
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell–cell interactions via non-covalent click chemistry†
Chad
Plumet
a,
Achmet Said
Mohamed
a,
Tanguy
Vendeuvre
b,
Brigitte
Renoux
a,
Jonathan
Clarhaut
ab and
Sébastien
Papot
 *a
*a
aUniversity of Poitiers, UMR CNRS 7285, Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP), rue Michel-Brunet, TSA 51106, 86073 Poitiers Cedex 9, France. E-mail: sebastien.papot@univ-poitiers.fr
bCHU de Poitiers, 2 rue de la Miléterie, CS 90577, Poitiers, F-86021, France
First published on 7th June 2021
Abstract
Metabolic glycoengineering with unnatural sugars became a valuable tool for introducing recognition markers on the cell membranes via bioorthogonal chemistry. By using this strategy, we functionalized the surface of tumor and T cells using complementary artificial markers based on both β-cyclodextrins (β-CDs) and adamantyl trimers, respectively. Once tied on cell surfaces, the artificial markers induced cell–cell adhesion through non-covalent click chemistry. These unnatural interactions between A459 lung tumor cells and Jurkat T cells triggered the activation of natural killer (NK) cells thanks to the increased production of interleukin-2 (IL-2) in the vicinity of cancer cells, leading ultimately to their cytolysis. The ready-to-use surface markers designed in this study can be easily inserted on the membrane of a wide range of cells previously submitted to metabolic glycoengineering, thereby offering a simple way to investigate and manipulate intercellular interactions.
Introduction
In nature, intercellular recognition is typically achieved by means of complementary adhesion molecules present on the surface of each cell partner. During the last decade, metabolic glycoengineering, in which unnatural monosaccharides bearing bioorthogonal functional groups are metabolically incorporated into cell-surface glycans,1–3 emerged as a powerful strategy for manipulating cellular interactions.4 As early as 2009, Gartner and Bertozzi were the first to use this approach for the construction of 3-dimensional micro-tissues, by conjugating single stranded DNA nucleotides at the membrane of azido-labelled Jurkat T cells.5 Cells bearing complementary DNA sequences led to the formation of aggregates with well-defined interconnectivities. More recently, Iwasaki and co-workers used the thiol–ene reaction to introduce aptamers on the surface of methacryloyl-functionalized macrophages, enabling them to recognize human T lymphoblasts in vitro.6 The manipulation of cell–cell interactions was also investigated by forming covalent bonds between cells through bioorthogonal ligation reactions.7–10 Within this framework, the strain-promoted azide–alkyne cycloaddition (SPAAC) reaction was employed by Ma and Cai to bind tumor cells and T cells.9 Intercellular covalent bonding triggered an increased cytotoxicity of T cells for tumor cells, highlighting the potential of this approach for the development of novel cancer immunotherapy. In 2016, Qu and co-workers reported an elegant strategy enabling the manipulation of cellular adhesion via photo-responsive host–guest recognition.11 By using a photoisomerizable azobenzene dimer as a reversible cell binder, they were able to control the assembly of tumor cells, previously functionalized on the surface by β-cyclodextrins (β-CD) via copper(I)-catalysed azide–alkyne cycloaddition (CuAAC). Herein, we present the development of complementary cell surface markers allowing unnatural cell–cell adhesion through non-covalent click chemistry (Fig. 1).12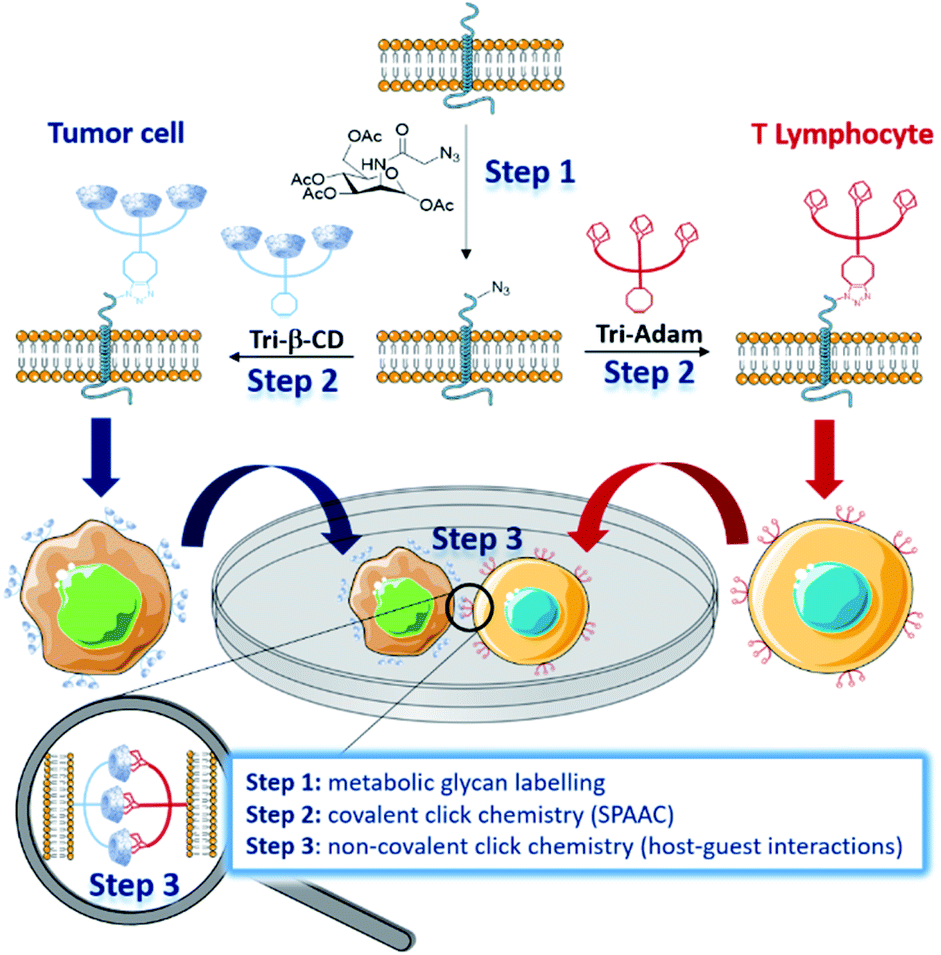 | ||
| Fig. 1 Principle of cell surface engineering with complementary artificial recognition markers based on host–guest pairs. Step 1: metabolic glycan labelling with Ac4ManNAz resulting in azide tag incorporation at cell surfaces. Step 2: introduction of the complementary artificial markers Tri-β-CD and Tri-Adam on the cell membranes via the bioorthogonal SPAAC ligation reaction. Step 3: cell–cell adhesion through non-covalent click chemistry. | ||
We designed β-CD and adamantyl trimers (Tri-β-CD and Tri-Adam, respectively) bearing a dibenzocyclooctyne (DBCO) that can be easily attached on the surface of azido-labelled cells by the SPAAC reaction. When installed on the membrane of cells that do not recognize naturally, these artificial recognition markers promoted cell–cell adhesion through host–guest interactions.
As proof of principle, we coated tumor cells and T lymphocytes with Tri-β-CD and Tri-Adam respectively, and we demonstrated that their forced interaction activated natural killer (NK) cells to kill tumor cells. This strategy, that combines metabolic glycan labelling with both covalent and non-covalent click chemistry, provides a simple way to manipulate cell–cell interactions, hence facilitating the study of resulting biological processes.
Results and discussion
The two artificial cell surface markers Tri-β-CD and Tri-Adam were constructed from the same molecular platform 1.13 Based on a gallic acid core, the latter one enables indeed the attachment of both a DBCO and three copies of the recognition unit in only a few synthetic steps (Scheme 1). Thus, the adamantyl moieties were first introduced on the primary amines of 1 by nucleophilic substitution using the N-hydroxysuccinimide ester 2. Cleavage of the tert-butoxycarbonyl protecting group (Boc) followed by coupling with the activated ester of DBCO 3 afforded Tri-Adam (56% yield over three steps). On the other hand, Tri-β-CD was prepared via a four-step strategy from compound 1. The reaction between 1 and the N-hydroxysuccinimide ester 4 allowed the introduction of three terminal alkynes on the molecular platform. After removal of the Boc protecting group, the platform was then functionalized with three equivalents of azido-β-CD 5 through the CuAAC reaction. Finally, the DBCO moiety was coupled with the remaining primary amine using the precursor 3, thereby providing Tri-β-CD with an overall yield of 41%.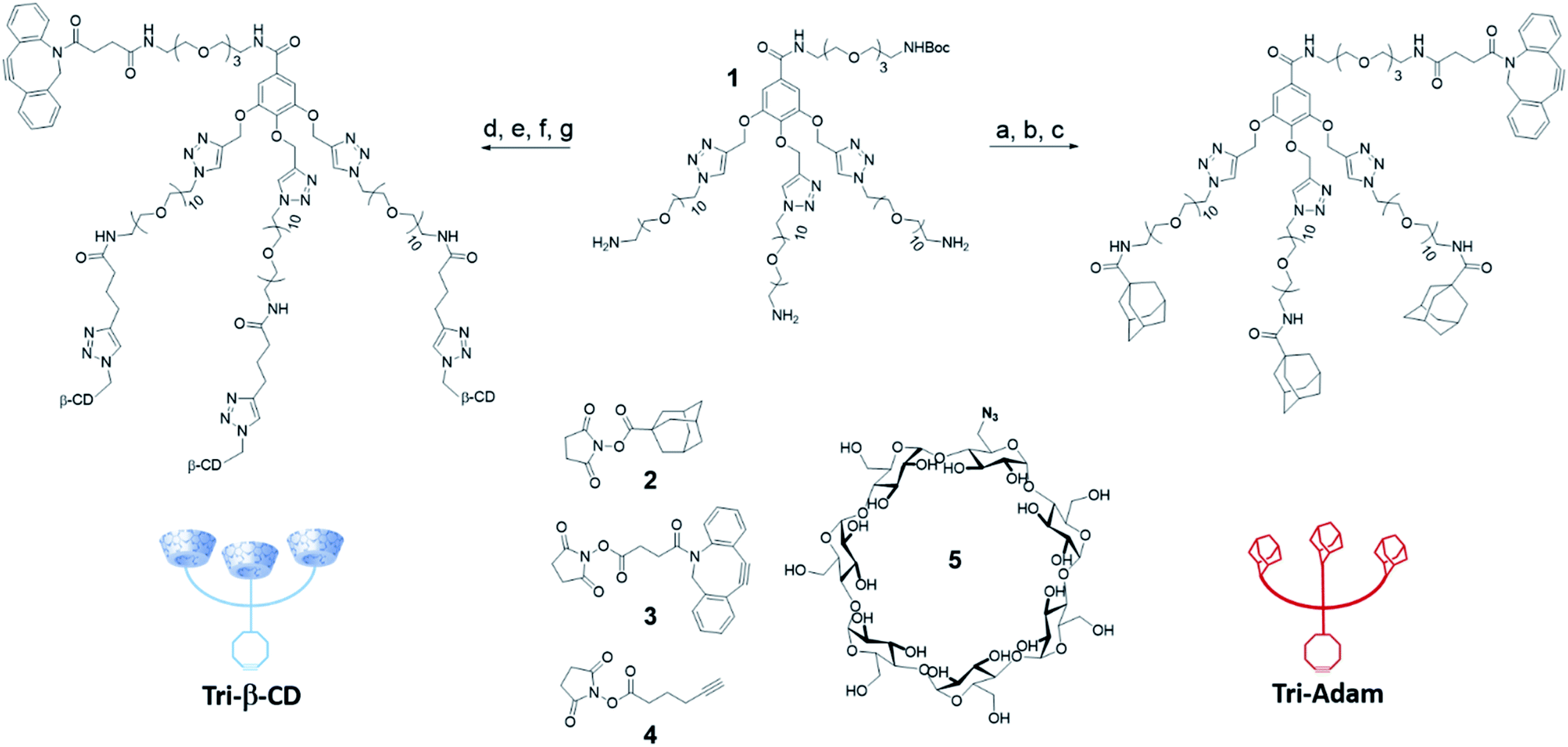 | ||
| Scheme 1 Synthesis of the artificial cell surface markers Tri-β-CD and Tri-Adam. (a) 2, Et3N, DMF, r.t., 12 h, 78%; (b) TFA/CH2Cl2 (20/80), 0 °C then r.t., 1 h, 95%; (c) 3, DMF, r.t., 12 h, 76%; (d) 4, Et3N, DMSO, r.t., 12 h, 77%; (e) TFA/CH2Cl2 (20/80), 0 °C then r.t., 3 h, 97%; (f) 5, Cu(MeCN)4PF6, tris-(3-hydroxypropyltriazolylmethyl)amine (THPTA), DMSO, r.t., 4 h, 66%; (g) 3, Et3N, DMSO, r.t., 2 h, 84%. | ||
We next investigated the ability of the complementary host/guest pair Tri-β-CD/Tri-Adam to trigger cell–cell adhesion. For this purpose, A549 human cancer cells and human Jurkat T lymphocytes were first treated for three days with tetraacetylated N-azidoacetyl-D-mannosamine (Ac4ManNAz) in order to install azides within cell surface glycoconjugates (see the ESI†). A549 and Jurkat cells were then incubated for thirty minutes with Tri-β-CD and Tri-Adam, respectively, for binding the artificial surface markers on the cell membrane via the SPAAC reaction. Such a procedure of cell surface engineering did not affect the viability of cells for at least forty eight hours post-functionalization. Furthermore, it is worth mentioning that with the trimeric structure of our artificial markers, each click reaction permits the introduction of three recognition units (host/guest), hence multiplying the potential interactions between complementary cells.
Once modified as described above, A549 (green) and Jurkat (red) cells were incubated together in order to analyze the effect of the artificial markers Tri-β-CD and Tri-Adam on cell recognition (Fig. 2). Thus, Jurkat T cells were seeded on A549 adherent cells (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Jurkat:A549 ratio) for ten minutes, then the supernatant was removed. The adherent cells were next washed and fixed prior to monitoring intercellular interactions by 3D confocal microscopy. As shown in Fig. 2a, Jurkat T cells remained bound to A549 cells when both cell lines were previously submitted to metabolic labelling with Ac4ManNAz followed by the bioorthogonal introduction of either Tri-Adam or Tri-β-CD on the surface glycans. In contrast, when Jurkat cells were not modified beforehand by the full procedure of cell engineering, they did not adhere to A549 cells, as testified by the absence of red fluorescence. 3D imaging analysis confirmed the establishment of contacts between modified cells and showed that each A549 tumor cell interacted with several Jurkat cells (Fig. 2b).
:1 Jurkat:A549 ratio) for ten minutes, then the supernatant was removed. The adherent cells were next washed and fixed prior to monitoring intercellular interactions by 3D confocal microscopy. As shown in Fig. 2a, Jurkat T cells remained bound to A549 cells when both cell lines were previously submitted to metabolic labelling with Ac4ManNAz followed by the bioorthogonal introduction of either Tri-Adam or Tri-β-CD on the surface glycans. In contrast, when Jurkat cells were not modified beforehand by the full procedure of cell engineering, they did not adhere to A549 cells, as testified by the absence of red fluorescence. 3D imaging analysis confirmed the establishment of contacts between modified cells and showed that each A549 tumor cell interacted with several Jurkat cells (Fig. 2b).
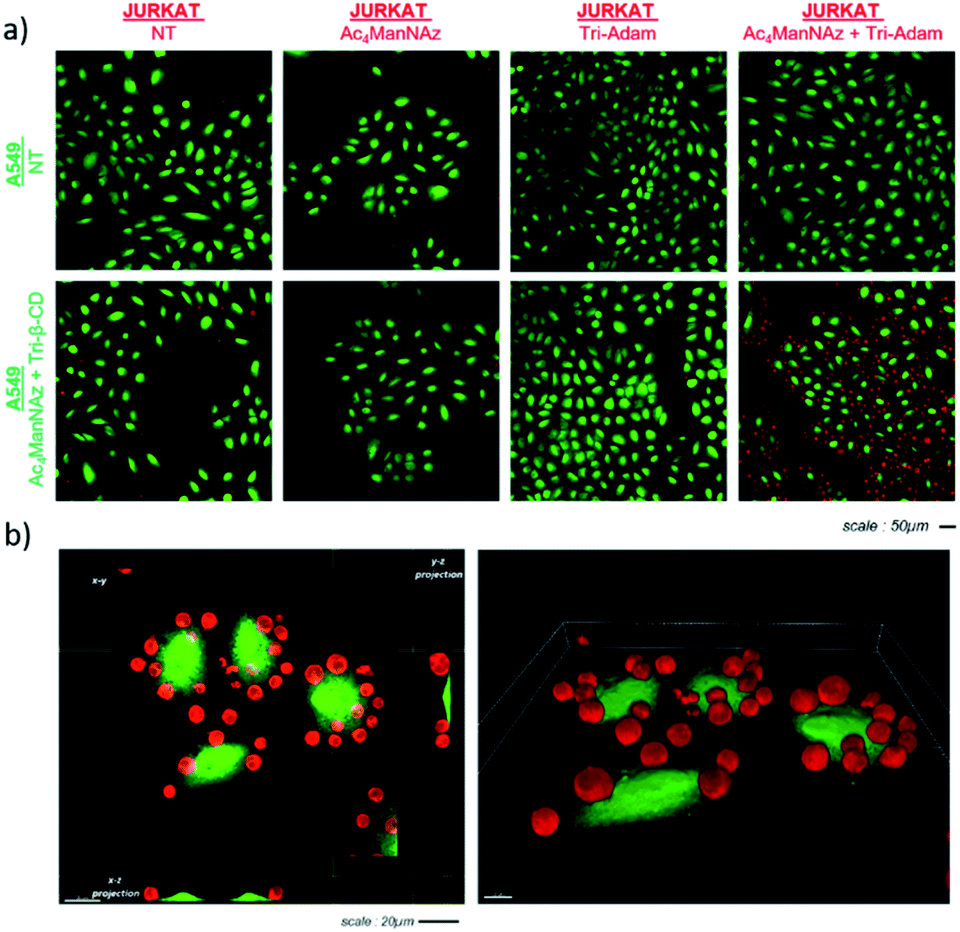 | ||
| Fig. 2 Confocal microscopy imaging of cellular recognition. (a) A549 adherent tumor cells (green) and Jurkat T cells (red) (1:5 ratio) were incubated together for 10 minutes and washed with PBS prior to imaging; (b) 3D imaging of cell–cell interactions. | ||
The interactions between both modified A549 and Jurkat T cells were also investigated by electron microscopy (Fig. 3). These experiments confirmed the adhesion of Jurkat T cells on the surface of A549 tumor cells (Fig. 3c). Interestingly, the images suggested that interactions between the two cell types induced the remodeling of the actin cytoskeleton with the formation of filaments14 (Fig. 3d).
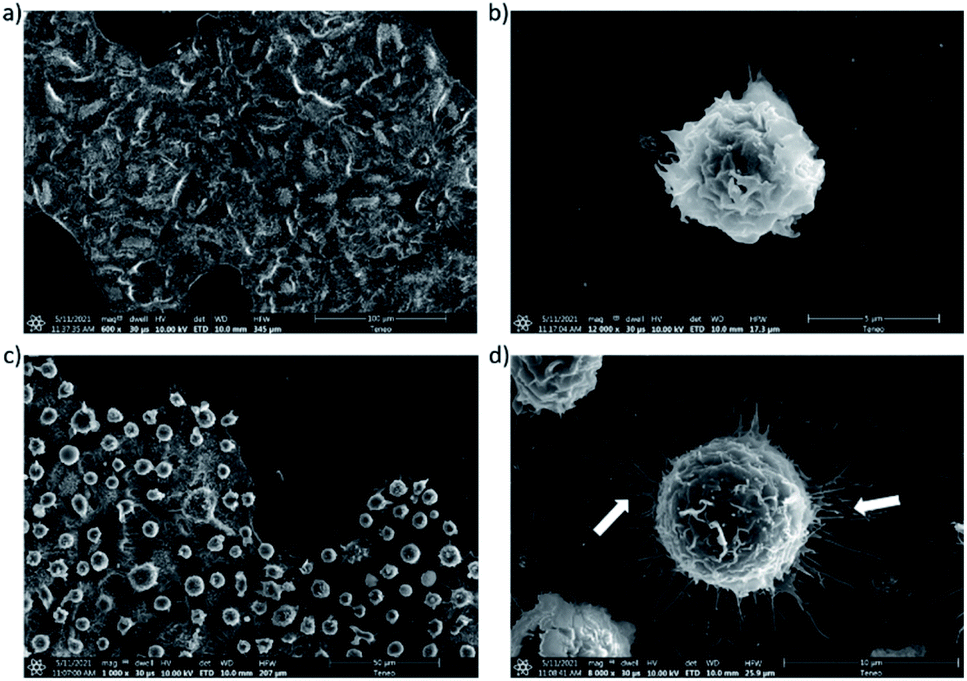 | ||
| Fig. 3 Electron microscopy imaging of cellular recognition. (a) Imaging of A549 cells bearing Tri-β-CD artificial surface markers; (b) imaging of Jurkat T cells functionalized with Tri-Adam markers; (c) and (d) imaging of Jurkat T cell on the surface of A549 cells at two different magnifications, showing the appearance of filaments (arrows). | ||
Overall, these results demonstrate that the functionalization of the cell surface with complementary artificial recognition markers, based on host–guest pairs (e.g. cyclodextrin–adamantyl), induces cell–cell adhesion via non-covalent click chemistry. Since such a bioorthogonal conjugation enables tying of cells that do not recognize naturally, it provides a valuable opportunity for studying and manipulating intercellular interactions. Under these circumstances, we decided to pursue our investigations by exploring the potential of our artificial surface markers within the framework of the destruction of cancer cells (Fig. 4a).
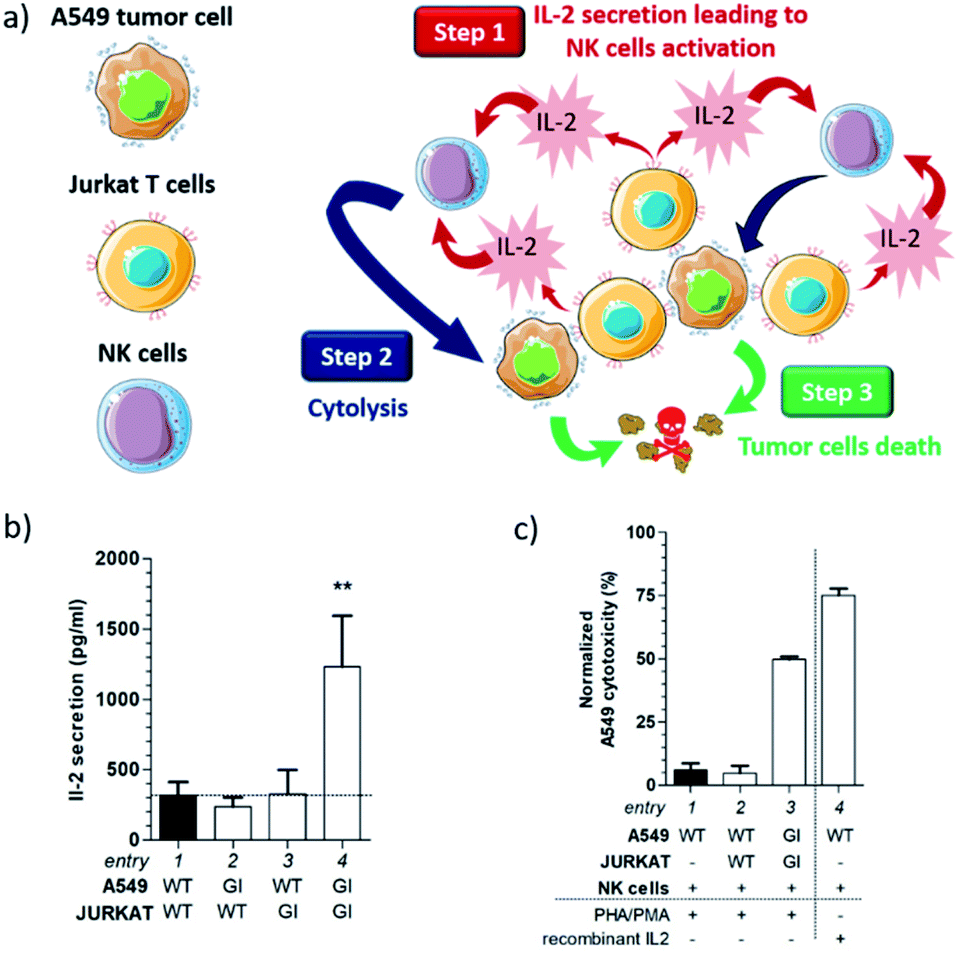 | ||
| Fig. 4 (a) Potential cell interaction network engendered by an unnatural cell recognition leading to the destruction of tumor cells; (b) Concentration of IL-2 secreted by Jurkat T cells when incubated with A549 cells in the presence of PHA and PMA (GI: glycoengineered cells, WT: wild-type cells); (c) NK cells-mediated cytotoxicity for A549 cells directed by cell–cell interactions. | ||
Indeed, Jurkat T cells readily secrete cytokines such as interleukin-2 (IL-2) when stimulated by lectins.15 Therefore, we postulated that the accumulation of Jurkat T cells at the surface of A549 cells could generate an IL-2 local concentration which may be sufficient to activate natural killer (NK) cells (step 1). Once activated by IL-2, NK cells could then initiate the lysis of cancer cells through the exocytosis of perforin (step 2),16 leading ultimately to tumor cell death (step 3).
To study this hypothesis, Tri-Adam-modified Jurkat T cells were seeded on A549 adherent cells previously functionalized with Tri-β-CD. After ten minutes of incubation, the culture medium was washed with PBS in order to remove non-adherent cells. Remaining cells were treated for forty eight hours with phytohemagglutinin (PHA) and phorbol 12-myristate 13-acetate (PMA) to stimulate IL-2 production by Jurkat T cells.17 The supernatant was then harvested and the amount of secreted IL-2 was analyzed by human IL-2 enzyme-linked immunosorbent assay (ELISA).
As shown in Fig. 4b, the IL-2 production was similar under almost all conditions (Fig. 4b, entries 1–3), except when both modified Jurkat and A549 cells were incubated together (Fig. 4b, entry 4). In the latter case, the IL-2 concentration was approximately 4-fold higher than that measured in all the control experiments. These results demonstrated a direct correlation between cell recognition promoted by non-covalent click chemistry and the increased IL-2 concentration. Indeed, the presence of Tri-β-CD and Tri-Adam on the cell surface prevented Jurkat T cells from being washed away during the experimental protocol, hence enabling the release of a larger IL-2 amount in the vicinity of A549 adherent cells.
We next investigated whether such an IL-2 production can trigger the lysis of tumor cells by NK cells. Toward this end, A549 and NK cells were incubated for four hours with the supernatants collected from previous experiments and the resulting cytotoxicity for cancer cells was monitored using a calcein-release assay (Fig. 4c, for the full experimental procedure see the ESI†). In this study, the basal toxicity of NK cells for A549 cells was used as the control experiment (Fig. 4c, entry 1). Thus, when unmodified Jurkat and A549 cells were incubated together, no supplementary toxicity was observed for tumor cells (Fig. 4c, entry 2). In contrast, the incubation of both glycoengineered Jurkat and A549 cells triggered a significant cytotoxic effect (Fig. 4c, entry 3) that was correlated with the increased concentration of IL-2 secreted by T cells (Fig. 4b, entry 4).
To prove that the killing of tumor cells was the consequence of NK cell activation by the cytokine, we conducted a positive control experiment in which A549 and NK cells were incubated in the presence of recombinant IL-2 (Fig. 4c, entry 4). Under these conditions, strong toxicity toward tumor cells was recorded, highlighting the role of IL-2 in the process of A549 cell destruction by NK cells. Taken altogether, these results showed that the interaction between A549 and Jurkat cells, induced by the presence on their surface of the complementary cell recognition markers Tri-β-CD and Tri-Adam, was necessary to launch the observed cytotoxic activity. This study also indicated that non-covalent click chemistry can be a powerful tool for modulating the behavior of cellular networks.
Conclusions
In summary, we designed complementary artificial recognition markers that can be easily introduced on the surface of glycoengineered cells via the SPAAC reaction. These artificial markers led to unnatural intercellular interactions via non-covalent click chemistry. We demonstrated that such cell–cell contacts offered the possibility to manipulate cell networks with potential medicinal applications. Since metabolic glycan labelling using azido-sugars has been applied to remodel the membranes of a wide range of cells, the Tri-β-CD and Tri-Adam recognition markers can be useful for associating cells that cannot recognize each other naturally. Therefore, these ready-to-use artificial markers should greatly facilitate the study of intercellular interactions, providing a better comprehension of biological mechanisms associated with cell adhesion, as well as new opportunities for the development of cell-based therapies.Author contributions
C. P. synthesised, purified and characterised the artificial markers. J. C. designed and conducted in vitro biological experiments. A. S. M. and B. R. supervised the synthesis work and analysed the data. T. V. analysed biological data and provide advices for clinical transfer. S. P. designed the study and wrote the manuscript. All the authors regularly discussed the results of the study and jointly decided on the directions to be followed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank La Ligue contre le Cancer (Comités Vienne, Charente, Charente-Maritime and Deux-Sèvres), the Région Nouvelle Aquitaine and the program PAUSE for financial support of this study. The authors also thank Jérémie Rousseau for his precious help in the design of figures and artworkNotes and references
- This field was first pioneered in 1992 by Kayser et al.: H. Kayser, C. C. Geilen, C. Paul, R. Zeitler and W. Reutter, FEBS Lett., 1992, 301, 137 Search PubMed.
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930 Search PubMed.
- (a) C. Agatemor, M. J. Buettner, R. Ariss, K. Muthiah, C. T. Saeui and K. J. Yarema, Nat. Rev. Chem., 2019, 3, 605 Search PubMed; (b) H. Wang and D. J. Mooney, Nat. Rev. Chem., 2020, 12, 1102–1114 Search PubMed.
- C. M. Csizmar, J. R. Petersburg and C. R. Wagner, Cell Chem. Biol., 2018, 25, 931 Search PubMed.
- Z. J. Gartner and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4606 Search PubMed.
- S. Sugimoto, R. Moriyama, T. Mori and Y. Iwasaki, Chem. Commun., 2015, 51, 17428 Search PubMed.
- H. Koo, M. Choi, E. Kim, S. K. Hahn, R. Weissleder and S. H. Yun, Small, 2015, 11, 6458 Search PubMed.
- H. Koo, S. K. Hahn and S. H. Yun, Bioconjugate Chem., 2016, 27, 2601 Search PubMed.
- W. Li, H. Pan, H. He, X. Meng, Q. Ren, P. Gong, X. Jiang, Z. Liang, L. Liu, M. Zheng, X. Shao, Y. Ma and L. Cai, Small, 2019, 15, 1804383 Search PubMed.
- H. Pan, W. Li, Z. Chen, Y. Luo, W. He, M. Wang, X. Tang, H. He, L. Liu, M. Zheng, Z. Liang, Y. Ma and L. Cai, Bioact. Mater., 2021, 6, 951 Search PubMed.
- P. Shi, E. Ju, Z. Yan, N. Gao, J. Wang, J. Hou, Y. Zhang, J. Ren and X. Qu, Nat. Commun., 2016, 7, 13088 Search PubMed.
- C. L. Schreiber and B. D. Smith, Nat. Rev. Chem., 2019, 3, 393 Search PubMed.
- R. Châtre, J. Lange, E. Péraudeau, P. Poinot, S. Lerondel, A. Le Pape, J. Clarhaut, B. Renoux and S. Papot, J. Controlled Release, 2020, 327, 19–25 Search PubMed.
- (a) M. Fritzsche, R. A. Fernandes, V. T. Chang, H. Colin-York, M. P. Clausen, J. H. Felce, S. Galiani, C. Erkenkämper, A. M. Santos, J. M. Heddleston, I. Pedroza-Pacheco, D. Waithe, J. Bernardino de la Serna, B. C. Lagerholm, T.-L. Liu, T.-L. Chew, E. Betzig, S. J. Davis and C. Eggeling, Sci. Adv., 2017, 3, e1603032 Search PubMed; (b) C. Nobile, D. Rudnicka, M. Hasan, N. Aulner, F. Porrot, C. Machu, O. Renaud, M.-C. Prévost, C. Hivroz, O. Schwartz and N. Sol-Foulon, J. Virol., 2010, 3, 2282 Search PubMed.
- E. E. Roosnek, M. C. Brouwer and L. A. Aarden, Eur. J. Immunol., 1985, 15, 652 Search PubMed.
- (a) C. Lehmann, M. Zeis and L. Uharek, Br. J. Haematol., 2001, 114, 660 Search PubMed; (b) T.-K. Yu, E. G. Caudell, C. Smid and E. A. Grimm, J. Immunol., 2000, 164, 6244 Search PubMed.
- T. Ohtsuka, Y. Kaziro and T. Satoh, Biochim. Biophys. Acta, 1996, 1310, 223 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01637g |
| This journal is © The Royal Society of Chemistry 2021 |