H bonds has emerged as a valuable complementary method for the synthesis of silylboronates (Scheme 1a).6,7 In 2008, Hartwig and co-workers reported a ground-breaking study on the borylation of trialkylhydrosilanes with bis(pinacolato)diboron [B2(pin)2] in the presence of an iridium-based catalyst. This reaction forms trialkylsilylboronates, which cannot be synthesized using the conventional reduction based on alkali metals.6 In 2020, our group reported that rhodium- and platinum-based catalysts can promote the borylation of trialkylhydrosilanes with B2(pin)2. Our method enables the synthesis of bulky and functionalized trialkylsilylboronates that are difficult to access via either the iridium-catalyzed borylation or the conventional reduction method (Scheme 1a).7
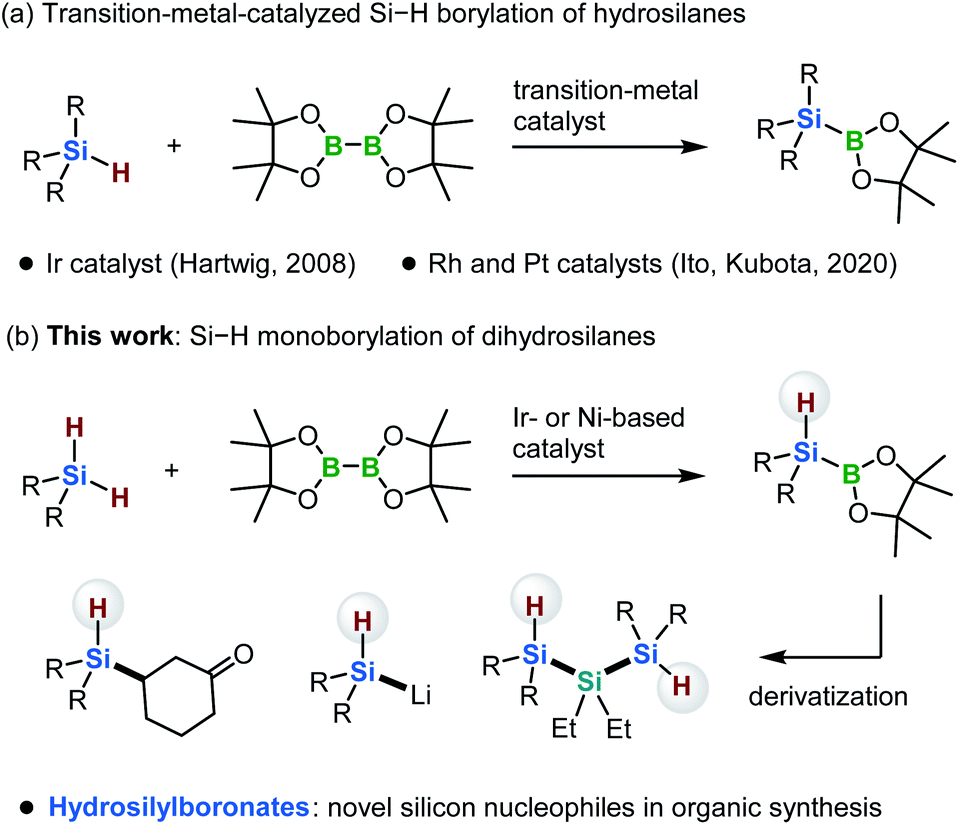 | ||
| Scheme 1 Transition-metal-catalyzed Si–H bond borylation of hydrosilanes. | ||
In the present study, we discovered that the monoborylation of a dihydrosilane Si–H bond can be achieved in the presence of iridium- or nickel-based catalysts, yielding hydrosilylboronates that bear a hydrogen atom at the silicon center (Scheme 1b). In 2004, Tokitoh and co-workers reported the first synthesis of diarylhydrosilylboronates via the insertion of a silylene into a H–B bond.8 Although this pioneering study is remarkable, the substituents on the silicon atom are limited to extremely bulky aryl moieties, such as mesityl and 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl groups, due to the highly reactive silylene species involved in the reaction.8 The monoborylation approach reported here is applicable to the synthesis of dialkylhydrosilylboronates and alkylarylhydrosilylboronates from easily accessible dihydrosilanes. These hydrosilylboronates are difficult to access by any other means. Furthermore, we demonstrate that these hydrosilylboronates can be used as novel silicon nucleophiles in the presence of a transition-metal catalyst or base. Moreover, we report the first 29Si{1H} NMR spectroscopic evidence for the formation of (t-Bu)2HSiLi, generated via the reaction of (t-Bu)2HSi–B(pin) with MeLi. Although the synthesis of disilylhydrosilyl lithium compounds has already been reported by Iwamoto, Kira and co-workers,5f this is the first example of the formation of dialkylhydrosily lithium species.
Results and discussion
We started by optimizing the reaction conditions for the borylation of di-tert-butylsilane (1a) with B2(pin)2 (2) in the presence of a variety of transition-metal catalysts (Table 1). Initially, we focused on an iridium-based catalytic system, as originally reported by Hartwig and co-workers (entry 1).6 The reaction using [Ir(cod)Cl]2/4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy) (L1) as the catalyst in cyclohexane at 80 °C proceeded smoothly to give the desired monoborylated product (3a) in 68% yield (entry 1). Notably, the di-borylated product was not detected. We also attempted the reaction with known Si–H bond borylation catalysts, such as [Rh(cod)Cl]2/1,3-dicyclohexylimidazolium chloride (ICy·HCl) (L2) and Pt/C.7 However, the rhodium-based catalyst generated a complex product mixture (3a: <1%; entry 2), and the platinum-based catalyst produced 3a in 36% yield (entry 3). In an effort to improve the iridium-based catalytic system, we decided to use N-heterocyclic carbene (NHC) ligands instead of dtbpy (L1).7 The use of ICy·HCl (L2) resulted in 43% yield of 3a (entry 4), whereas the use of L3, i.e., an NHC ligand that contains a pyridine moiety, provided a slightly higher yield than when dtbpy (L1) was employed (73%, entry 5).9 We found that 3a could be obtained in 71% yield (entry 6) even at a lower catalyst loading (0.5 mol%). Although there aren't any reports in the literature on the borylation of Si–H bonds using a nickel catalyst,10 we discovered that nickel-based catalysts are also effective for the monoborylation of 1a. While the formation of 3a was not observed when Ni(cod)2/1,3-dimesitylimidazolium chloride (IMes·HCl) (L4) was employed (3a: <1%; entry 7), the use of L2 instead of L4 afforded 3a in 50% yield (entry 8). However, the reproducibility of the reaction was unsatisfactory under these conditions (for details, see the ESI†). After an extensive screening of the reaction conditions, we found that when the reaction was carried out in n-octane at 120 °C, 3a was obtained in 54% yield with excellent reproducibility (entry 9; for details, see the ESI†). We also demonstrated that 3a can be isolated in 63% yield using column chromatography on silica gel (entry 6).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Ligand (mol%) | Base (mol%) | Yield (%) of 3ab |
| a Conditions: 1a (0.50 mmol), 2 (1.0 mmol), catalyst, ligand, and base in cyclohexane (1.0 mL) at 80 °C. b Determined by GC analysis of the reaction mixture using n-C13H28 as the internal standard. The isolated yield is shown in parentheses. c N,N-Dimethylformamide (DMF) was used as the solvent. d The reaction was carried out at the 1.0 mmol scale. e Average of four runs. f n-Octane was used as the solvent and the reaction was carried out at 120 °C. | ||||
| 1 | [Ir(cod)Cl]2 (2.5) | L1 (5) | None | 68 |
| 2c | [Rh(cod)Cl]2 (2.5) | L2 (10) | K(O-t-Bu) (20) | <1 |
| 3 | Pt/C (5 wt%) (5) | None | None | 36 |
| 4 | [Ir(cod)Cl]2 (2.5) | L2 (10) | K(O-t-Bu) (15) | 43 |
| 5 | [Ir(cod)Cl]2 (2.5) | L3 (5) | K(O-t-Bu) (10) | 73 |
| 6d | [Ir(cod)Cl]2 (0.5) | L3 (1) | K(O-t-Bu) (2) | 71 (63) |
| 7 | Ni(cod)2 (5) | L4 (10) | K(O-t-Bu) (10) | <1 |
| 8 | Ni(cod)2 (5) | L2 (10) | K(O-t-Bu) (10) | 50e |
| 9f | Ni(cod)2 (5) | L2 (10) | K(O-t-Bu) (10) | 54e |
|
||||

To highlight the practical utility of this borylation protocol, a gram-scale synthesis of 3a was carried out (Scheme 2). In the presence of the newly developed iridium-based catalyst, 3a was obtained in 70% yield (1.33 g) even when a lower catalyst loading was employed (0.5 mol%).
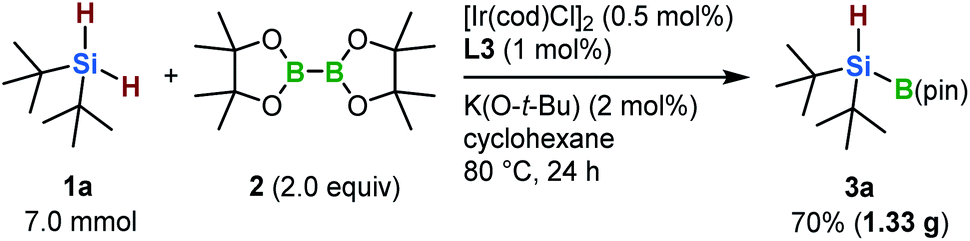 | ||
| Scheme 2 Gram-scale synthesis of 3a. | ||
The molecular structure of 3a was confirmed by single-crystal X-ray diffraction analysis (for details, see the ESI†). Although several conformers of 3a were observed in the disordered structure, the presence of a silicon–boron bond was confirmed unambiguously.
With the optimized conditions in hand, the synthesis of various dialkylhydrosilylboronates was investigated (Scheme 3). The dialkylhydrosilylboronate that bears two cyclohexyl groups (3b) was produced via both of the iridium- and nickel-catalyzed borylation reactions of the corresponding dihydrosilane (1b) (24% and 38% GC yields, respectively). In the case of nickel-catalyzed borylation, 3b was isolated by silica-gel column chromatography without significant decomposition (29% isolated yield). Next, the monoborylation of a dihydrosilane bearing a tertiary and a primary alkyl group (1c) was investigated. The desired dialkylhydrosilylboronate (3c) was obtained in low yield using the iridium-based catalyst (11% isolated yield, Scheme 3b). The nickel-based catalyst was more effective for the borylation of 1c, generating 3c in 43% isolated yield (Scheme 3b). We found that the sterically less hindered dialkyldihydrosilane 1d did not provide 3d under either set of conditions (Scheme 3c). We speculated that less hindered dihydrosilanes could potentially undergo fast dehydrogenative coupling with another dihydrosilane molecule, facilitated by the metal catalyst, to form silicon-based oligomers,11 thus impeding the desired borylation reaction.
 | ||
| Scheme 3 Si–H monoborylation of dihydrosilanes 1b–1d. Isolated yields are shown. GC yields are shown in parentheses. | ||
Furthermore, aryl-substituted dihydrosilanes were employed for the present monoborylation reaction (Table 2). Pleasingly, t-BuPhHSi–B(pin) (3e) was obtained from both the iridium- and nickel-based catalytic systems in 57% and 31% yield, respectively (entry 1). Unfortunately, sterically less hindered CyPhSiH2 (1f) and n-BuPhSiH2 (1g) did not produce the corresponding hydrosilylboronates (3f and 3g; entries 2 and 3). In these cases, the formation of oligosilanes produced by dehydrogenative homo-coupling was observed,11 suggesting that the presence of a bulky t-Bu group on the silicon atom is necessary for efficient Si–H monoborylation. Next, the steric effect of the aryl group was investigated (entries 4–6). para-Tolyl- and meta-tolyl-substituted hydrosilylboronates 3h and 3i were obtained in yields comparable to that of 3e (entries 4 and 5). However, ortho-tolyl-substituted hydrosilylboronate 3j was not obtained by the Si–H monoborylation using the iridium-based catalyst; this is probably due to a competing benzylic C–H borylation (entry 6).12 In contrast, the nickel-based catalyst afforded 3j in good yield (59% isolated yield; entry 6). Hydrosilylboronate 3k, which bears a 4-MeOC6H4 group, was also obtained in a yield comparable to that of 3e (entry 7). Notably, these hydrosilylboronates (3e and 3h–3k) show high stability toward air and moisture and can be isolated by flash column chromatography on silica gel. Although the monoborylation of diarylsilanes was also attempted, the desired silylboronates were not obtained (for details, see the ESI†).
|
|
|||
|---|---|---|---|
| Entry | Silane (1) | Product (3) | Isolated yield (%) |
| a [Ir] conditions: 1e–i (0.50 mmol), 2 (1.0 mmol), [Ir(cod)Cl]2 (0.50 mol%), and L1 (1.0 mol%) in cyclohexane (1.0 mL) at 80 °C for 24 h. [Ni] conditions: 1e–i (0.50 mmol), 2 (1.0 mmol), Ni(cod)2 (5.0 mol%), L2 (10 mol%), and K(O-t-Bu) (10 mol%) in n-octane (1.0 mL) at 120 °C for 24 h. | |||
| 1 |

|

|
[Ir]: 57% |
| [Ni]: 31% | |||
| 2 |

|
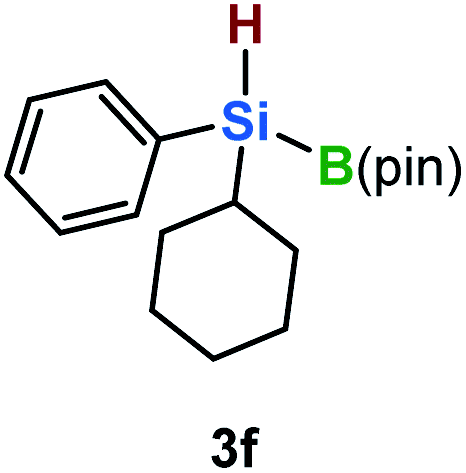
|
[Ir]: <1% |
| [Ni]: <1% | |||
| 3 |

|
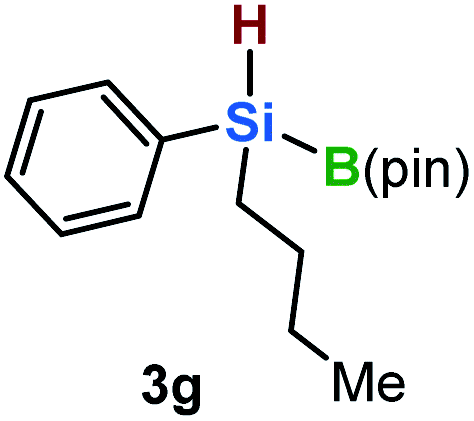
|
[Ir]: <1% |
| [Ni]: <1% | |||
| 4 |

|
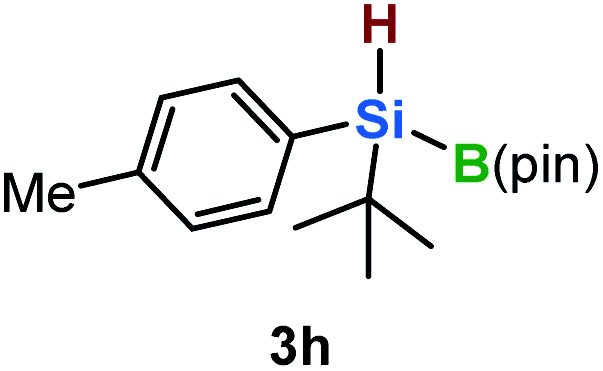
|
[Ir]: 61% |
| [Ni]: 29% | |||
| 5 |
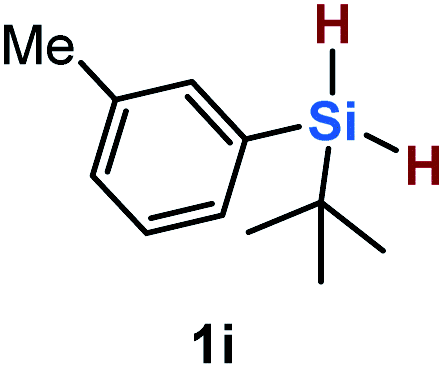
|
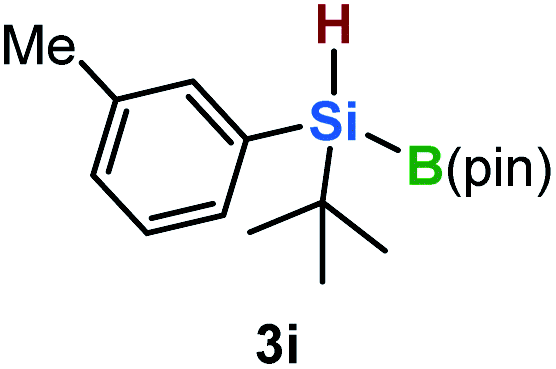
|
[Ir]: 58% |
| [Ni]: 31% | |||
| 6 |

|
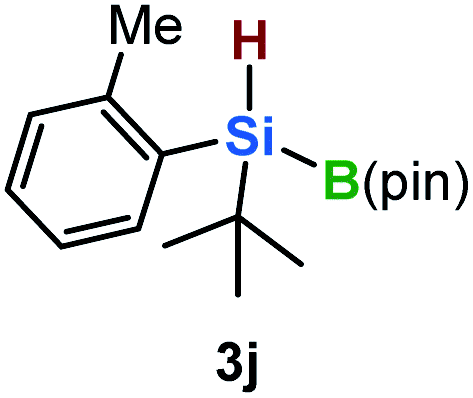
|
[Ir]: <1% |
| [Ni]: 59% | |||
| 7 |

|

|
[Ir]: 71% |
| [Ni]: 29% | |||

To demonstrate the synthetic utility of the newly synthesized hydrosilylboronates, a preliminary study of the activity of 3a in nucleophilic silylation reactions was conducted (Scheme 4). A copper(I)-catalyzed conjugated silylation of cyclohexenone (4) with 3a proceeded to form the desired β-silylated ketone (5) in 56% yield (Scheme 4).13 Furthermore, a nickel-catalyzed silylation of 2-methoxynaphthalene 6 with 3a produced the corresponding aryl silane (7) in 36% yield (Scheme 4).14 In addition to nucleophilic silylations, 3a could also be applied to a Si–H bond-functionalization reaction. For example, the chlorination of a Si–H bond in 3a, when treated with trichloroisocyanuric acid (8), furnished chlorosilylborane 9 in 76% yield (Scheme 4).15
 | ||
| Scheme 4 Selective transformations with 3a. | ||
Oligosilanes are currently of interest due to their unique optical, electronic, and photoreactive properties, which originate from their silicon–silicon bonds.16 Thus, we carried out silicon–silicon cross-coupling reactions between newly synthesized silylboronate 3a and various silyl chlorides, in the presence of an activating nucleophile (Scheme 5).7,17 The silicon–silicon coupling between 3a and triethylsilyl chloride in the presence of methyl lithium (MeLi) afforded the corresponding Si–H bond-bearing disilane (10a) in excellent yield (92%, Scheme 5a). Furthermore, the nucleophilic disilylation of both dichlorodiethylsilane and 1,2-dichlorotetramethyldisilane proceeded smoothly to form the desired trisilane (10b) and tetrasilane (10c) in 85% and 76% yield, respectively (Scheme 5b and c). The silicon–silicon coupling products can be used as building blocks for the construction of novel silicon-based compounds via further derivatizations based on Si–H bond functionalizations.
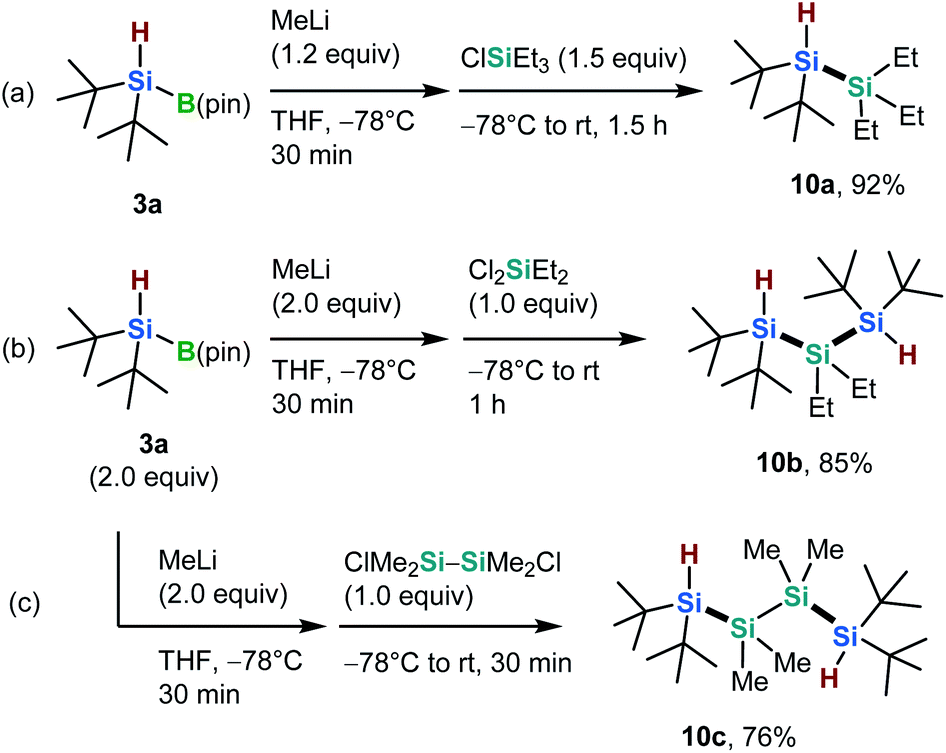 | ||
| Scheme 5 Silicon–silicon coupling reactions with 3a. | ||
Finally, we conducted in situ29Si{1H} NMR experiments to confirm the formation of a dialkylhydrosilyl anion in the reaction of 3a with MeLi. Kawachi and Tamao have reported the formation of Ph3SiLi during the reaction of Ph3Si–B(pin) with MeLi.17 More recently, we have reported the formation of i-Pr3SiLi during the reaction of i-Pr3Si–B(pin) with MeLi.7 Although Iwamoto, Kira and co-workers have already reported the generation of disilylhydrosilyllithium compounds in the reaction between disilyldihydrosilanes and alkyllithiums,5f to the best of our knowledge, the generation of a dialkylhydrosilyllithium species has not been reported so far.5f In the present study, we attempted to produce the dialkylhydrosilyl anion of 3avia treatment with MeLi in THF-d8 (Fig. 1). We observed a new 29Si signal (σ 14.2 ppm), which was attributed to silyllithium 11, in the 29Si{1H} NMR spectrum at room temperature (Fig. 1a). Furthermore, the 29Si–7Li coupling of 11 was observed at −95 °C (σ 11.8 ppm, quartet, J [29Si–7Li] = 50 Hz) (Fig. 1b). These results indicate that (t-Bu)2HSiLi (11) is generated in situ. This is in agreement with the reports from Kawachi and our group on the heterolytic cleavage and the formation of silyl anion species Ph3SiLi and i-Pr3SiLi.7,17 To the best of our knowledge, this is the first 29Si{1H} NMR spectroscopic evidence for the formation of a dialkylhydrosilyllithium species.
 | ||
| Fig. 1 29Si{1H} NMR spectra of (t-Bu)2HSi–Li (11) obtained from the reaction of 3a with MeLi: (a) 3a (0.1 mmol) with MeLi (0.15 mmol) in THF-d8 (0.2 M) after stirring for 1 h at −78 °C and further 1 h at rt. The 29Si{1H} NMR analysis was conducted at room temperature. Me4Si was used as an external standard to calibrate the 29Si{1H} NMR spectra. (b) The 29Si{1H} NMR spectrum of the mixture of 3a (0.1 mmol) and MeLi (0.15 mmol) in THF-d8 (0.2 M) analyzed at −95 °C. | ||
Conclusions
In conclusion, we have developed iridium- and nickel-catalyzed monoborylations of dihydrosilane Si–H bonds with bis(pinacolato)diboron to produce hydrosilylboronates. Notably, these molecules bear a hydrogen atom at the silicon center. Importantly, the newly synthesized silylboronates can be used in the presence of activating transition-metal catalysts or bases as novel silicon nucleophiles. Furthermore, the first 29Si{1H} NMR spectroscopic evidence for the formation of a (t-Bu)2HSiLi species was reported.Author contributions
K. K. and H. I. conceived and designed the study. T. T., K. K. and H. I. co-wrote the paper. T. T. performed the chemical experiments and analyzed the data. R. S. performed the preliminary experimental studies. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Japan Society for the Promotion of Science (JSPS) via KAKENHI grants 18H03907, 17H06370, 20H04795 and 21H01926; by the JST via CREST grant JPMJCR19R1; FOREST grant JPMJFR201I; and by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Initiative (WPI), MEXT, Japan. We would like to thank Satsuki Matsuura, Yu Ozawa, and Dr Mingoo Jin (Hokkaido University) for their help analyzing the X-ray crystallography data. We also thank Dr Y. Kumaki (High-Resolution NMR Laboratory, Faculty of Science, Hokkaido University) for his assistance with NMR measurements.Notes and references
- (a) Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2020 Search PubMed; (b) Silicon in Organic, Organometallics, and Polymer Chemistry, ed. M. A. Brook, Wiley-Interscience Publication, 2000 Search PubMed; (c) A. B. Cuenca, R. Shishido, H. Ito and E. Fernandez, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- (a) M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777–2778 RSC; (b) M. Suginome, T. Matsuda and Y. Ito, Organometallics, 2000, 19, 4647–4649 CrossRef CAS; (c) T. Ohmura, K. Masuda, H. Furukawa and M. Suginome, Organometallics, 2007, 26, 1291–1294 CrossRef CAS.
- (a) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef CAS; (b) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29–49 CrossRef CAS; (c) W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef CAS PubMed; (d) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- For reviews on silyl anions, see: (a) H.-W. Lerner, Coord. Chem. Rev., 2005, 249, 781–798 CrossRef CAS; (b) A. Sekiguchi, V. Y. Lee and M. Nanjo, Coord. Chem. Rev., 2000, 210, 11–45 CrossRef CAS; (c) A. Kawachi and K. Tamao, Bull. Chem. Soc. Jpn., 1997, 70, 945–955 CrossRef CAS.
- For examples of the generation of functionalized silyl lithium compounds, see: (a) K. Tamao, A. Kawachi and Y. Ito, J. Am. Chem. Soc., 1992, 114, 3989–3990 CrossRef CAS; (b) K. Tamao and A. Kawachi, Organometallics, 1995, 14, 3108–3111 CrossRef CAS; (c) A. Kawachi and K. Tamao, Organometallics, 1996, 15, 4653–4656 CrossRef CAS; (d) A. Kawachi and K. Tamao, J. Am. Chem. Soc., 2000, 122, 1919–1926 CrossRef CAS; (e) A. Kawachi, Y. Oishi, T. Kataoka and K. Tamao, Organometallics, 2004, 23, 2949–2955 CrossRef CAS; (f) T. Iwamoto, J. Okita, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2002, 124, 11604–11605 CrossRef CAS PubMed.
- T. A. Boebel and J. F. Hartwig, Organometallics, 2008, 27, 6013–6019 CrossRef CAS.
- R. Shishido, M. Uesugi, R. Takahashi, T. Mita, T. Ishiyama, K. Kubota and H. Ito, J. Am. Chem. Soc., 2020, 142, 14125–14133 CrossRef CAS.
- T. Kajiwara, N. Takeda, T. Sasamori and N. Tokitoh, Organometallics, 2004, 23, 4723–4734 CrossRef CAS.
- M. Peter and R. Breinbauer, Tetrahedron Lett., 2010, 51, 6622–6625 CrossRef.
- For examples of nickel-catalyzed C–H bond borylation reactions, see: (a) T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC; (b) H. Zhang, S. Hagihara and K. Itami, Chem. Lett., 2015, 44, 779–781 CrossRef CAS; (c) A. Das, P. K. Hota and S. K. Mandal, Organometallics, 2019, 38, 3286–3293 CrossRef CAS.
- For selected examples of the dehydrogenative coupling of silanes mediated by transition-metal catalysts, see: (a) H.-G. Woo and T. D. Tilley, J. Am. Chem. Soc., 1989, 111, 8043–8044 CrossRef CAS; (b) L. Rosenberg, C. W. Davis and J. Yao, J. Am. Chem. Soc., 2001, 123, 5120–5121 CrossRef CAS; (c) P. Diversi, F. Marchetti, V. Ermini and S. Matteoni, J. Organomet. Chem., 2000, 593–594, 154–160 CrossRef CAS; (d) T. Baumgartner and W. Wilk, Org. Lett., 2006, 8, 503–506 CrossRef CAS; (e) M. Itazaki, K. Ueda and H. Nakazawa, Angew. Chem., Int. Ed., 2009, 48, 3313–3316 CrossRef CAS.
- S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157–8160 CrossRef CAS.
- K.-S. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 2898–2900 CrossRef CAS.
- C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed.
- S. Varaprath and D. H. Stutts, J. Organomet. Chem., 2007, 692, 1892–1897 CrossRef CAS.
- (a) T. Karatsu, J. Photochem. Photobiol., C, 2008, 9, 111–137 CrossRef CAS; (b) H. Tsuji, J. Michl and K. Tamao, J. Organomet. Chem., 2003, 685, 9–14 CrossRef CAS.
- A. Kawachi, T. Minamimoto and K. Tamao, Chem. Lett., 2001, 30, 1216–1217 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2065033. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01440d |
| This journal is © The Royal Society of Chemistry 2021 |