
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme mediated incorporation of zirconium-89 or copper-64 into a fragment antibody for same day imaging of epidermal growth factor receptor†
Stacey E.
Rudd
a,
Jessica K.
Van Zuylekom
 b,
Anna
Raicevic
c,
Lesley A.
Pearce
c,
Carleen
Cullinane
bd,
Charlotte C.
Williams
c,
Timothy E.
Adams
c,
Rodney J.
Hicks
d and
Paul S.
Donnelly
*a
b,
Anna
Raicevic
c,
Lesley A.
Pearce
c,
Carleen
Cullinane
bd,
Charlotte C.
Williams
c,
Timothy E.
Adams
c,
Rodney J.
Hicks
d and
Paul S.
Donnelly
*a
aSchool of Chemistry and Bio21 Molecular Science, Biotechnology Institute University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: pauld@unimelb.edu.au
bResearch Division, Peter MacCallum Cancer Centre, Melbourne, Victoria 3000, Australia
cCSIRO Manufacturing, Parkville, Victoria 3052, Australia
dSir Peter MacCallum Department of Oncology, University of Melbourne, Parkville, Victoria, Australia
First published on 25th May 2021
Abstract
Identification of tumors which over-express Epidermal Growth Factor Receptor (EGFR) is important in selecting patients for anti-EGFR therapies. Enzymatic bioconjugation was used to introduce positron-emitting radionuclides (89Zr, 64Cu) into an anti-EGFR antibody fragment for Positron Emission Tomography (PET) imaging the same day as injection. A monovalent antibody fragment with high affinity for EGFR was engineered to include a sequence that is recognized by the transpeptidase sortase A. Two different metal chelators, one for 89ZrIV and one for 64CuII, were modified with a N-terminal glycine to enable them to act as substrates in sortase A mediated bioconjugation to the antibody fragment. Both fragments provided high-quality PET images of EGFR positive tumors in a mouse model at 3 hours post-injection, a significant advantage when compared to radiolabeled full antibodies that require several days between injection of the tracer and imaging. The use of enzymatic bioconjugation gives reproducible homogeneous products with the metal complexes selectively installed on the C-terminus of the antibody potentially simplifying regulatory approval.
Introduction
Human epidermal growth factor receptor (EGFR) (also known as erbB1) is a 170 kDa type I transmembrane protein tyrosine kinase involved in signal transduction, cell growth and division.1 A wide range of malignant tumors over-express EGFR including bladder, lung, breast and colorectal cancers, where the overexpression is associated with resistance to chemotherapy. Therapies that target EGFR, such as the antibody cetuximab and tyrosine kinase inhibitors such as gefitinib, can be remarkably effective but identification of patients most likely to respond to these treatments is essential.2–4 Unfortunately, biopsies for EGFR expression do not always reflect the EGFR burden of heterogenous tumors and do not always predict a likely response to anti-EGFR therapy.5,6 An alternative approach to identify EFGR positive tumors is to perform diagnostic Positron Emission Tomography (PET) imaging with radiolabeled antibodies. Most research on PET imaging of EGFR has focused on the use of radiolabeled anti-EGFR full IgG antibodies.7,8 Such antibody-based probes take several days to clear from the blood and accumulate in the tumor due to their large molecular weight (∼150 kDa) as well as interactions between their Fc (fragment crystallisable) region and Fc-gamma receptors on a myriad of cell types. It is therefore necessary to image the patient several days after injection of the tracer. The several days required between injection and imaging is not always practical in a clinical setting. Radiolabled smaller engineered antibody fragments such as a Fab (fragment antigen-binding) (∼55 kDa) and F(ab′)2 fragments (∼110 kDa) retain the selectivity to the antigen, but clear from the blood and accumulate in tumors more rapidly as they lack the Fc region.9,10 This earlier time point imaging is often compromised by lower tumor uptake and higher kidney uptake when compared to tracers that use full IgG antibodies.11 Nonetheless, the potential of imaging on the same day as injection is an important consideration when developing a tracer for routine clinical use.In this work, we aimed to produce conjugates that enable same-day imaging of tumor EGFR expression using a recombinant Fab of an anti-EGFR murine monoclonal antibody, Fab528 (∼52 kDa)12 that was engineered for site-specific radiolabeling with either of the positron emitting radionuclides zirconium-89 (t1/2 = 79 h, βmean+ = 0.396 MeV) or copper-64 (t1/2 = 12.7 h, βmean+ = 0.278 MeV). Labeling of Fabs with metal ions requires the incorporation of specifically designed metal chelators. A conventional approach to attach chelators to Fabs is to use metal-binding chelators bearing N-hydroxysuccinimydyl esters that react with the amine functional group of lysine residues on the Fab. The multiple lysine residues present on Fabs leads to heterogeneous products with various numbers of chelators attached. Furthermore, modifications involving residues close to the antigen binding site can compromise biological activity by blocking interaction with the receptor. In this context it is preferable to achieve controlled site-specific conjugation. Several methodologies for site-specific incorporation of chelators have been developed including the incorporation of unnatural amino acids with biorthogonal functional groups for pre-targeting applications,8,13 or additional cysteine residues for subsequent maleimide conjugation chemistry.14,15 Enzymatic bioconjugations with enzymes such as transglutaminase,16–18 engineered galactosyltransferase,19–23 formylglycine generating enzyme24 and sortase A25 have also been investigated.
The bacterial transpeptidase, Staphylococcus aureus sortase A (SrtA) can be used for enzyme-mediated, site-specific incorporation of substrates onto suitably engineered target proteins.26 The enzyme acts by recognising a short –LPETG– amino acid motif, cleaving the threonine–glycine bond and forming a thio-acyl intermediate from a cysteine residue located in the active site.27,28 The enzyme then accepts an incoming nucleophilic N-terminal glycine to form a new amide bond. The incoming glycine can be sourced from the cleaved peptide or from another peptide in solution containing the required N-terminal glycine, allowing for modification of the C-terminus of the substrate protein (Fig. 1).29–31 SrtA mediated bioconjugation has been used to install positron-emitting radionuclides into the variable region segment of a camelid heavy chain-only antibody (∼15 kDa) to enable imaging of inflammation in mouse models.32–34 In our previous work, we used SrtA bioconjugation to install a copper-64 complex into a single-chain variable fragment antibody (∼30 kDa) to allow imaging of activated platelets in a mouse model of thrombosis, but this work is the first time the approach has been used for attaching a copper chelator to EGFR-targeting Fab.25
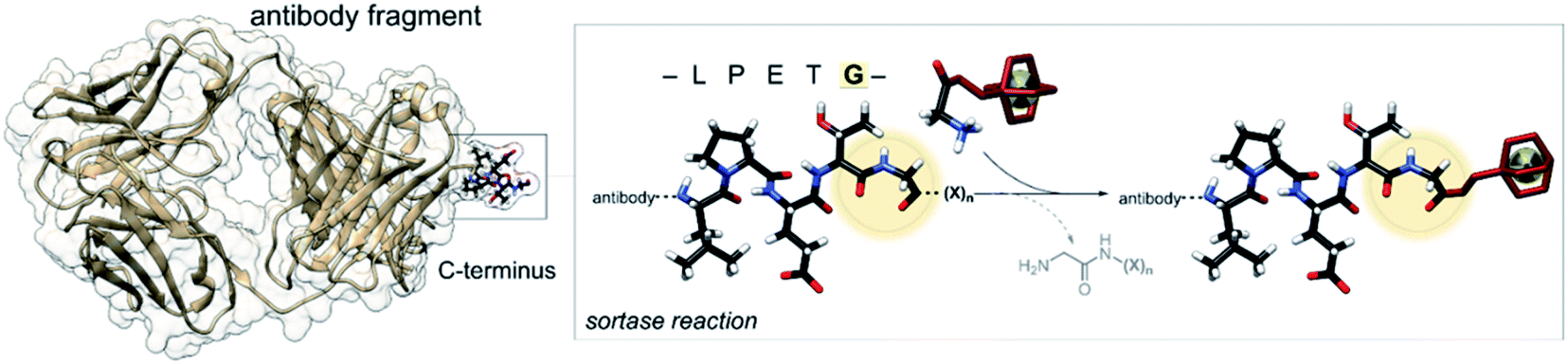 | ||
| Fig. 1 Schematic representation of the enzyme-mediated bioconjugation of a chelator to a Fab using SrtA at the C-terminal recognition sequence (LPETG). | ||
To label with zirconium-89 we prepared a squaramide ester derivative of desferrioxamine B, H3DFOSq with a terminal N-glycine residue to enable it to act as a substrate for SrtA (Fig. 2). A DFOSq derivative was used for zirconium-89 labeling as the conjugates are easier to prepare, easier to radiolabel and often have superior solubility when compared to derivatives prepared with the commonly used desferrioxamine B isothiocyanate.35 In addition, the [89Zr]ZrIV complexes that are formed are more resistant to ligand exchange.35,36 To develop the [64Cu]CuII-based agents a derivative of the macrobicyclic sarcophagine (sar = 3,6,10,13,16,19-hexaazabicyclo-[6.6.6]icosane) cage amine ligand, MeCOSar (Fig. 2), with an N-terminal glycine residue was used as radiolabeling can be achieved rapidly at room temperature to give high radiochemical yields and the complexes formed are stable in vivo.37–41 The new conjugates were evaluated in an EGFR positive A431 xenograft model and compared to conjugates of the full-length anti-EGFR IgG antibody, cetuximab, radiolabeled using the same metal chelators.
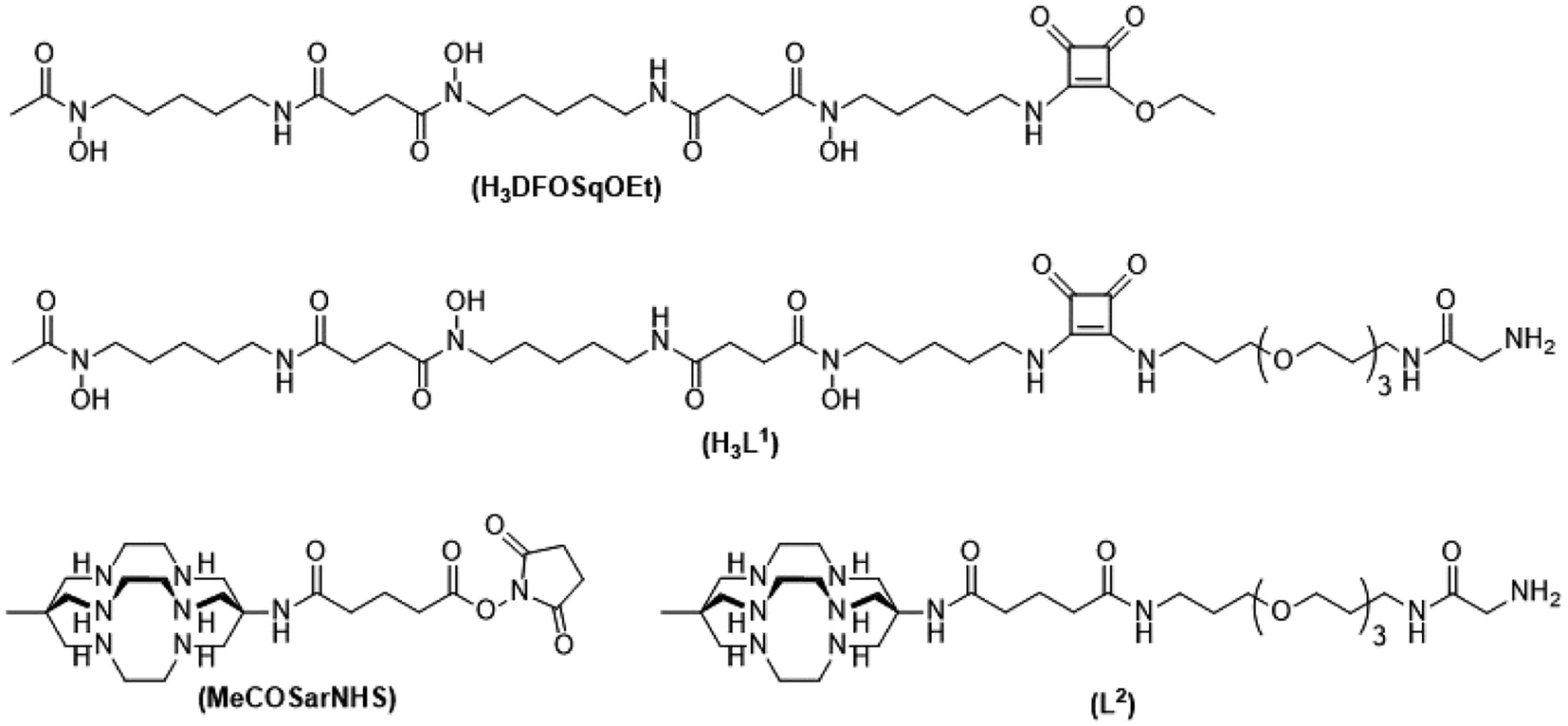 | ||
| Fig. 2 Chemical structures of H3DFOSqOEt, H3L1, MeCOSarNHS and L2. | ||
Results
Synthesis of metal chelators designed to be substrates for Srt A
The two ligands, H3DFOSq and MeCOSar, were modified to act as substrates for SrtA by incorporation of a short oligoethylene glycol (OEG) linker followed by a single N-terminal glycine residue to give H3L1 and L2 (Fig. 2). The site-specific labeling of target proteins by SrtA-mediated bioconjugation results in the reformation of LPETG consensus sequence and this leads to the potential for the enzyme to initialise further reactions. The presence of a synthetic OEG linker adjacent to a glycine residue in the substrate produces a product where the new bioconjugate contains a LEPTG-OEG sequence, impeding further reactivity and improving isolated yields.The synthesis of H3DFOSq-OEG-Gly (H3L1) required a selective single protection of one of the two primary amines present in 4,7,10-trioxa-1,13-tridecanediamine with a tert-butoxycarbonyl group (t-Boc) to give mono-t-Boc protected OEG3 diamine (1) (Fig. 3). Protection of the hydroxyamic acid functional groups in H3DFOSq with iron(III) was followed by reaction of this iron(III) complex with the mono-protected diamine 1 to give [Fe(DFOSq-OEG-NH2(t-Boc))] (2). Deprotection with trifluoracetic acid followed by reaction with t-Boc protected N-hydroxy succinimidyl ester of glycine (t-BocGlyOSu) and a final deprotection resulted in [Fe(DFOSq-OEG-Gly)] ([FeL1]). The iron(III) was removed with a large excess of disodium ethylenediamine tetraacetic acid (Na2H2EDTA) to give H3L1.
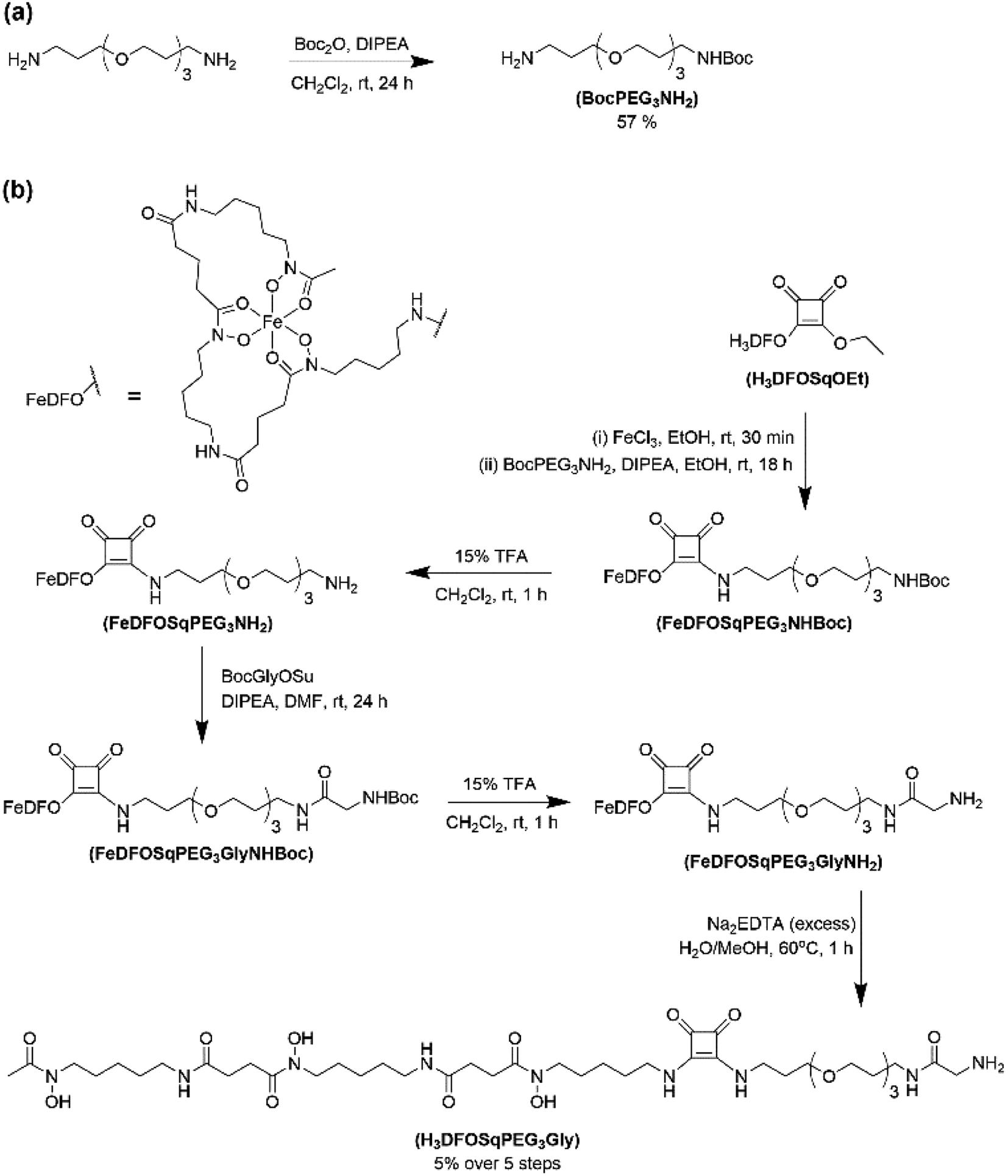 | ||
| Fig. 3 Reaction scheme for the synthesis of (a) compound 1 and (b) H3L1. | ||
A suitably modified variant of MeCOSar with an OEG linker and a terminal NH2-glycine residue, MeCOSarOEG-Gly (L2, Fig. 2) was prepared from the N-hydroxysuccinimide-activated (t-Boc)4–5MeCOSar-NHS ester as reported previously.40
Assembly and expression of anti-EGFR 528 Fab fragments
The murine monoclonal antibody 528 binds to the ligand-binding domain III of the EGFR in a similar manner to cetuximab. Monovalent 528 antibody fragments retain high affinity (KD ∼3 nM) for the EGFR.42 To generate both LPETG-His6 and LPETG-FLAG tagged Fab versions of 528 (Fab528), a DNA cassette was synthesised that facilitated the biosynthesis, assembly and secretion of tagged Fabs from a single mRNA following transfection of mammalian cells with the relevant expression vector. Preparations of each monovalent Fab were purified by a combination of affinity and gel filtration chromatography. Analysis of Fab528-LPETG-His6 and Fab528-LPETG-FLAG by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with samples prepared under non-reducing/reducing conditions revealed that the majority of both Fabs were in the form of LC:HC disulphide-linked dimers. The yield of recombinant Fabs was in the range of 8–10 mg per litre of transfected FreestyleTM-293 cells.Purification of mixtures containing MeCOSar and its conjugates was difficult using conventional His6-tag labeled proteins and immobilized metal affinity chromatography was complicated by low recovered yields. This is due to the sarcophagine chelator, which has extremely high affinities for first row transition metal ions, removing the NiII from the purification column. To circumvent this problem, the His6 tags in the vectors encoding Fab528-LPETG-His6, and SrtA-His6 were replaced with FLAG tags. As a result, superior yields were obtained when Fab528-LPETG-FLAG and SrtA-FLAG were used in MeCOSar conjugation reactions.
Site-specific enzymatic incorporation of chelators into an anti-EGFR 528 fragment
The coupling of the H3DFOSq derivative, H3L1, to Fab528-LPETG-His6 was catalysed by SrtA-His6 (60 μM) in the presence of Fab528-LPETG-His6 (20 μM) and H3L1 (2 mM). Unreacted Fab528-LPETG-His6, SrtA-His6 and the resulting cleavage peptide were removed using affinity chromatography followed by centrifuge membrane filtration to remove unreacted H3L1. The Fab528-LPET-H3L1 conjugate (H3L1-Fab528, 75% yield) was analysed by LC-ESIMS which confirmed a single addition of H3DFOSq to the protein.The coupling of the MeCOSar derivative, L2, to Fab528-LPETG-FLAG was catalysed by SrtA-FLAG (60 μM), in the presence of Fab528-LPETG-FLAG (13 μM) and L2 (2 mM). Purification by FLAG-affinity chromatography followed by centrifuge membrane filtration allowed isolation of Fab528-LPET-L2 conjugate (L2-Fab528 90% yield) with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of protein to chelator confirmed by LC-ESIMS.
:1 ratio of protein to chelator confirmed by LC-ESIMS.
PET imaging of EGFR-positive tumors with an anti-EGFR 528 fragment radiolabeled with either zirconium-89 or copper-64
The H3L1-Fab528 conjugate was radiolabeled with zirconium-89 in HEPES buffer at pH 7.4 and purified using a size exclusion column to give [89Zr][ZrL1]-Fab528 with radiochemical yield of >60% and a radiochemical purity of ≥99% by radio-SEHPLC (0.7 MBq μg−1) [89Zr][ZrL1]-Fab528 (2.2 MBq, ∼3 μg) was administered to EGFR-positive A431 tumor-bearing NSG mice via tail vein injection (n = 3). PET/CT images were acquired at 1, 3 and 18 h post-administration. Inspection of the PET images (Fig. 4a) reveals excellent tumor uptake of [89Zr][ZrL1]-Fab528 even at 1 h post-administration.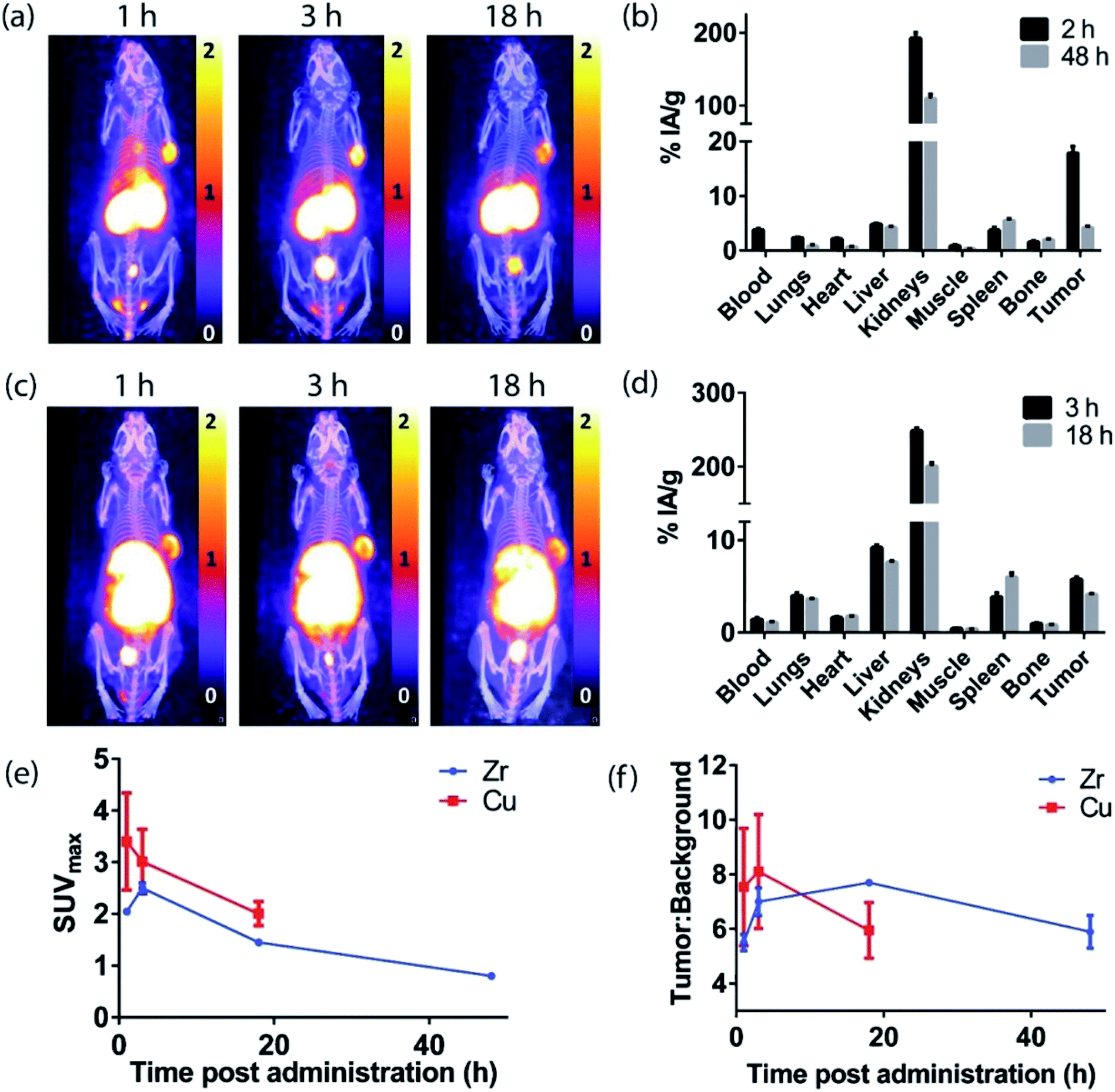 | ||
| Fig. 4 A431 xenograft tumor-bearing NSG mice following administration with radiolabeled Fab528. (a) PET/CT MIPs (scale given in SUV) following administration with [89Zr][ZrL1]-Fab528. (b) Ex vivo biodistribution data following administration with [89Zr][ZrL1]-Fab528. (c) PET/CT MIPs following administration with [64Cu][CuL2]-Fab528. (d) Ex vivo biodistribution data following administration with [64Cu][CuL2]-Fab528. (e) SUVmax values and (f) tumor:background ratios for both tracers. All values are given as mean ± SEM (n = 3). | ||
Quantification of tumor uptake by calculating SUVmax (where SUVmax = maximum standardised uptake value, eqn (1), where cROI is the concentration of the highest pixel within the region of interest selected, ID is the injected activity and BW is body weight) resulted in a tumor SUVmax = 2.05 ± 0.05 at 1 h post-administration that further increased to SUVmax = 2.50 ± 0.10 at 3 h. There is significant retention of the [89Zr][ZrL1]Fab528 in the tumor out to 18 h post-administration (Fig. 4e). The high degree of kidney uptake and retention is expected for Fab fragments43 but still permits PET images of high quality with good contrast between tumor to background (tumor:background ratio = 5.50 ± 0.30 and 7.00 ± 0.50 at 1 and 3 h respectively, where background is defined as an area of the mediastinum or gut, Fig. 4f).
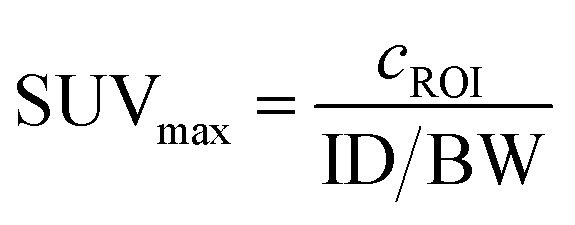 | (1) |
The tumor uptake and biodistribution of [89Zr][ZrL1]-Fab528 was also assessed at 2 and 48 h post-administration by ex vivo tissue analysis. Following intravenous injection of [89Zr][ZrL1]-Fab528 (2.2 MBq, ∼3 μg) the mice were euthanised at either 2 h or 48 h after administration and the amount of radioactivity in the tumor and major organs was quantified (Fig. 4b). The high tumor uptake of [89Zr][ZrL1]-Fab528 was confirmed (18 ± 1% IA g−1 at 2 h post-administration) reducing to 4.2 ± 0.3% IA g−1 at 48 h. The initial kidney uptake (192 ± 8% IA g−1 at 2 h) reduced to 109 ± 6% IA g−1 at 48 h. Importantly, the degree of liver, spleen and bone uptake was relatively low (<6% IA g−1) at both 2 and 48 h.
The L2-Fab528 conjugate was radiolabeled with copper-64 in PBS (pH 7.4) at room temperature. The reaction was complete within 1 h to give [64Cu][CuL2]-Fab528 with >95% radiochemical purity by radio-SEHPLC and used without further purification (0.18 MBq μg−1). [64Cu][CuL2]-Fab528 was administered to EGFR-positive A431 tumor-bearing NSG mice via tail vein injection (2.3 MBq, ∼13 μg each, n = 3). Mice underwent PET/CT imaging at 1, 3 and 18 h post-administration (Fig. 4c), with excellent tumor uptake evident at 1 h (SUVmax = 3.4 ± 0.9) and retention at 18 h (SUVmax = 2.0 ± 0.2). High kidney uptake and retention was also observed with [64Cu][CuL2]-Fab528, while tumor:background ratios remained high (7.54 ± 2.16 and 8.11 ± 2.10 at 1 and 3 h, respectively). Mice were euthanised at 3 and 18 h post-injection, dissected and the activity in major organs was quantified (Fig. 4d). Uptake of [64Cu][CuL2]-Fab528 was observed in the tumor at all timepoints, with the highest at 3 h (5.76 ± 0.25% IA g−1), decreasing to 4.12 ± 0.11% IA g−1 by 18 h. Accumulation in off-target organs was also observed, with particularly high kidney uptake (249 ± 4% IA g−1 at 3 h) clearing to 201 ± 5% IA g−1 at 18 h. Liver uptake evident in the images was confirmed (9.2 ± 0.3% IA g−1 decreasing to 7.6 ± 0.1% IA g−1 at 3 & 18 h, respectively). Spleen uptake (maximum observed 6.0 ± 0.5% IA g−1 at 18 h) and bone (<1% IA g−1 at all timepoints) remained low throughout the duration of the study.
PET imaging of EGFR positive tumors with cetuximab radiolabeled with either zirconium-89 or copper-64
The H3DFOSq-cetuximab conjugate was prepared by treating the antibody with H3DFOSqOEt35 (Fig. 2) at room temperature overnight. Analysis of the purified conjugate by mass spectrometry revealed an average of ∼3.9 chelators per antibody. Radiolabeling of H3DFOSq-cetuximab with zirconium-89 (HEPES buffer, pH 7) and purification by size-exclusion chromatography allowed isolation of [89Zr][ZrDFOSq]-cetuximab in a radiochemical yield of ∼64% and >95% radiochemical purity with a specific activity of 0.64 MBq μg−1. [89Zr][ZrDFOSq]-cetuximab (2.8 MBq, ∼5 μg) was administered to EGFR-positive A431 tumor-bearing Balb/c nu/nu mice via tail vein injection (n = 3) and PET images were acquired at 24, 48 and 120 h post-administration. The PET images show excellent tumor uptake at 24 h post-administration (SUVmax = 11.7 ± 1.4) (Fig. 5a and b) with an excellent tumor to background ratio of 19.6 ± 1.7 that increased to 24.4 ± 4.0 at 48 h post-administration (Fig. 5c). The high degree of tumor uptake was confirmed by quantification of radioactivity in the tumor and major organs at 24 and 120 h post-administration (Fig. 5d). The tumor uptake at 24 h post-administration (51 ± 2% IA g−1) was consistent with the high tumor uptake observed in the PET images. The uptake in the liver was 28 ± 2% IA g−1 at 24 h post-administration and was similar at 120 h post-administration. The uptake in bone, 10.0 ± 1.1% IA g−1 at 24 h post-administration increased to 21.1 ± 1.5% IA g−1 at 120 h and this increase was evident in the epiphysis of the long bones in the PET images, likely due to the epiphyseal growth plates and the age of the mice.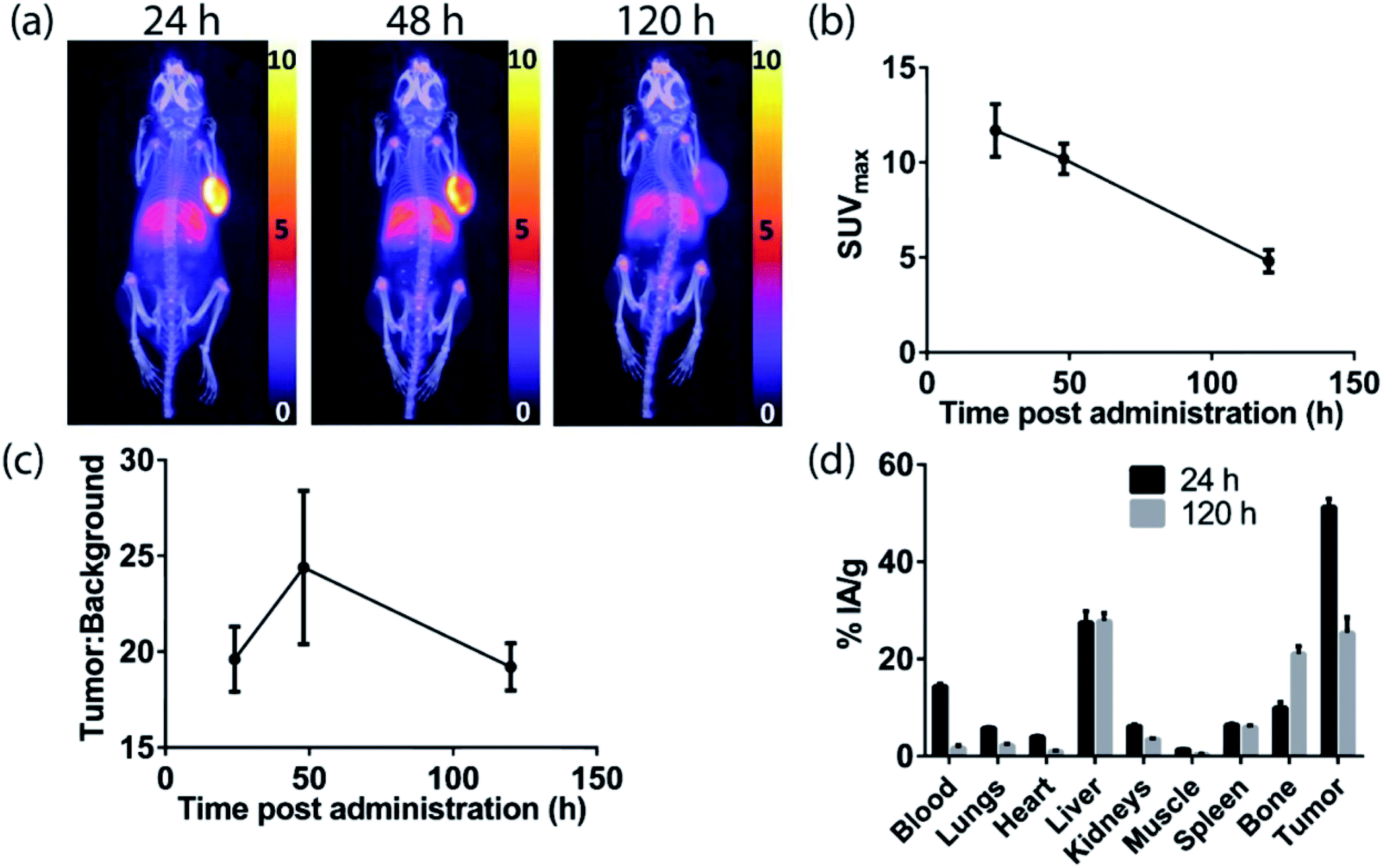 | ||
| Fig. 5
In vivo results following administration of [89Zr][ZrDFOSq]-cetuximab in A431 xenograft tumor-bearing Balb/c nu/nu mice. (a) PET/CT MIP images (scale given in SUV). (b) SUVmax values (c) tumor:background ratios and (d) ex vivo biodistribution results. All values are given as mean ± SEM (n = 3). | ||
The sarcophagine chelator, MeCOSar, was attached to cetuximab by treating the antibody with the N-hydroxysuccinimdyl ester MeCOSar-NHS39,40(Fig. 2) for 30 min at 37 °C (carbonate buffer, pH 9). Analysis of the MeCOSar-cetuximab conjugate by mass spectrometry revealed an average of ∼3.6 MeCOSar chelators per antibody. Radiolabeling MeCOSar-cetuximab with copper-64 in ammonium acetate buffer, and purification with size-exclusion chromatography allowed isolation of [64Cu][CuMeCOSar]-cetuximab (40% radiochemical yield, >95% radiochemical purity by radio-iTLC, specific activity of 0.1 MBq μg−1). [64Cu][CuMeCOSar]-cetuximab (2.1 MBq, ∼19 μg) was administered to A431 tumor-bearing NSG mice via tail vein injection (n = 3). PET images were acquired at 1, 3 and 18 h post-administration (n = 3). The images show that whilst tumor uptake was evident at 1 h post-administration the best values of SUVmax (17.6 ± 0.8) and tumor:background ratios (14.4 ± 1.3) were at 24 h (Fig. 6a–c).
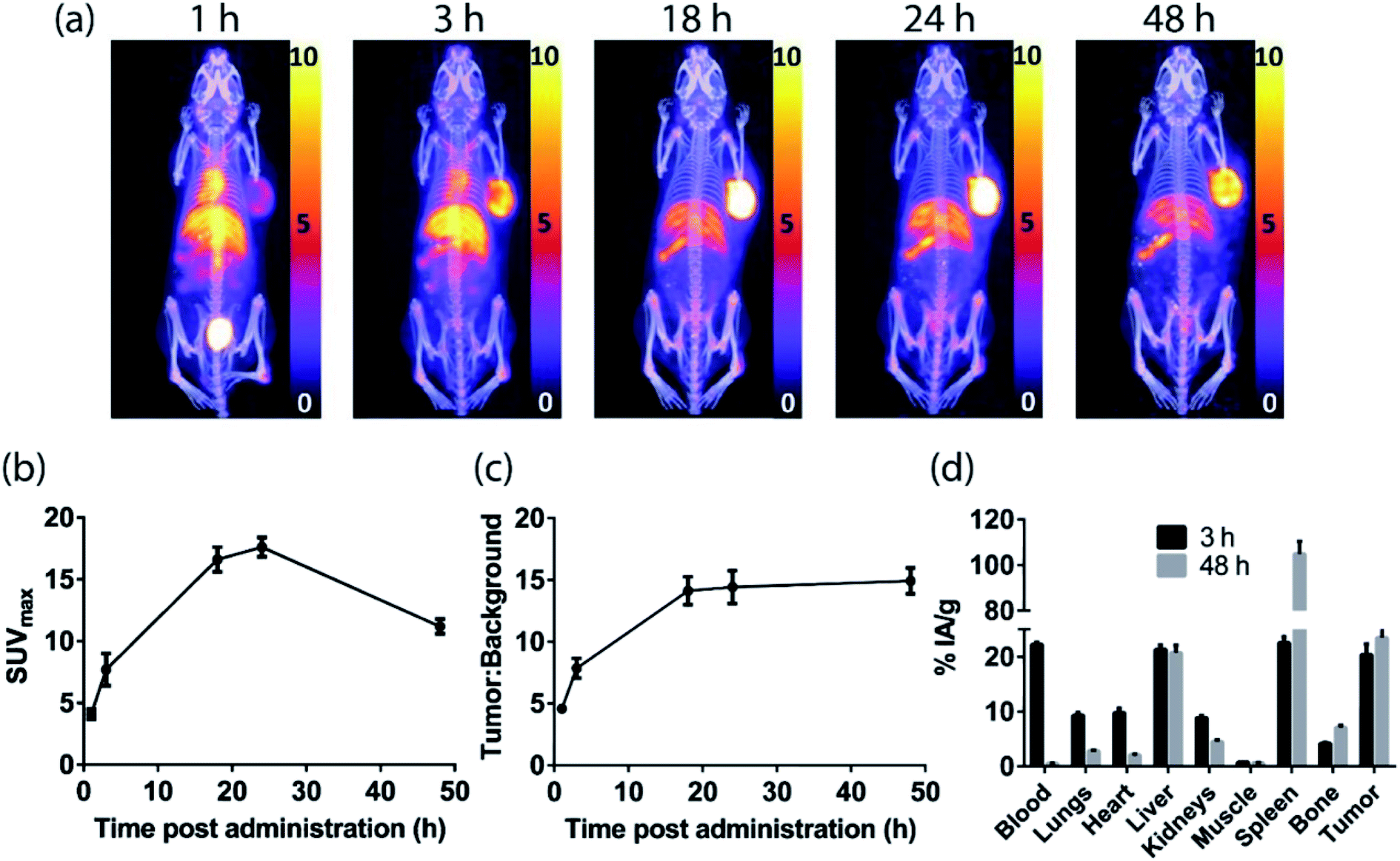 | ||
| Fig. 6
In vivo results following administration of [64Cu][CuMeCOSar]-cetuximab in A431 xenograft tumor-bearing NSG mice. (a) PET/CT MIP images (scale given in SUV). (b) SUVmax values (c) tumor:background ratios and (d) ex vivo biodistribution results. All values are given as mean ± SEM (n = 3). | ||
The high tumor uptake and retention was confirmed by an ex vivo biodistribution study (Fig. 6d) where tumor uptake at 3 h post-administration was 20 ± 2% IA g−1 and was still 23 ± 2% IA g−1 at 48 h post-administration. The liver uptake was 21.3 ± 0.8% IA g−1 at 3 h, and this remained steady throughout the rest of the study. The uptake of the tracer in the spleen uptake increased from 23 ± 1% IA g−1 at 3 h to 105 ± 5% IA g−1 at 48 h. A significant amount of radioactivity was evident in bone in the images at later timepoints, (18, 24 and 48 h) and the ex vivo biodistribution study confirms this with the bone uptake at 3 h 4.2 ± 0.2% IA g−1 increasing to 7.1 ± 0.4% IA g−1 at 48 h post-administration.
Discussion
Two positron-emitting radionuclides with different radioactive half-lives, zirconium-89 (t1/2 = 79 h, βmean+ = 0.396 MeV) and copper-64 (t1/2 = 12.7 hours, βmean+ = 0.278 MeV) were investigated. The relatively long radioactive half-life and amenable positron emission energy of zirconium-89 have seen the radionuclide emerge as the radionuclide of choice for PET imaging with full IgG antibodies.15,44,45 It was therefore of interest to directly compare the imaging characteristics of [89Zr][ZrDFOSq]-cetuximab with that of the smaller fragment, [89Zr][ZrL1]-Fab528. A potential disadvantage of zirconium-89 is that β+-emission leads to the formation of 89mY, which itself decays rapidly through γ-emission (920 keV, t1/2 = 15.7 s) to the stable 89Y isotope. This relatively high-energy γ-emission adds to radiation exposure. From a practical consideration, 89Zr needs more shielding for safe transport and handling than copper-64. The shorter radioactive half-life of copper-64 better suited for PET imaging with antibody fragments than zirconium-89, although imaging at least 24 hours after administration remains feasible.46 A potential advantage of using copper-64 based imaging agents is that they can be used to predict dosimetry for the same conjugate labeled with copper-67, a potentially therapeutic β−-emitting radionuclide (t1/2 = 62 h).44,45,47–49Two variants of an anti-EGFR Fab528 containing a LPETG SrtA recognition sequence were developed, one with a His6-tag and another with a FLAG tag to assist in purification.50 A derivative of H3DFOSq, suitably modified to act as a substrate for SrtA by the addition of a single N-terminal glycine residue separated from the chelator by an oligoethylene glycol spacer (H3L1), allowed the enzymatic incorporation of H3L1 into anti-EGFR Fab528 to give H3L1-Fab528 in good yields. This new construct was radiolabeled with zirconium-89 to give [89Zr][ZrL1]-Fab528 in good radiochemical yield and purity. To enzymatically incorporate a copper-binding ligand into Fab528, a derivative of a sarcophagine ligand featuring a similar oligoethylene glycol-glycine, L2, was used. The resulting conjugate could be radiolabeled with copper-64 at room temperature to give [64Cu][CuL2]-Fab528.
Both [89Zr][ZrL1]-Fab528 and [64Cu][CuL2]-Fab528 were evaluated in an A431 xenograft mouse model. The A431 cell line has very high levels of EGFR expression. While this may not be representative of most clinical situations, the model is well-established and provides a good basis for the comparison of different tracers. It is interesting to note that previous imaging studies have shown that this high-expression cell line has only medium-level uptake of EGFR-binding antibodies in vivo, with other medium-level EGFR expression lines showing even greater uptake.51 In our study, both Fab528 agents displayed rapid clearance from the blood and significant uptake in the tumor at just 1 h post-administration of the tracer with SUVmax = 2.05 ± 0.05 and 3.40 ± 0.94 and tumor:background ratios of 5.50 ± 0.30 and 7.54 ± 2.16, respectively. There is a further increase in the tumor:background ratio at 3 hours post-administration to 7.00 ± 0.50 and 8.11 ± 2.10. The majority of the background non-tumor bound radioactivity is in the kidneys. This is to be expected as small proteins such as Fab fragments generally undergo renal clearance, and is consistent with the renal uptake and retention of a 64Cu-labeled cetuximab Fab fragment reported previously.52 F(ab)2 fragments of cetuximab have also been imaged with copper-64 although only 24 h imaging results were reported, with significant uptake in kidney and liver.10,53 In the case of [64Cu][CuL2]-Fab528, there is also evidence of uptake in the liver and this is confirmed by the ex vivo biodistribution studies that reveal a slightly higher liver uptake (9.19 ± 0.30% IA g−1 at 3 h) than for the [89Zr][ZrL1]-Fab528 (4.79 ± 0.22% IA g−1 at 2 h). Liver uptake when using 64Cu-based imaging agents is often attributed to ‘free’ Cu, but this is unlikely in the case of [64Cu][CuL2]-Fab528. No ‘free’ copper was present on administration, and release of copper from MeCOSar in vivo has not been observed with other MeCOSar-based agents in mice,40,41 or humans54 nor with [64Cu][CuMeCOSar]-cetuximab, where liver uptake was antibody-mediated (Fig. 6). The differences in tumor and non-target uptake of the two Fab tracers highlights that modification of antibodies even with relatively low molecular weight metal complexes can alter biodistribution. The amount of antibody administered can also affect biodistribution and tumor uptake. In these studies picomole amounts of antibody conjugate were injected ([64Cu][CuMeCOSar]-cetuximab 127 pmol, [89Zr][ZrDFOSq]-cetuximab 33 pmol, [64Cu][CuL2]-Fab528 260 pmol, [89Zr][ZrL1]-Fab528 60 pmol). It is likely that the number of receptors expressed in the tumour far outweighs the molecules of tracer administered, so the extent of tumour uptake is likely to reflect the clearance and permeability of the tracer rather than saturation of receptors or the number of receptors present.55 Full IgG antibodies can also experience anomalous biodistribution in immune compromised mice due to Fc mediated interactions.56
It is also notable that, in addition to the rapid tumor uptake, both agents display significant retention in the tumor, even at 18 h after administration (Fig. 4). It is possible that this antibody fragment may display sufficient affinity and residualization within the tumor to be used for radionuclide therapy. Many therapeutic radiopharmaceuticals have a mixture of renal and hepatobiliary clearance, with the former generally required to reduce circulation of unbound agent through bone marrow, which contributes to myelotoxicity, and the latter, generally slower excretion still maintaining bioavailability for tumor uptake. Sharing radiation dose between these two non-target organs, both of which are relatively radioresistant, also increases the therapeutic index.
The radiolabeled full IgG antibodies, [89Zr]ZrDFOSq-cetuximab and [64Cu]CuMeCOSar-cetuximab were radiolabeled under mild, room temperature conditions and no evidence of aggregation was observed by size exclusion chromatography. The conjugates showed excellent tumor uptake in the images, but at much later timepoints than the Fab528. This high tumor uptake evidenced by the SUVmax values from the images at 24 h (11.7 ± 1.4 and 17.6 ± 0.8 for 89Zr and 64Cu, respectively) was confirmed by the ex vivo measurements (89Zr at 24 h = 51 ± 2% IA g−1, 64Cu at 48 h = 23.5 ± 1.6% IA g−1). The tumor uptake of [64Cu]CuMeCOSar-cetuximab is consistent with other 64Cu cetuximab studies using DOTA.57,58 Potential advantages of using a MeCOSar chelator rather than DOTA include that MeCOSar-cetuximab can be radiolabeled at room temperature and that sarcophagine ligands form CuII complexes that are more stable in vivo than complexes with DOTA (incubation of [67Cu][Cu(sar)]2+ in blood plasma for 174 h revealed <2% of the copper(II) dissociated from the complex).37,59 A previous study with a DFO-N-succinimide conjugated to cetuximab and radiolabeled with 89ZrIV exhibited much lower tumor uptake in A431 xenografts (<8% IA mL−1) than what we observed using DFOSq-cetuximab (up to 51 ± 2% IA g−1), although a different mouse strain was used.51
The lack of kidney uptake in the cetuximab images is unsurprising for these large proteins, which typically undergo hepatic rather than renal clearance. The radioactivity in the liver remained constant over the study period and similar levels were observed for both Zr (27.5 ± 2.3% IA g−1 at 24 h) and Cu (21.3 ± 0.8% IA g−1 at 3 h). Off-target uptake can compromise image quality if tumors are close to organs of high uptake (e.g. liver and kidney), and this has been previously demonstrated for liver metastases when using a Zr-labeled cetuximab in humans.60 [64Cu]Cu-cetuximab also exhibits high liver uptake in mouse models in our study and others.57 Uptake in bone was evident with both conjugates (89Zr at 24 h = 10 ± 1% IA g−1, 64Cu at 48 h = 7.1 ± 0.4% IA g−1). It is often assumed that all bone uptake for zirconium labeled conjugates is due to ‘free’ 89ZrIV but the significant bone uptake following administration of the 64Cu conjugate highlights that other factors can also contribute. Immune compromised mice can give anomalous biodistribution due to Fc-mediated interactions in innate immune effector cells within the bone marrow.56 A limitation of this study is that different immunodeficient mouse strains were used for [89Zr]ZrDFOSq-cetuximab (Balb/c nu/nu) and [64Cu]CuMeCOSar (NSG) and this will produce different off-target distribution, including in the spleen.56 The high spleen uptake of [64Cu]CuMeCOSar-cetuximab in the NSG mice model is similar to a what was reported in a previous study with 89Zr-labeled cetuximab.56
Although an extremely high tumor uptake (51 ± 2% IA g−1) was observed with 89Zr-cetuximab at late timepoints, the requirement for a patient to return for imaging 24 h or more after administration adds complexities with respect to management of patients. With 64Cu-cetuximab, although tumor uptake was observed at very early timepoints (SUVmax = 4.1 at 1 h), optimal tumor:background ratio was obtained at 24 h–48 h post administration. Both [64Cu]CuMeCOSar-cetuximab and [89Zr]ZrDFOSq-cetuximab displayed significant retention in the tumor. The high tumor uptake and retention of [64Cu]CuMeCOSar-cetuximab suggests treatment with a 67Cu variant is worth investigating as a potential agent for radionuclide therapy.
Methods
General
All reagents and solvents were obtained from standard commercial sources and unless otherwise stated were used as received. 1H and 13C spectra were recorded with a Varian FT-NMR 400, Varian FT-NMR 500 or Varian FT-NMR 600 spectrometer (Varian, California USA). 1H NMR spectra were acquired at 400, 500 or 600 MHz and 13C NMR spectra were acquired at 125.7 MHz. All NMR spectra were recorded at 25 °C and all 13C NMR was obtained as proton decoupled 13C NMR, unless otherwise stated. The reported chemical shifts (in parts per million) are referenced relative to residual solvent signal. ESI-MS were recorded on an Agilent 6510 ESI-TOF LC/MS Mass Spectrometer (Agilent, California USA) or a Thermo Scientific Exactive Plus OrbiTrap LC/MS (Thermo Fisher Scientific, Massachusetts, USA) and calibrated to internal references. Protein samples were analysed using an Agilent 6220 ESI-TOF LC/MS mass spectrometer coupled to an Agilent 1200 LC system (Agilent, Palo Alto, CA). All data were acquired and reference mass corrected via a dual-spray electrospray ionisation (ESI) source. Acquisition was performed using the Agilent Mass Hunter Acquisition software version B.02.01 (B2116.30). Protein desalting and chromatographic separation was performed using an Agilent Poroshell C18 2.1 × 75 mm 5 μm column or Phenomenex Jupiter C5 5 5 μm 300 Å 2.1 × 50 mm column using 5% (v/v) acetonitrile ported to waste (0–5 min). Upon desalting of sample the flow was ported back into the ESI source for subsequent gradient elution with (5% (v/v) to 100% (v/v)) acetonitrile/0.1% formic acid over 8 min at 0.25 mL min−1. Analysis was performed using Mass Hunter version B.06.00 with BioConfirm software. Protein concentrations were determined using a Merck Direct Detect® Spectrometer or a Thermo Fisher Scientific NanoDrop instrument (model NanoDrop One). All protein yields were calculated using the volume of solution obtained and the protein concentration of these solutions. 64Cu was supplied as no carrier added [64Cu]CuCl2 in 0.02 M HCl by RAPID Laboratories, Medical Technology and Physics, Sir Charles Gairdner Hospital, Western Australia, or as no carrier added [64Cu]CuCl2 in 0.1 M HCl by Austin Health Molecular Imaging & Therapy, Victoria, Australia. 89Zr was supplied as no carrier added [89Zr]Zr(ox)4 in 1 M oxalic acid by RAPID PET Laboratories, Radiometals Section, Medical Technology & Physics, Sir Charles Gairdner Hospital, Nedlands, WA, Australia, or by PerkinElmer Inc, BV Cyclotron VU, De Boelelaan 1081, HV Amsterdam, The Netherlands. For analysis of radioactive samples, size exclusion HPLC was performed on a Shimadzu SCL-10A VP/LC-10 AT VP system with a Shimadzu SPD-10A VP UV detector followed by a radiation detector (Ortec model 276 photomultiplier base with preamplifier, Ortec 925-SCINT ACE mate preamplifier, BIAS supply and SCA, Bicron 1M 11/2 photomultiplier tube) using a Phenomenex Yarra SEC-3000 3 μm 4.6 × 300 mm column (0.35 mL min−1 flow rate, 0.1 M phosphate buffer pH 6.8 mobile phase). Radio-iTLC were performed using a Raytest Rita-Star TLC scanner and silica gel impregnated glass fiber iTLC plates.Chemical synthesis of H3L1 and L2 are described in the ESI.†
H3DFOSq-cetuximab
Cetuximab (100 μL, 1.8 mg, 12 nmol) was diluted in borate buffer (150 μL, 0.1 M, pH 9.0) and milli-Q H2O (35 μL). H3DFOSqOEt (12.3 μL, 10 mg mL−1 in DMSO, 0.18 μmol) was added and the reaction mixture allowed to stand at ambient temperature for 19 h. The mixture was then filtered using an Amicon 50 kDa MWCO centrifuge filter membrane. The membrane was washed (2× 400 μL 4% DMSO in HEPES buffer 10 mM pH 7.4, then 1× 400 μL HEPES buffer 10 mM pH 7.4), and the resulting protein solution collected to give H3DFOSq-cetuximab (1.058 mg, 59%, ESI Fig. 2†).MeCOSar-cetuximab
Cetuximab (30 μL, 0.4 mg) was diluted in carbonate buffer (30 μL, 0.1 M, pH 9.0). MeCOSarNHS (0.6 μL, 29 mg mL−1 in DMSO, 13 molar equivalents) was added and the reaction mixture allowed to stand at ambient temperature for 0.5 h. The mixture was then filtered using an Amicon 100 kDa MWCO centrifuge filter membrane. The membrane was washed (2× 400 μL 10 mM ammonium acetate buffer pH 6.0), and the resulting protein solution collected to give MeCOSar-cetuximab (166 μg, 42%, ESI Fig. 3†).Recombinant Fab528 fragments
A DNA template encoding a Fab fragment of the mouse IgG2a anti-EGFR monoclonal antibody, 528, codon-optimised for translation in human cells, was synthesised by Genscript (Piscataway, New Jersey) and sub-cloned into the mammalian expression vector, pCAGGS. The encoded Fab528 comprised the complete 528 light chain (LC), fused in-frame with a furin cleavage site, the foot-and-mouth disease virus (FMDV) 2A peptide, and the signal peptide, variable and CH1 constant regions of the 528 heavy chain (HC) (ESI Fig. 4†). A glycine–serine linker (Gly4Ser)2 was introduced between the C-terminus of the CH1 domain and a SrtA recognition sequence (LPETGG), followed by an alanine spacer (Ala3) and either a His6 (HHHHHH) or FLAG (DYKDDDDK) affinity tag. Mammalian expression vectors encoding Fab528-LPETG-His6 and Fab528-LPETG-FLAG were transiently transfected into suspension-adapted cultures of FreestyleTM-293 cells (Thermo Fisher Scientific) using linear polyethyleneimine (PEI, Polysciences, Inc., Warrington, PA) and secreted Fabs purified by immobilized metal ion affinity chromatography (IMAC; Fab528-LPETG-His6) or M2monoclonal antibody anti-FLAG immunoaffinity chromatography (Fab528-LPETG-FLAG) followed by gel filtration.61Recombinant SrtA-His6 and FLAG-SrtA
The expression and purification of His-tagged, recombinant SrtA (SrtA-His6) from E. coli has been described previously.40,42 To enable the expression and purification of a FLAG-tagged version of SrtA, the coding region of SrtA was excised from the SrtA-His6 vector and subcloned in-frame into a pET28a vector modified to include an N-terminal FLAG tag. Expression of SrtA-FLAG was induced using standard protocols and purified by M2 anti-Flag immunoaffinity chromatography.H3L1-Fab528
Fab528-LPETG-His6 (300 μg, 5.99 nmol) was diluted in SrtA reaction buffer (137 μL, for a final concentration of 50 mM tris, 150 mM NaCl, 10 mM CaCl2, pH 7.5), followed by the addition of H3L1 (50 mM in DMSO, 11.5 μL, 575 nmol). SrtA-His6 (327 μg, 17.3 nmol) was then added. The reaction mixture was incubated for 18.5 h at 37 °C with gentle shaking (250 rpm), then quenched by cooling to 4 °C. The mixture was then incubated with Qiagen NiNTA resin (75 μL) in PBS (50 mM, 0.7 mL) at 4 °C with shaking for 1 h. The supernatant was collected and filtered on an Amicon 10 kDa MWCO centrifuge filter, and washed on the filter (2× 400 μL HEPES buffer, 10 mM, pH 7.4) to give H3L1-Fab528 (224 μg, 75%, ESI Fig. 5†).L2-Fab528
Fab528LPETG-FLAG (300 μg) was diluted in SrtA reaction buffer (144 μL, for a final concentration of 50 mM tris, 150 mM NaCl, 10 mM CaCl2, pH 7.5), followed by the addition of L2 (50 mM in DMSO, 18 μL). SrtA-FLAG (500 μg) was then added. The reaction mixture was incubated overnight at 37 °C with gentle shaking (500 rpm), then quenched by cooling to 4 °C. The mixture was then incubated with anti-DYKDDDDK affinity resin in binding buffer (PBS pH 7.4, 1 mL) at 4 °C with shaking for 3 h. The supernatant was collected and filtered on an Amicon 10 kDa MWCO centrifuge filter and washed on the filter (2× 400 μL ammonium acetate buffer, 100 mM, pH 6) to give L2-Fab528 (269 μg, 90%, ESI Fig. 6†).[89Zr]ZrDFOSq-cetuximab
A solution of 89Zr in 1 M oxalic acid (140 μL, 100 MBq) was diluted with milli-Q H2O (140 μL), then neutralised through sequential addition of small aliquots of Na2CO3 (total 113 μL, 1 M) to pH 7. HEPES buffer (130 μL, 1 M, pH 7.4) was then added. H3DFOSq-cetuximab (9.5 μL, 100 μg) was added to the buffered 89Zr solution and left to stand at ambient temperature for 75 min. The reaction mixture was then purified using a PD-10 column conditioned with PBS and 0.5% sodium gentisate. Four 500 μL fractions were collected, and the fraction with the highest activity (28.9 MBq) selected. Radiochemical purity was confirmed by radio-SEHPLC, the remaining fraction was diluted further (600 μL of PBS/gentisate) and six doses (2.9 MBq in 120 μL each) were drawn up into 0.3 mL insulin syringes. Each dose was administered via tail vein injection into A431 xenograft tumor-bearing nude mice.[89Zr]ZrL1-Fab528
A solution of 89ZrIV in 1 M oxalic acid (141 μL, 160 MBq) was diluted with milli-Q H2O (141 μL), then neutralised through sequential addition of small aliquots of Na2CO3 (total 118 μL, 1 M) to pH 7. HEPES buffer (200 μL, 1 M, pH 7.4) was then added. H3L1-Fab528 (50 μg) was added to a portion of the buffered 89ZrIV solution (125 μL, 20 MBq) and left to stand at ambient temperature for 45 min. Radio-SEHPLC indicated approximately 70% labeling, and so a further portion of 89ZrIV (375 μL, 60 MBq) was added with a further 20 μg of H3L1-Fab528. After a further 40 min, the reaction mixture was purified using a PD-10 column conditioned with PBS and 0.5% sodium gentisate. Four 500 μL fractions were collected, and the fraction with the highest activity (27.2 MBq) selected. Radiochemical purity was confirmed by radio-SEHPLC, the remaining fraction was diluted further (1.02 mL of PBS/gentisate) and six doses (2.2 MBq, 3 μg 150 μL each) were drawn up into 0.3 mL insulin syringes. Each dose was administered via tail vein injection into A431 xenograft tumor-bearing NSG mice.[64Cu]CuMeCOSar-cetuximab
A solution of [64Cu]CuCl2 in 0.1 M HCl (24 μL, 40 MBq) was buffered to pH 6 with ammonium acetate (48 μL, 0.1 M). MeCOSar-cetuximab (92 μL, 152 μg) was added and the mixture left to stand at ambient temperature for 30 min. The reaction mixture was then purified using a PD-10 column conditioned with PBS. Radiochemical purity was confirmed by radio-iTLC, and the sample was diluted further with PBS and six doses (2.1 MBq, 19 μg, 175 μL each) were drawn up into 0.3 mL insulin syringes. Each dose was administered via tail vein injection into A431 xenograft tumor-bearing NSG mice.[64Cu]CuL2-Fab528
A solution of [64Cu]CuCl2 in 0.02 M HCl (17 μL, 33 MBq) was buffered to pH 7 with PBS (100 μL). L2-Fab528 (148 μL, 188 μg) was added and the mixture left to stand at ambient temperature for 1 h. Radiochemical purity was confirmed by radio-SEHPLC and the reaction mixture was used without further purification. The mixture was further diluted with PBS and six doses of [64Cu]CuL2-Fab528 (2.3 MBq, 13 μg, 200 μL each) were drawn up into 0.3 mL insulin syringes. Each dose was administered via tail vein injection into A431 xenograft tumor-bearing NSG mice.Mice imaging
All animal experiments were performed with the approval of the Peter MacCallum Cancer Centre Animal Experimentation Ethics Committee and in accordance with the Australian code for the care and use of animals for scientific purposes, 8th Edition, 2013. Balb/c nude and NSG mice were sourced from Animal BioResources, (Moss Vale, New South Wales). 3 × 106 A431 cells (American Type Culture Collection) were implanted subcutaneously onto the right flank of the mice and once tumors were well established, the animals were used for imaging and biodistribution studies. For PET-CT imaging, animals were anaesthetised with 2.5% isoflurane and placed in the imaging chamber of a PerkinElmer/Sofie Biosciences small animal G8 PET/CT. A CT scan was performed followed by a 10 min PET acquisition. Images were acquired using the G8 acquisition engine software and reconstructed using a 3D ML-EM algorithm. Quantification was performed using VivoQuant software, Version 3.0 (inviCRO Imaging Services and Software). After imaging the final time point mice were euthanized and tissues excised, weighed and counted using a Capintec (Captus 4000e) gamma counter. Separate cohorts of mice were harvested for earlier biodistribution time points.Conclusions
The effective use of EGFR targeting therapies relies on personalised medicine where diagnostic molecular imaging can provide relatively quick selection of suitable patients as well as rapid assessment of clinical responses. The antibody fragments used in this study were genetically modified to incorporate a SrtA recognition sequence to allow enzyme mediated site selective incorporation of either copper-64 or zirconium-89. For each radionuclide the respective chelators were also modified by the inclusion of a N-terminal glycine, so the chelator is also able to act as substrate for SrtA. The use of SrtA to attach the chelator has the added benefit of potentially simplifying regulatory approval for clinical use of these tracers, as the mixtures obtained with non-specific attachment methods can be challenging to reproduce and characterise. The Fab528 conjugates are potentially easier to manufacture reproducibly in terms of chelator-to-antibody ratio.Both [64Cu][CuL2]-Fab528 and [89Zr][ZrL1]-Fab528 were able to give high quality PET images with high tumor uptake when imaging 3 hours post-injection in A431 EGFR-positive tumors. The shorter time between injection and imaging represents a potential advantage of the smaller Fab528 agents when compared to the radiolabeled full antibody agents that require several days between injection and imaging. The high kidney uptake and retention using the Fab528 conjugates may be an acceptable trade-off for the liver uptake in the full IgG conjugates in terms of radiation dose, but this remains to be confirmed. The shorter radioactive half-life of copper-64 is perhaps better suited to imaging within this relatively short time frame than zirconium-89. Another potential advantage of using copper-64 based imaging agents is that they can be used to predict dosimetry for the same conjugate labeled with copper-67, a potentially therapeutic β−-emitting radionuclide (t1/2 = 62 h).49,62 The ability to obtain suitable images in 1–3 hours post injection suggest diagnostic imaging with 68Ga, a positron-emitting radionuclide that is available from a generator and has a radioactive half-life of 68 minutes, could be feasible. The DFOSq chelator can also be used to coordinate to 68GaIII.63 The major advantage of the Fab528 conjugates presented is that high-quality PET images can be acquired three hours after administration and this could translate to same day imaging in humans improving the feasibility of diagnostic EGFR PET imaging.
Author contributions
S. E. Rudd conducted the chemical synthesis, enzyme-mediated bioconjugations, chemical characterisations and radiolabelling and wrote the initial manuscript draft. J. K. Van Zuylekom conducted the mouse experiments and PET data analysis. A. Raicevic and L. A. Pearce expressed the 528 protein and Sortase enzyme. C. Cullinane, C. C. Williams, T. E. Adams, R. J. Hicks and P. S. Donnelly conceptualized the research, directed the project and finalized the manuscript. All authors contributed discussions and manuscript editing.Conflicts of interest
Stacey E. Rudd and Paul S. Donnelly are inventors on intellectual property relating to the use of DFOSq that have been licenced from the University of Melbourne to Telix Pharmaceuticals. Paul S. Donnelly is an inventor of intellectual property relating to this area of research that has been licensed from the University of Melbourne to Clarity Pharmaceuticals. Paul S. Donnelly serves on the Scientific Advisory board and has a financial interest in Clarity Pharmaceuticals. Rodney J. Hicks has shares in Telix Pharmaceuticals.Acknowledgements
Funding from the Australian Research Council and the National Health and Medical Research Council. The G8 PET/CT scanner was purchased with the support of a grant from the Australian Cancer Research Foundation. We thank the Mass Spectrometry and Proteomics Facility at Bio21 Institute, University of Melbourne. We also thank Professor Roger I. Price, Dr Ali Asad and Thien Dinh for provision of 89Zr/64Cu. Susan Jackson and Rachael Walker for expert technical assistance with mouse models, Stewart Nuttall for pET28a-FLAG vector, Stephan Eckert and Laura Castelli for help with vector cloning, the Fermentation Team (National Biologics Facility) for scale-up cell culture and transfections, Jo Caine for help with protein purification, and Dr Mark Richardson for assistance with figures. The National Biologics Facility is supported by Therapeutic Innovation Australia through the National Collaborative Research Infrastructure Strategy.References
- Y. Yarden and M. X. Sliwkowski, Nat. Rev. Mol. Cell Biol., 2001, 2, 127–137 Search PubMed.
- R. Pirker, Curr. Opin. Oncol., 2015, 27, 87–93 Search PubMed.
- R. S. Herbst, M. W. Redman, E. S. Kim, T. J. Semrad, L. Bazhenova, G. Masters, K. Oettel, P. Guaglianone, C. Reynolds, A. Karnad, S. M. Arnold, M. Varella-Garcia, J. Moon, P. C. Mack, C. D. Blanke, F. R. Hirsch, K. Kelly and D. R. Gandara, Lancet Oncol., 2018, 19, 101–114 Search PubMed.
- T. Mizukami, N. Izawa, T. E. Nakajima and Y. Sunakawa, Drugs, 2019, 79, 633–645 Search PubMed.
- F. Penault-Llorca, A. Cayre, L. Arnould, F. Bibeau, M. Bralet, P. Rochaix, J. Savary and J. Sabourin, Oncol. Rep., 2006, 16, 1173–1179 Search PubMed.
- M. Francoual, M. C. Etienne-Grimaldi, J. L. Formento, D. Benchimol, A. Bourgeon, M. Chazal, C. Letoublon, T. André, N. Gilly, J. R. Delpero, P. Lasser, J. P. Spano and G. Milano, Annals of Oncology, 2006, 17, 962–967 Search PubMed.
- P. M. R. Pereira, L. Abma, K. E. Henry and J. S. Lewis, Cancer Lett., 2018, 419, 139–151 Search PubMed.
- D. Zeng, Y. Guo, A. G. White, Z. Cai, J. Modi, R. Ferdani and C. J. Anderson, Mol. Pharm., 2014, 11, 3980–3987 Search PubMed.
- S. M. Larson, J. A. Carrasquillo, K. A. Krohn, J. P. Brown, R. W. McGuffin, J. M. Ferens, M. M. Graham, L. D. Hill, P. L. Beaumier and K. E. Hellström, J. Clin. Invest., 1983, 72, 2101–2114 Search PubMed.
- N. S. Turker, P. Heidari, R. Kucherlapati, M. Kucherlapati and U. Mahmood, Theranostics, 2014, 4, 893–903 Search PubMed.
- T. Olafsen and A. M. Wu, Semin. Nucl. Med., 2010, 40, 167–181 Search PubMed.
- J. D. Sato, T. Kawamoto, A. D. Le, J. Mendelsohn, J. Polikoff and G. H. Sato, Mol. Biol. Med., 1983, 1, 511–529 Search PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 Search PubMed.
- J. N. Tinianow, H. S. Gill, A. Ogasawara, J. E. Flores, A. N. Vanderbilt, E. Luis, R. Vandlen, M. Darwish, J. R. Junutula, S.-P. Williams and J. Marik, Nucl. Med. Biol., 2010, 37, 289–297 Search PubMed.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, Nucl. Med. Biol., 2013, 40, 3–14 Search PubMed.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, G. F. Filzen, C. J. O'Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 Search PubMed.
- P. Dennler, A. Chiotellis, E. Fischer, D. Brégeon, C. Belmant, L. Gauthier, F. Lhospice, F. Romagne and R. Schibli, Bioconjugate Chem., 2014, 25, 569–578 Search PubMed.
- F. Lhospice, D. Brégeon, C. Belmant, P. Dennler, A. Chiotellis, E. Fischer, L. Gauthier, A. Boëdec, H. Rispaud, S. Savard-Chambard, A. Represa, N. Schneider, C. Paturel, M. Sapet, C. Delcambre, S. Ingoure, N. Viaud, C. Bonnafous, R. Schibli and F. Romagné, Mol. Pharm., 2015, 12, 1863–1871 Search PubMed.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 Search PubMed.
- B. M. Zeglis, C. B. Davis, D. Abdel-Atti, S. D. Carlin, A. Chen, R. Aggeler, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2014, 25, 2123–2128 Search PubMed.
- P. Adumeau, D. Vivier, S. K. Sharma, J. Wang, T. Zhang, A. Chen, B. J. Agnew and B. M. Zeglis, Mol. Pharm., 2018, 15, 892–898 Search PubMed.
- B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2002, 277, 20833–20839 Search PubMed.
- B. Ramakrishnan, E. Boeggeman, M. Manzoni, Z. Zhu, K. Loomis, A. Puri, D. S. Dimitrov and P. K. Qasba, Bioconjugate Chem., 2009, 20, 1383–1389 Search PubMed.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 Search PubMed.
- B. M. Paterson, K. Alt, C. M. Jeffery, R. I. Price, S. Jagdale, S. Rigby, C. C. Williams, K. Peter, C. E. Hagemeyer and P. S. Donnelly, Angew. Chem., Int. Ed., 2014, 53, 6115–6119 Search PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760 Search PubMed.
- H. Ton-That, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424 Search PubMed.
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. L. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 Search PubMed.
- M. W. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707 Search PubMed.
- M. W.-L. Popp and H. L. Ploegh, Angew. Chem., Int. Ed., 2011, 50, 5024–5032 Search PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 Search PubMed.
- M. Rashidian, E. J. Keliher, A. M. Bilate, J. N. Duarte, G. R. Wojtkiewicz, J. T. Jacobsen, J. Cragnolini, L. K. Swee, G. D. Victora, R. Weissleder and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6146–6151 Search PubMed.
- M. Rashidian, L. Wang, J. G. Edens, J. T. Jacobsen, I. Hossain, Q. Wang, G. D. Victora, N. Vasdev, H. Ploegh and S. H. Liang, Angew. Chem., Int. Ed., 2016, 55, 528–533 Search PubMed.
- M. Rashidian, J. R. Ingram, M. Dougan, A. Dongre, K. A. Whang, C. LeGall, J. J. Cragnolini, B. Bierie, M. Gostissa, J. Gorman, G. M. Grotenbreg, A. Bhan, R. A. Weinberg and H. L. Ploegh, J. Exp. Med., 2017, 214, 2243–2255 Search PubMed.
- S. E. Rudd, P. Roselt, C. Cullinane, R. J. Hicks and P. S. Donnelly, Chem. Commun., 2016, 52, 11889–11892 Search PubMed.
- C. B. Jacobsen, R. Raave, M. O. Pedersen, P. Adumeau, M. Moreau, I. E. Valverde, I. Bjoernsdottir, J. B. Kristensen, M. F. Grove, K. Raun, J. McGuire, V. Goncalves, S. Heskamp, F. Denat and M. Gustafsson, Nucl. Med. Biol., 2020, 82–83, 49–56 Search PubMed.
- N. M. Di Bartolo, A. M. Sargeson, T. M. Donlevy and S. V. Smith, J. Chem. Soc., Dalton Trans., 2001, 2303–2309 Search PubMed.
- S. D. Voss, S. V. Smith, N. DiBartolo, L. J. McIntosh, E. M. Cyr, A. A. Bonab, J. L. J. Dearling, E. A. Carter, A. J. Fischman, S. T. Treves, S. D. Gillies, A. M. Sargeson, J. S. Huston and A. B. Packard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17489–17493 Search PubMed.
- K. Alt, B. M. Paterson, K. Ardipradja, C. Schieber, G. Buncic, B. Lim, S. S. Poniger, B. Jakoby, X. Wang, G. J. O'Keefe, H. J. Tochon-Danguy, A. M. Scott, U. Ackermann, K. Peter, P. S. Donnelly and C. E. Hagemeyer, Mol. Pharm., 2014, 11, 2855–2863 Search PubMed.
- B. M. Paterson, P. Roselt, D. Denoyer, C. Cullinane, D. Binns, W. Noonan, C. M. Jeffery, R. I. Price, J. M. White, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2014, 43, 1386–1396 Search PubMed.
- B. M. Paterson, G. Buncic, L. E. McInnes, P. Roselt, C. Cullinane, D. S. Binns, C. M. Jeffery, R. I. Price, R. J. Hicks and P. S. Donnelly, Dalton Trans., 2015, 44, 4901–4909 Search PubMed.
- M. P. Madej, G. Coia, C. C. Williams, J. M. Caine, L. A. Pearce, R. Attwood, N. A. Bartone, O. Dolezal, R. M. Nisbet, S. D. Nuttall and T. E. Adams, Biotechnol. Bioeng., 2012, 109, 1461–1470 Search PubMed.
- T. M. Behr, D. M. Goldenberg and W. Becker, Eur. J. Nucl. Med., 1998, 25, 201–212 Search PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 Search PubMed.
- L. E. McInnes, S. E. Rudd and P. S. Donnelly, Coord. Chem. Rev., 2017, 352, 499–516 Search PubMed.
- C. J. Anderson, J. M. Connett, S. W. Schwarz, P. A. Rocque, L. W. Guo, G. W. Philpott, K. R. Zinn, C. F. Meares and M. J. Welch, J. Nucl. Med., 1992, 33, 1685–1690 Search PubMed.
- S. J. DeNardo, G. L. DeNardo, D. L. Kukis, S. Shen, L. A. Kroger, D. A. DeNardo, D. S. Goldstein, G. R. Mirick, Q. Salako, L. F. Mausner, S. C. Srivastava and C. F. Meares, J. Nucl. Med., 1999, 40, 302–310 Search PubMed.
- P. J. Blower, J. Chem. Soc., Dalton Trans., 2015, 44, 4819–4844 Search PubMed.
- C. Cullinane, C. M. Jeffery, P. D. Roselt, E. M. van Dam, S. Jackson, K. Kuan, P. Jackson, D. Binns, J. van Zuylekom, M. J. Harris, R. J. Hicks and P. S. Donnelly, J. Nucl. Med., 2020, 61, 1800–1805 Search PubMed.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 Search PubMed.
- H. J. Aerts, L. Dubois, L. Perk, P. Vermaelen, G. A. van Dongen, B. G. Wouters and P. Lambin, J. Nucl. Med., 2009, 50, 123–131 Search PubMed.
- R. Chakravarty, S. Goel, H. F. Valdovinos, R. Hernandez, H. Hong, R. J. Nickles and W. Cai, Bioconjugate Chem., 2014, 25, 2197–2204 Search PubMed.
- L. K. van Dijk, C.-B. Yim, G. M. Franssen, J. H. A. M. Kaanders, J. Rajander, O. Solin, T. J. Groenroos, O. C. Boerman and J. Bussink, Contrast Media Mol. Imaging, 2016, 11, 65–70 Search PubMed.
- R. J. Hicks, P. Jackson, G. Kong, R. E. Ware, M. S. Hofman, D. A. Pattison, T. A. Akhurst, E. Drummond, P. Roselt, J. Callahan, R. Price, C. M. Jeffery, E. Hong, W. Noonan, A. Herschtal, L. J. Hicks, A. Hedt, M. Harris, B. M. Paterson and P. S. Donnelly, J. Nucl. Med., 2019, 60, 777–785 Search PubMed.
- R. M. Reilly, R. Kiarash, J. Sandhu, Y. W. Lee, R. G. Cameron, A. Hendler, K. Vallis and J. Gariépy, J. Nucl. Med., 2000, 41, 903 Search PubMed.
- S. K. Sharma, A. Chow, S. Monette, D. Vivier, J. Pourat, K. J. Edwards, T. R. Dilling, D. Abdel-Atti, B. M. Zeglis, J. T. Poirier and J. S. Lewis, Cancer Res., 2018, 78, 1820–1832 Search PubMed.
- W. Cai, K. Chen, L. He, Q. Cao, A. Koong and X. Chen, Eur. J. Nucl. Med. Mol. Imaging, 2007, 34, 850–858 Search PubMed.
- L. A. M. Wen Ping Li, D. A. Capretto, C. D. Sherman and a. C. J. Anderson, Cancer Biother.Radiopharm., 2008, 23, 158–171 Search PubMed.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 Search PubMed.
- C. W. Menke, E. C. Gootjes, M. C. Huisman, N. C. T. v. Grieken, D. J. Vugts, C. Roth, E. R. Mulder, R. C. Schuit, R. Boellaard, O. S. Hoekstra, G. A. M. S. v. Dongen and H. M. W. Verheul, J. Clin. Oncol., 2014, 32, 11102 Search PubMed.
- T. C. Elleman, T. Domagala, N. M. McKern, M. Nerrie, B. Lönnqvist, T. E. Adams, J. Lewis, G. O. Lovrecz, P. A. Hoyne, K. M. Richards, G. J. Howlett, J. Rothacker, R. N. Jorissen, M. Lou, T. P. J. Garrett, A. W. Burgess, E. C. Nice and C. W. Ward, Biochemistry, 2001, 40, 8930–8939 Search PubMed.
- O. Keinänen, K. Fung, J. M. Brennan, N. Zia, M. Harris, E. van Dam, C. Biggin, A. Hedt, J. Stoner, P. S. Donnelly, J. S. Lewis and B. M. Zeglis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28316–28327 Search PubMed.
- A. Noor, J. K. Van Zuylekom, S. E. Rudd, K. Waldeck, P. D. Roselt, M. B. Haskali, M. P. Wheatcroft, E. Yan, R. J. Hicks, C. Cullinane and P. S. Donnelly, J. Med. Biochem., 2020, 63, 9258–9270 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical synthesis of H3L1 and L2, supplementary Fig. 1–6. See DOI: 10.1039/d1sc01422f |
| This journal is © The Royal Society of Chemistry 2021 |