
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLibrary construction of stereochemically diverse isomers of spirooliganin: their total synthesis and antiviral activity†
Ru-Bing
Wang‡
a,
Shuang-Gang
Ma‡
 *a,
Cooper S.
Jamieson‡
b,
Rong-Mei
Gao
c,
Yun-Bao
Liu
a,
Yong
Li
a,
Xiao-Jing
Wang
a,
Yu-Huan
Li
c,
Kendall N.
Houk
*b,
Jing
Qu
*a and
Shi-Shan
Yu
*a
*a,
Cooper S.
Jamieson‡
b,
Rong-Mei
Gao
c,
Yun-Bao
Liu
a,
Yong
Li
a,
Xiao-Jing
Wang
a,
Yu-Huan
Li
c,
Kendall N.
Houk
*b,
Jing
Qu
*a and
Shi-Shan
Yu
*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, No.1 Xian Nong Tan Street, Beijing, 100050, People's Republic of China. E-mail: mashuanggang@imm.ac.cn; qujing@imm.ac.cn; yushishan@imm.ac.cn
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: houk@chem.ucla.edu
cInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 1 Tian Tan Xi Li, Beijing, 100050, People's Republic of China
First published on 10th April 2021
Abstract
The construction of libraries of stereoisomers of natural products serves as an important approach to investigating the correlation between the stereostructure and biological activity. However, the total synthesis and isomerzation of polycyclic scaffolds with multiple chrial centers are rare. Spirooliganin (1), a new skeleton natural product isolated from the plant Illicium oligandrum, was structurally characterized by comprehensive analysis of NMR spectroscopic data and ECD which revealed an unprecedented 5–6–6–6–7 polycyclic framework with six chiral centers. Here we report a 17-step total synthesis to prepare a library of stereochemically diverse isomers of spirooliganin, including 16 diastereoisomers and 16 regioisomers. In addition to a regioselective hetero-Diels–Alder cycloaddition, the synthetic strategy involves a photo-induced stereoselective Diels–Alder reaction, which gives only the abnormal trans-fused product as rationalized by density functional theory calculations. Preliminary biological evaluation showed that spirooliganin and regioisomers 39 exhibited potent inhibition of Coxsackievirus B3. It also revealed the pharmacophore effect of the D-ring (16R,18R,24R, and 26R) for their antiviral activities.
Introduction
Natural products continue to be a major source of novel lead compounds or pharmacophores for medicinal chemistry due to their diverse structure and biological activities. Bioactive natural products often possess one or multiple chiral centers and spatially defined arrangements of functional groups. Given the important relationship between stereochemistry and bioactivity, the creation of stereoisomer libraries of natural products is envisaged to maximally screen the potential of novel frameworks in drug discovery.Examples of the construction of stereoisomer libraries of natural products containing multiple chiral centers has been reported in the past few decades, such as annonaceous acetogenins, a class of fatty acid-derived natural products from members of the Annonaceae family,1–4 cladosporin, an isocoumarin-based metabolite from fungus,5 spirotryptostatin, a prenyl-containing alkaloid from Aspergillus fumigatus,6–8 and cocaine, a member of the tropane alkaloids isolated from the leaves of Erythroxylon coca.9 However, stereoisomeric variation in these studies was limited to scaffolds with long carbon-chain skeletons or polyketide macrolides10,11 that contain multiple oxygenated chiral centers, or molecules with two vicinal stereogenic centers.9,12–15 Due to the synthetic challenge, there exists no stereoisomer library constructed for natural products with a rigid, polycyclic fused ring scaffold and multiple resulting stereocenters. Thus, the stereostructure–activity relationship (SSAR) for many of these natural products is unknown.
Inspired by the potential antiviral activity of prenylated C6–C3 compounds,16–19 which are characteristic constituents of plants from the genus Illicium, we isolated and characterized spirooliganin (1) from the stem bark of I. oligandrum.20 The compound 1 features an unprecedented linear tetracycle (6–6–6–7, rings B, C, D, and E, Fig. 1). Remarkably, this molecule exhibits potent activity against Coxsackievirus B3 (CVB3), with an IC50 of 2.1 μM and a selectivity index (SI) value of 5.2. Here we report the construction of a library of stereoisomers of spirooliganin, including 16 diastereoisomers and 16 regioisomers, via a 17-step total synthesis. The synthetic strategy involves a regioselective hetero-Diels–Alder cycloaddition and an abnormal photochemical trans-selective Diels–Alder reaction, which has been rationalized by density functional theory calculations. The antiviral activity of the stereoisomer library was evaluated against CVB3, thus revealing the relationship between the stereostructure and antiviral activities of this analogue of natural products.
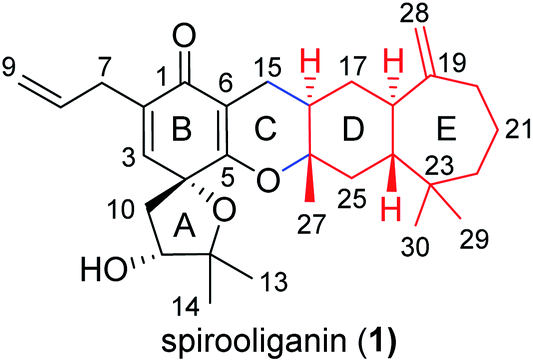 | ||
| Fig. 1 Structure of spirooliganin, a novel prenylated C6–C3 compound, isolated from I. oligandrum. | ||
Results and discussion
Structural elucidation of spirooliganin
Spirooliganin (1), a colorless oil, was determined to have the molecular formula C30H42O4 on the basis of 13C NMR data and the (+)-HRESIMS ion peak at m/z 467.3160 [M + H]+ (calcd 467.3156), suggesting ten degrees of unsaturation. The IR spectrum displayed absorptions characteristic of hydroxy (3437 cm−1) and carbonyl (1671 cm−1) groups. Analysis of the 13C NMR spectrum (Table S1†) with the aid of DEPT and HSQC spectra revealed the presence of four double bonds and one cross-conjugated carbonyl (δC 185.2) group in the downfield region (δC 186.0–109.0 ppm) and five methyls, eight methylenes, four methines (one oxygenated), and four quaternary carbons (three oxygenated) in the upfield region (δC 86.0–19.0 ppm). Analysis of the degree of unsaturation indicates the presence of five rings (rings A–E) in 1. The planar structure of 1 (Fig. 1) was determined by examination of the 2D NMR spectra, which was described as follows. Four proton-bearing structural fragments a–d, as depicted by bold bonds (Fig. S1A†), were readily established by interpretation of the HSQC and 1H–1H COSY spectra. Subsequently, a series of key HMBC correlations (Fig. S1B†) led to the determination of the planar structure of the unprecedented linearly fused tetracyclic (6–6–6–7) skeleton bearing a 1-oxaspiro[4.5]deca-6,9-dien-8-one moiety. The stereochemistry (4R,11R,16R,18R,24R, and 26S) of the chiral centers in rings A and D was unambiguously determined from the NOESY spectrum and a series of NOE experiments (Fig. S1B, S18 and S19†), as well as circular dichroism (CD) spectroscopy showing a negative Cotton effect at 255 nm and a positive Cotton effect at 309 nm. The full details of the structural elucidation can be found in the ESI.†Strategy for the isomer library construction of spirooliganin
Structurally, spirooliganin contains a spiro-type prenylated C6–C3 unit (C1–C14, black in Fig. 1) and a himachalane-type sesquiterpenoid with a bicyclo[5.4.0]undecane skeleton (C16–C30, red in Fig. 1), which are fused through a methylene (C15) and an oxygen bridge involved in a pyran ring (C-ring). Four of the six chiral centers in spirooliganin are located on the linearly fused tetracycle (B, C, D and E rings) and two on the oxaspiro ring (A and B rings). To construct a library of spirooliganin stereoisomers for a better understanding of its SSAR, the total synthesis was envisioned to diversify the introduction of stereocenters in each step. We conceived a route to spirooliganin that succinctly delivers a wide diversity of isomers of 1 (Fig. 2).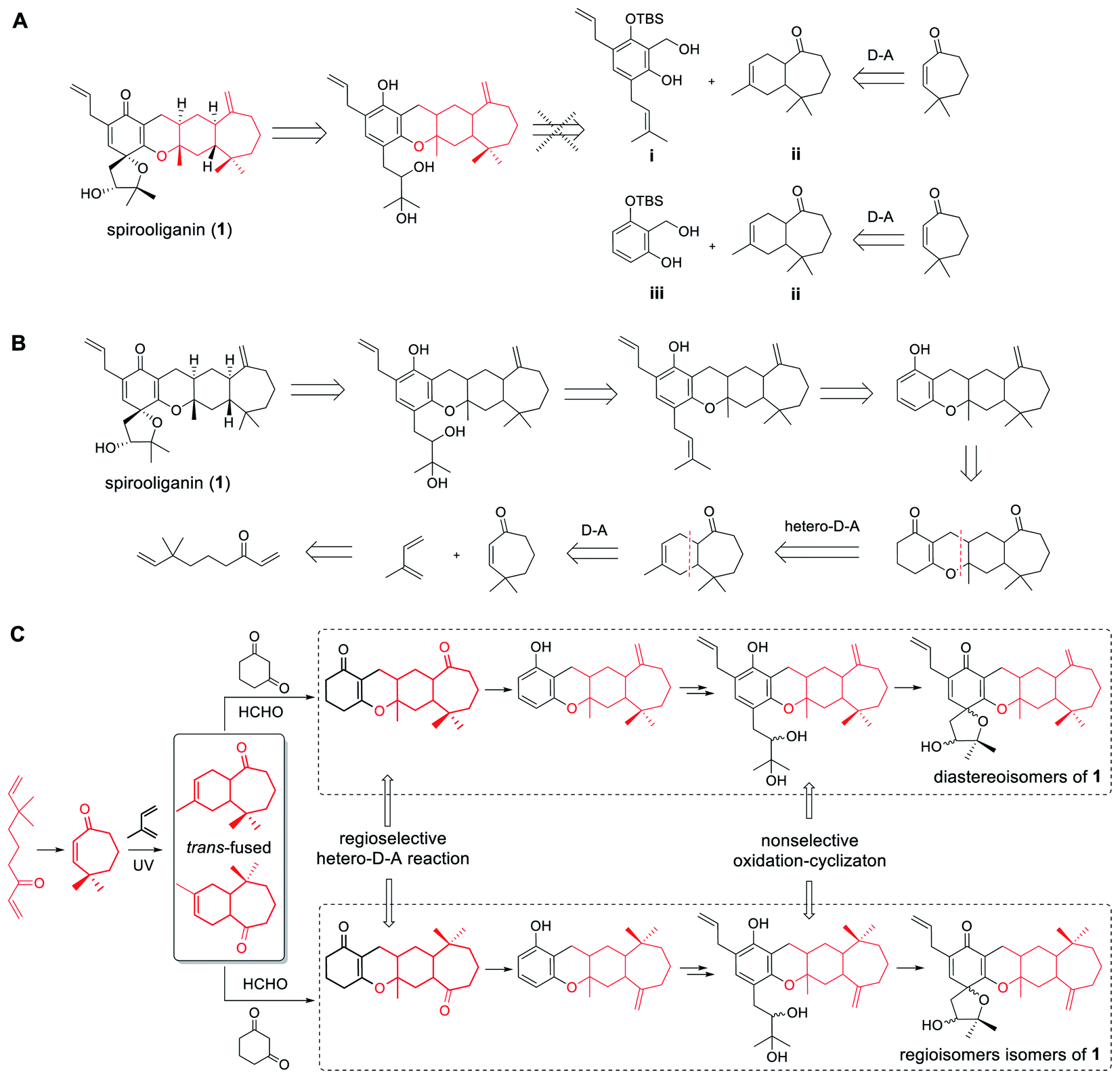 | ||
| Fig. 2 General strategy and retrosynthetic analysis to produce stereochemically diverse isomers of spirooliganin (1). (A) Unrealized convergent retrosynthetic strategy for the construction of spirooliganin. (B) Linear retrosynthetic strategy to achieve the synthesis of isomers of spirooliganin. (C) General synthetic strategy of stereochemically diverse isomers of spirooliganin. | ||
The general synthetic strategy includes three main transformations: (1) use of a nonselective oxidation–cyclizaton to diversify the two chiral centers on the oxaspiro ring while the characteristic prenylated C6–C3 unit was retained, (2) use of a non-regioselective Diels–Alder reaction between 4,4-dimethylcyclohept-2-en-1-one and isoprene to give two kinds of cycloadditon product including regioisomers resulting from the opposite location of the exocyclic double bond and a gem-dimethyl group to maximize the isomers of 1, and (3) use of a regioselective hetero-Diels–Alder reaction of an o-quinone methide with himachalane cyclohexene to obtain the linearly fused 6–6–6–7-tetracycle, which matches the basic skeleton of 1 and produces diverse stereoisomers of tetracyclic intermediates. We firstly intended to install the basic skeleton of 1 using a convergent strategy21–23 of i and ii. However, this was not achieved, even after simplification of the starting material (iii to ii) (Fig. 2A). Therefore, we transferred to another linear synthetic strategy to achieve the synthesis of isomers of spirooliganin (Fig. 2B). Finally, library construction of stereo isomers of spirooliganin was realized as shown in Fig. 2C.
Total syntheses of 16 diastereoisomers and 16 regioisomers of spirooliganin
The total synthesis of the polycyclic fused skeleton of spirooliganin 1 started from the commercially available methyl 3,3-dimethylpent-4-enoate 2, which was reduced to the corresponding alcohol 3 by LiAlH4 in an almost quantitative yield. A substitution reaction of 3 with iodine, imidazole and triphenylphosphine afforded 4 in 90% yield. Compound 4 was treated with n-BuLi and acetylmethylene triphenylphosphorane to generate an ylide intermediate,24 which was directly used for the subsequent Wittig reaction with formaldehyde to afford α,β-unsaturated ketone 5 in the two steps with 80% yield. Compound 5 was refluxed in dichloromethane over a Grubbs II catalyst to give intramolecular ring-closure product 6 in 85% yield. Diels–Alder cycloaddition of compound 6 with isoprene (also as the solvent) under ultraviolet (UV) 365 nm irradiation generated a pair of trans-fused regioisomers 7a and 7b (Fig. 3) in a ratio of 46.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :53.2, which were separated by using reverse phase C18 preparative HPLC to give two pairs of racemates 7a and 7b.25,26 Their structures were determined based on extensive spectroscopic evidence (Fig. S29–S40†) and also supported by the X-ray crystal structures of the products from subsequent steps (Fig. 3). The trans-fused product rac-7a and rac-7b served as key intermediates in the synthesis of stereoisomers and regioisomers of spirooliganin, respectively. A three-component hetero-Diels–Alder cycloaddition of rac-7a, formaldehyde and 1,3-cyclohexanedione was used to construct the 6–6–6–7 tetracyclic core according to a reported strategy.27 Two pairs of enantiomers (rac-8a and rac-8b) were obtained (Fig. 3). To ensure the complete conversion of rac-7a, 1,3-cyclohexanedione was strictly controlled to be added in portions after the reaction was heated to 100 °C. The reaction from rac-7b was same as that from rac-7a. The hetero-Diels–Alder reaction afforded a cis-fused 6–6 ring system, resulting in the assembly of the key tetracyclic core, the stereochemistry of which was confirmed by X-ray crystal structures of rac-8a, rac-8b, rac-8c and rac-8d (Fig. 3, Fig. S5–S8 and Tables S21–S28†).
:53.2, which were separated by using reverse phase C18 preparative HPLC to give two pairs of racemates 7a and 7b.25,26 Their structures were determined based on extensive spectroscopic evidence (Fig. S29–S40†) and also supported by the X-ray crystal structures of the products from subsequent steps (Fig. 3). The trans-fused product rac-7a and rac-7b served as key intermediates in the synthesis of stereoisomers and regioisomers of spirooliganin, respectively. A three-component hetero-Diels–Alder cycloaddition of rac-7a, formaldehyde and 1,3-cyclohexanedione was used to construct the 6–6–6–7 tetracyclic core according to a reported strategy.27 Two pairs of enantiomers (rac-8a and rac-8b) were obtained (Fig. 3). To ensure the complete conversion of rac-7a, 1,3-cyclohexanedione was strictly controlled to be added in portions after the reaction was heated to 100 °C. The reaction from rac-7b was same as that from rac-7a. The hetero-Diels–Alder reaction afforded a cis-fused 6–6 ring system, resulting in the assembly of the key tetracyclic core, the stereochemistry of which was confirmed by X-ray crystal structures of rac-8a, rac-8b, rac-8c and rac-8d (Fig. 3, Fig. S5–S8 and Tables S21–S28†).
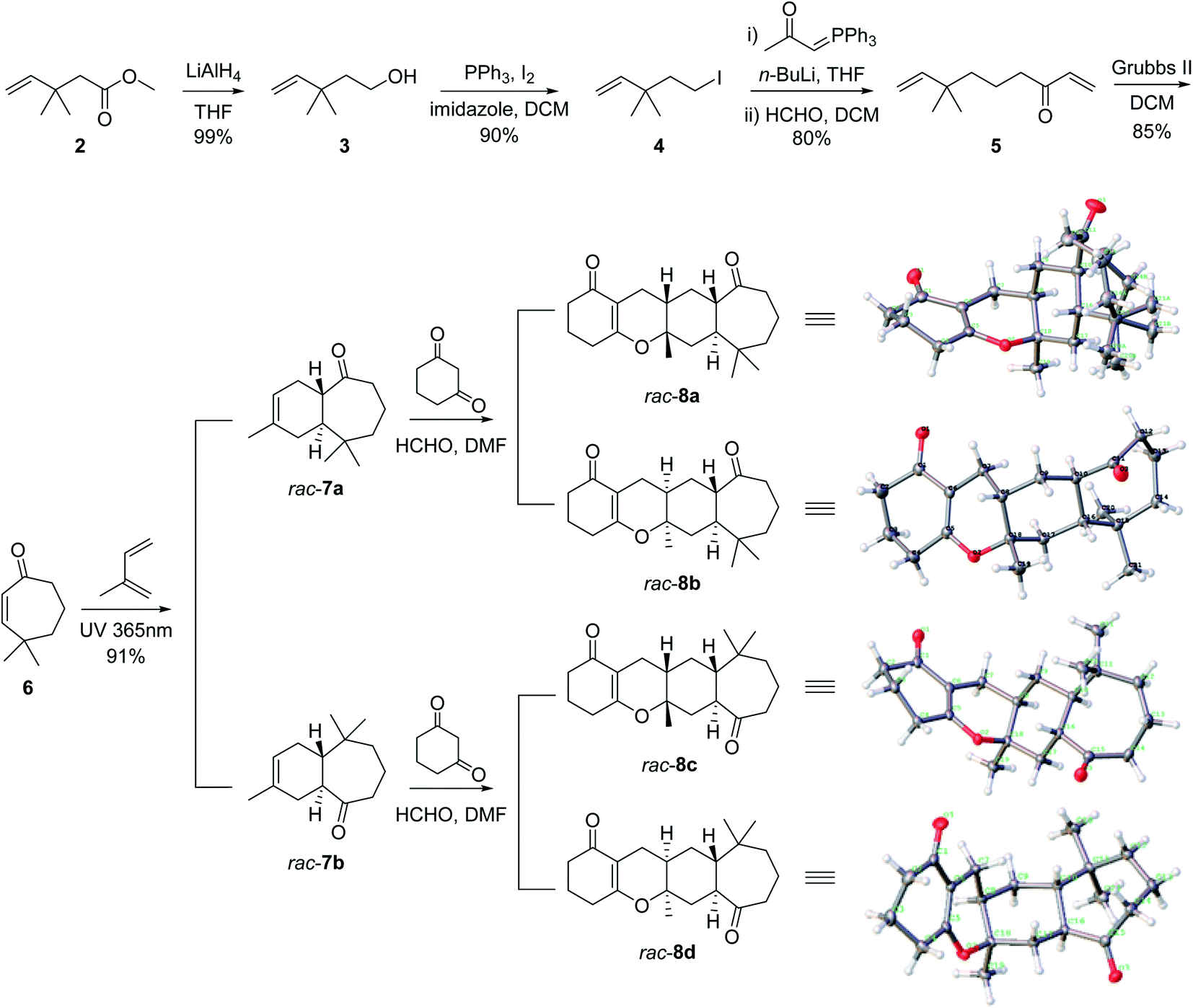 | ||
| Fig. 3 Construction of the linear tetracyclic core for spirooliganin. The crystal structures of the key tetracyclic intermediates rac-8a, rac-8b, rac-8c and rac-8d were shown. rac-8c and rac-8d would be used in the same synthetic route to give spirooliganin regioisomers. | ||
With the tetracyclic core in place, our attention was focused on the formation of the B ring by aromatization (Fig. 4). We expected to accomplish selective aromatization of the B ring in one step based on the slight activity difference between the α,β-unsaturated carbonyl and saturated carbonyl group. Many reported methods (Pd/C, I2/CH3OH, I2/DMSO/CH3NO2, DDQ, and NaH/PhSO2CH3) were attempted,27–29 such as (NaH/PhSO2CH3) reported by Xie et al. as a practical method for the synthesis of spirooliganones.27 However, these attempts gave at best a trace amount of the desired product. We speculated that this might be due to the presence of carbonyl on the seven-membered ring, which was accompanied by β-keto sulfoxide substitution when the reaction was heated to 100 °C using methyl benzenesulfinate for aromatization. We thus attempted an alternative strategy by protecting the carbonyl before aromatization. The reaction of rac-8a with glycol and trimethyloxymethane catalyzed by p-TsOH in benzene under violent reflux gave the corresponding protected racemates. The following aromatization of rac-9a not only converted the α,β-unsaturated ketone ring (B ring) into the corresponding phenol but also completely removed the protection of the carbonyl in two steps with 60% yield for rac-10a.
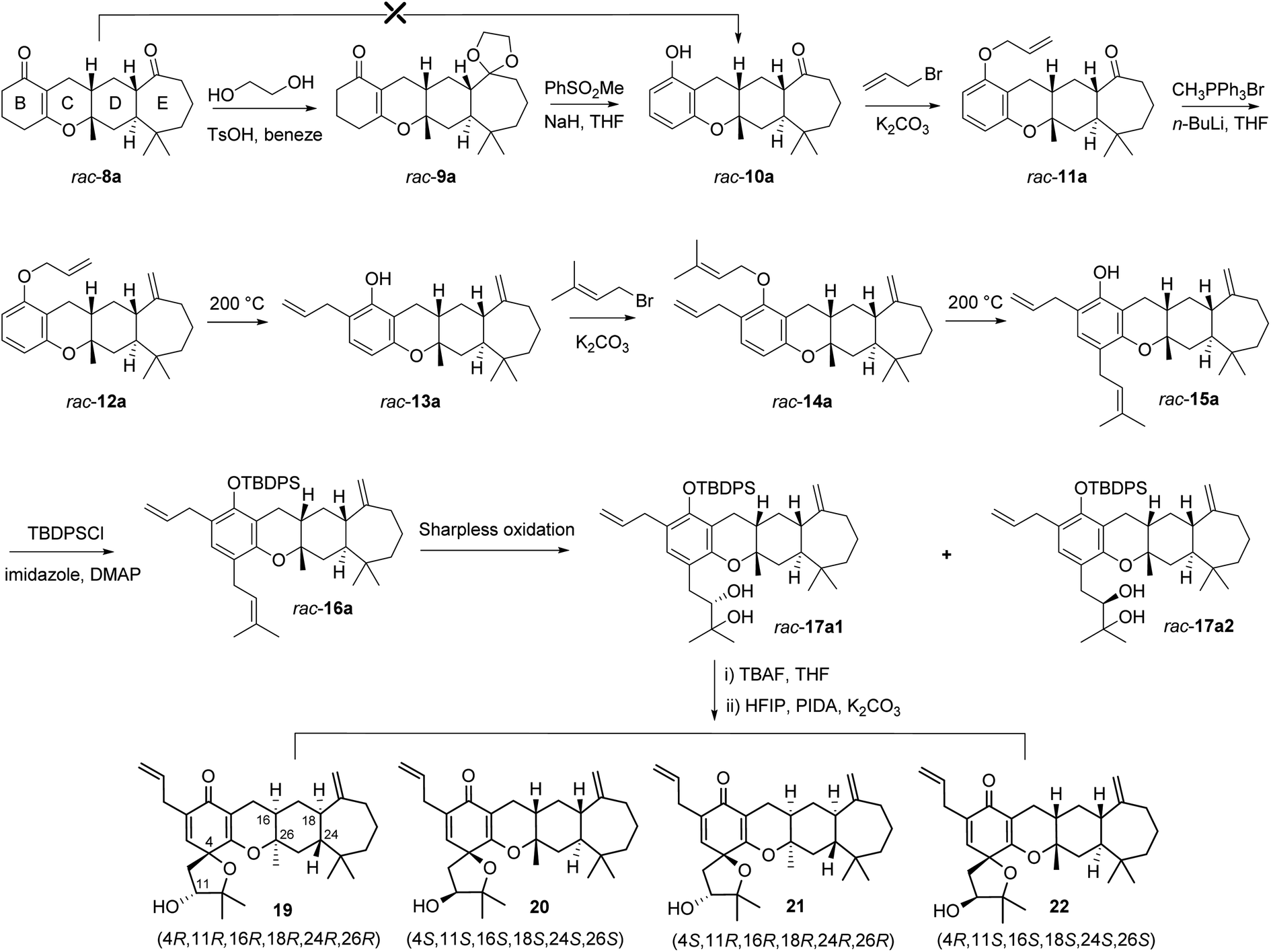 | ||
| Fig. 4 Syntheses of spirooliganin diastereoisomers from rac-8a. The four diastereoisomers 19, 20, 21, and 22 formed via rac-17a1 were shown. | ||
With racemic phenols rac-10a in hand, the allylic and prenyl side chains of the B ring were introduced by a general alkylation/Claisen rearrangement reaction sequence. Treatment of rac-10a with allyl bromide and K2CO3 in refluxing acetone efficiently delivered allylic ether rac-11a in 95% yield, followed by a Wittig reaction using the ylide of methyltriphenylphosphonium bromide to give rac-12a in 90% yield. This intermediate was converted to o-allylic phenols rac-13a with high regioselectivity in 95% yield when heated in N,N-diethylaniline. Similarly, treatment of rac-13a with prenyl bromide and K2CO3 in refluxing acetone efficiently produced rac-14a, which was then heated in N,N-diethylaniline to give the p-prenyl phenols rac-15a with high regioselectivity in 75% yield over two steps. rac-15a was protected by tert-butyldiphenylsilyl chloride (TBDPSCl) to produce rac-16a in 96% yield, which was subjected to Sharpless dihydroxylation with AD-mix-β at 10–15 °C for 36 h, and two pairs of racemic diols, rac-17a1 (24% yield) and rac-17a2 (21% yield), were generated with poor stereoselectivity and 30% conversion. We attempted to prolong the reaction time and increase the reaction temperature, but tetrol derivatives were observed as the main product from HRMS data, which may be due to the oxidation of both the isopentenyl and allyl groups. The structures of the intermediates (rac-10a, rac-11a, rac-12a, rac-13a, rac-14, and rac-15a) were confirmed by a series of 1D and 2D-NMR spectroscopic analyses (Fig. S63–S66, S79–S82, S95–S98, S111–S114, S127–S130, and S143–S146†), and their stereochemistry was the same as that of rac-8a. The stereochemistry of two pairs of racemic diols (rac-17a1 and rac-17a2) was verified based on the absolute configuration of the final products 19–26 (Fig. S191–S230†).
Followed by the preparation of diols, a tandem oxidative dearomatization/cyclization reaction was adopted to construct the oxaspiro ring moiety. Treatment of rac-17a1 with TBAF in THF afforded the deprotected compounds, which was reacted with iodobenzene diacetate (PIDA) in hexafluoroisopropanol (HFIP) in the presence of K2CO3 to generate two pairs of scalemic enantiomers 19/20 in 20% yield and 21/22 in 22% yield after purification by silica column chromatography and preparative HPLC. The enantiomers were further separated by chiral HPLC to afford single diastereoisomers, including 19, the 26-epimer of spirooliganin.
We also prepared the other 12 diastereoisomers (23–34) of spirooliganin from rac-17a2 and rac-8b, as well as 16 regioisomers (35–50) from rac-8c and rac-8d (Fig. 5). The synthetic route was same as that used for diastereoisomer synthesis from rac-8a. The structures of the final products, including the absolute configurations, were unambiguously characterized by comprehensive NMR spectroscopic analysis, a series of NOE experiments, ECD analysis, and Mosher's method (Fig. S2, S3, and S191–S369, Tables S12–S20†). Moreover, the X-ray crystal structure of a pair of enantiomers, 31 and 32, obtained from methanol containing trace H2O also confirmed the fascinating highly linearly fused tetracyclic (6–6–6–7) skeleton bearing a 1-oxaspiro [4.5]deca-6,9-dien-8-one moiety and six dense chiral centers (Fig. 6).
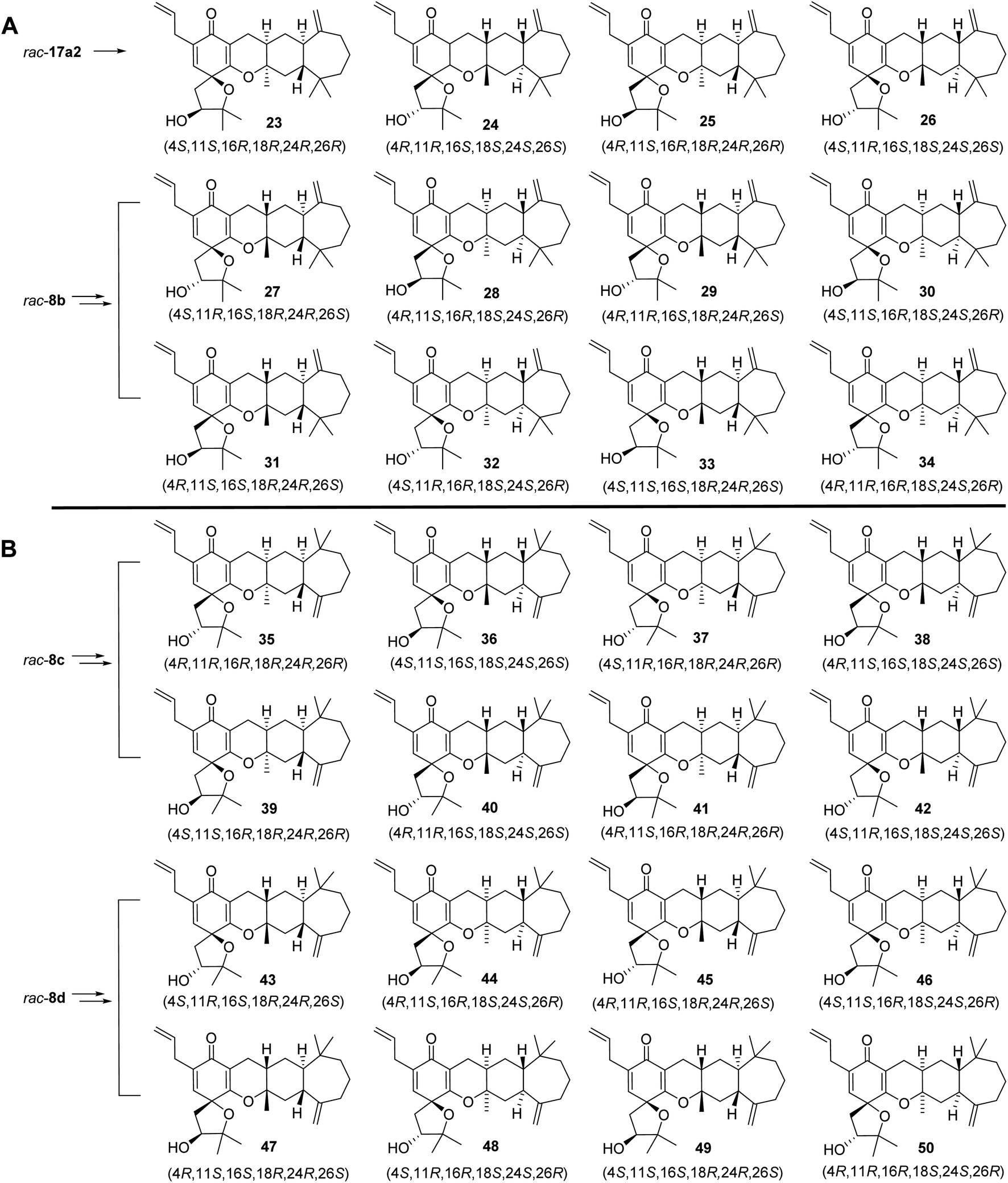 | ||
| Fig. 5 Stereochemically diverse structures of isomers of spirooliganin. (A) Structures of the other 12 diastereoisomers of spirooliganin from rac-17a2 and rac-8b. (B) Structures of 16 regioisomers from rac-8c and rac-8d. | ||
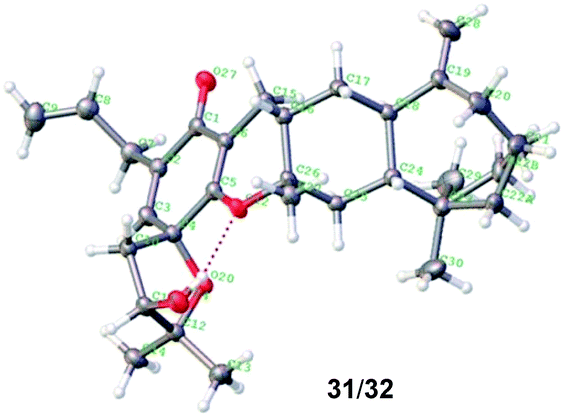 | ||
| Fig. 6 The X-ray crystal structure of a pair of enantiomers 31/32. | ||
Abnormal photo-induced trans-selective Diels–Alder reaction
The observation that the Diels–Alder cycloaddition of compound 6 with isoprene under UV yielded 100% trans-fused products intrigued us to investigate the mechanism of this uncommon trans-photochemical cycloaddition reaction by density functional theory calculations at the ωB97X-D/def2-QZVPP//ωB97X-D/def2-TZVP level of theory. Since medium ring trans-cycloalkenes are known to be highly reactive in cycloadditions,30 we hypothesized that the well-known photochemical cis, trans isomerization31 of cycloheptenones would lead to a reactive trans-cycloheptenone. The reactivity of trans-cycloheptenones is derived from distorting the structure towards the cycloaddition transition state geometry. To verify this, we calculated the molecular orbitals (MOs) of isoprene, cis-, and trans-cycloheptenones (Fig. 7A). As has been known and explained32 the highest occupied MO of isoprene has very similar coefficients at the two termini and is expected to react with low regioselectivity. The lowest unoccupied MOs (LUMOs) of cis- and trans-cyclohepetenones have similar orbital energies and support the idea that trans-cycloheptenone reactivity is distortion accelerated and not accelerated by improved orbital interactions. Alkene isomerization rotates the reacting p-orbitals out of conjugation from the ketone and results in a slight increase in the LUMO energy (+0.04 eV), despite the fact that this distortion of the alkene should intrinsically lower the alkene LUMO energy.33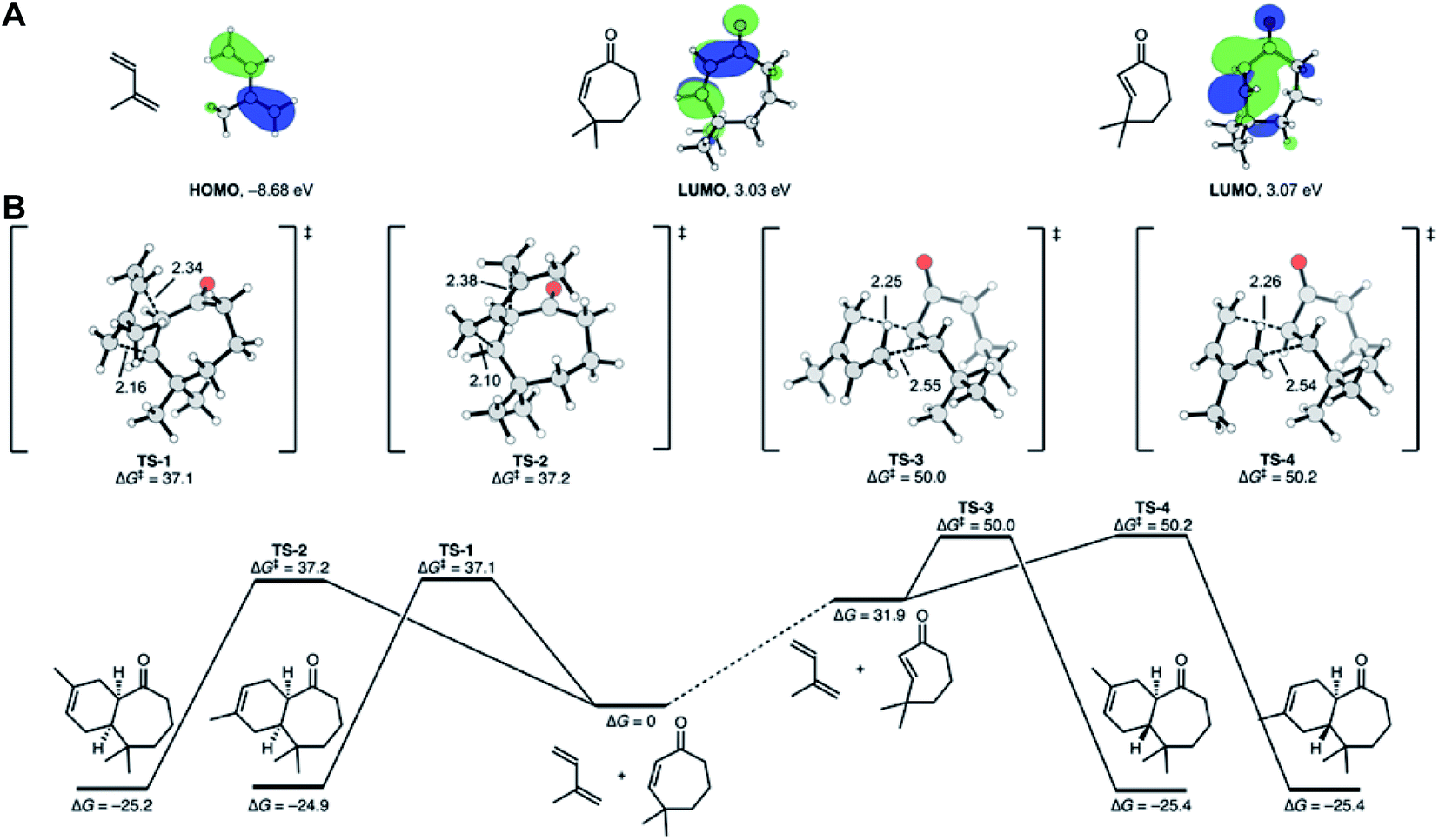 | ||
| Fig. 7 DFT calculations for the intermolecular [4 + 2] cycloaddition reaction. (A) Energies and shapes of the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) of isoprene, cis-cycloheptenone and trans-cycloheptenone. (B) Gibbs free energy surface of cycloadditions from cis- and trans-cycloheptenones. Relative to reactants, TS-3 and TS-4 have low barriers of 18.1 and 18.3 kcal mol−1 and readily take place, whereas TS-1 and TS-2 are too high in energy to proceed under the experimental conditions. | ||
We located the transition states for the cycloadditions of both the cis- and trans-cycloheptenones (Fig. 7B). The cis-cycloheptenone can react via endo or exo transition states to form two regiosomeric cis-fused cycloheptenone adducts. The lowest energy transition states for the regioisomeric cycloadditions are TS-1 and TS-2. These transition states are endo and have free energies of activation of 37.1 and 37.2 kcal mol−1, respectively. There is a minute (0.1 kcal mol−1) preference for C-1 of isoprene to react at the β carbon of the α,β-unsaturated ketone, in agreement with the frontier molecular orbital prediction. The barriers for these cycloadditions are very high, and under mild reaction conditions would not be observed.
Isomerization of cis-cycloheptenone to trans-cycloheptenone is uphill thermodynamically by 31.9 kcal mol−1. The trans-cycloheptenone can, similarly, undergo cycloadditions to form two regiosomeric trans-fused cycloheptenone adducts. In this case, the lowest energy transition states TS-3 and TS-4 are exo with respect to the cycloheptenone ketone and have low barriers of 18.1 and 18.3 kcal mol−1 from the trans-cycloheptenone, respectively. The transition states are slightly more asynchronous than those from the cis-cycloheptenone, but still clearly concerted. Now, there is a small preference for the opposite regioselectivity compared to the cis-enone reaction. The barriers for these cycloadditions of trans-cycloheptenone are nearly 20 kcal mol−1 lower than that of cis-cycloheptenone.
These calculations rationalize why the cis-fused adduct was not observed and showed the origins of stereochemistry and regioselectivity. In short, the cis-cycloheptenone is unreactive in cycloadditions, but the trans-cycloheptenone is highly reactive and leads by a concerted cycloaddition to the trans-fused product.
Antiviral screening and stereostructure–activity relationship
With spirooliganin (1) and the 32 isomer library in hand, we were able to investigate the influence of the stereostructure on their biological activity. Compound 1 and its 16 diastereoisomers (Table 1, Groups A and B) and 16 regioisomers (Table 1, Groups C and D) were evaluated for their antiviral inhibitory activities against CVB3 in African green monkey kidney cells (Vero cells), which were infected with CVB3 and measured with CPE assay.34 The preliminary biological study showed that compound 1 and its regioisomer 39 displayed the most potent activities against CVB3 among all the isomers, with IC50 values of 2.1 μM and 3.7 μM and SI values of 5.2 and 4.3, respectively, which are better than those of the positive control ribavirin (IC50 1577.5 μM and SI = 5.2) but less effective than those of pleconaril. Diastereoisomer 23 and regioisomers 35, 37, 41, 42, and 47 also exhibited good inhibition against CVB3, with IC50 values ranging from 8.6–11.1 μM. Among the 16 diastereoisomers, compounds 21, 23, and 25, with the same D-ring configuration (16R,18R,24R, and 26R), showed a better selectivity index than their corresponding enantiomers 22, 24, and 26, which have the opposite D-ring configuration (16S,18S,24S, and 26S). Interestingly, a similar trend was observed for the regioisomers. With a D-ring configuration of 16R,18R,24R, and 26R, compounds 35, 37, 39 and 41 exhibited significant inhibitory activities against CVB3, with IC50 values ranging from 3.7 to 11.1 μM, although 42 also showed a similar level of activity compared to 41. This indicated the crucial role of the D-ring configuration for their anti-CVB3 activity, while the configuration of the A-ring may be less important. Notably, natural spirooliganin (1) exhibited excellent inhibitory effects against CVB3 (IC50 2.1 μM), while its synthetic 26-epimer (19) was much less active (IC50 > 33.3 μM), suggesting that the 26S configuration of the methyl group on the D-ring plays an important role in inhibiting CVB3.| Groups | Compounds | CEsb | Configuration | TC50c (μM) | IC50 (μM) | SId | |
|---|---|---|---|---|---|---|---|
| A ring | D ring | ||||||
| a TC50 and IC50 data represent the mean values for three independent determinations. b Cotton effects at λmax 255 nm. c Cytotoxic concentration required to inhibit Vero cell growth by 50%. d The SI value equaled TC50/IC50. e The SI could not be determined under the test conditions. f Positive control. g nM. | |||||||
| 1 | (−) | 4R,11R | 16R,18R,24R,26S | 11.1 ± 1.5 | 2.1 ± 1.1 | 5.2 | |
| A | 19 | (−) | 4R,11R | 16R,18R,24R,26R | 57.7 ± 5.4 | >33.3 | —e |
| 20 | (+) | 4S,11S | 16S,18S,24S,26S | 57.7 ± 2.8 | >33.3 | —e | |
| 21 | (+) | 4S,11R | 16R,18R,24R,26R | 57.7 ± 9.1 | 33.3 ± 4.2 | 1.7 | |
| 22 | (−) | 4R,11S | 16S,18S,24S,26S | 57.7 ± 5.3 | >33.3 | —e | |
| 23 | (+) | 4S,11S | 16R,18R,24R,26R | 48.1 ± 5.2 | 11.1 ± 2.5 | 4.3 | |
| 24 | (−) | 4R,11R | 16S,18S,24S,26S | 48.1 ± 6.3 | >11.1 | —e | |
| 25 | (−) | 4R,11S | 16R,18R,24R,26R | 57.7 ± 7.0 | 33.3 ± 2.2 | 1.7 | |
| 26 | (+) | 4S,11R | 16S,18S,24S,26S | 57.7 ± 6.6 | >33.3 | —e | |
| B | 27 | (+) | 4S,11R | 16S,18R,24R,26S | >100.0 | >33.3 | —e |
| 28 | (−) | 4R,11S | 16R,18S,24S,26R | >100.0 | >33.3 | —e | |
| 29 | (−) | 4R,11R | 16S,18R,24R,26S | 48.1 ± 5.2 | >11.1 | —e | |
| 30 | (+) | 4S,11S | 16R,18S,24S,26R | 23.1 ± 4.2 | >11.1 | —e | |
| 31 | (−) | 4R,11S | 16S,18R,24R,26S | 57.7 ± 1.0 | 22.6 ± 3.3 | 2.6 | |
| 32 | (+) | 4S,11R | 16R,18S,24S,26R | 100.0 ± 8.8 | 33.3 ± 2.9 | 3.0 | |
| 33 | (+) | 4S,11S | 16S,18R,24R,26S | 48.1 ± 4.4 | >11.1 | —e | |
| 34 | (−) | 4R,11R | 16R,18S,24S,26R | 57.7 ± 2.4 | >33.3 | —e | |
| C | 35 | (−) | 4R,11R | 16R,18R,24R,26R | 23.1 ± 4.1 | 11.11 ± 1.35 | 2.1 |
| 36 | (+) | 4S,11S | 16S,18S,24S,26S | 33.3 ± 3.2 | >11.11 | —e | |
| 37 | (+) | 4S,11R | 16R,18R,24R,26R | 19.3 ± 3.7 | 11.1 ± 2.3 | 1.7 | |
| 38 | (−) | 4R,11S | 16S,18S,24S,26S | 19.3 ± 2.4 | >11.1 | —e | |
| 39 | (+) | 4S,11S | 16R,18R,24R,26R | 16.0 ± 2.2 | 3.7 ± 1.2 | 4.3 | |
| 40 | (−) | 4R,11R | 16S,18S,24S,26S | 16.0 ± 1.8 | >3.7 | —e | |
| 41 | (−) | 4R,11S | 16R,18R,24R,26R | 19.3 ± 4.1 | 8.6 ± 1.3 | 2.2 | |
| 42 | (+) | 4S,11R | 16S,18S,24S,26S | 48.1 ± 6.2 | 8.6 ± 1.5 | 5.6 | |
| D | 43 | (+) | 4S,11R | 16S,18R,24R,26S | >100.0 | >33.3 | —e |
| 44 | (−) | 4R,11S | 16R,18S,24S,26R | >100.0 | >33.3 | —e | |
| 45 | (−) | 4R,11R | 16S,18R,24R,26S | 57.7 ± 6.9 | >33.3 | —e | |
| 46 | (+) | 4S,11S | 16R,18S,24S,26R | 57.7 ± 5.2 | >33.3 | —e | |
| 47 | (−) | 4R,11S | 16S,18R,24R,26S | 48.1 ± 4.1 | 11.11 | 4.3 | |
| 48 | (+) | 4S,11R | 16R,18S,24S,26R | 57.7 ± 4.4 | >33.3 | —e | |
| 49 | (+) | 4S,11S | 16S,18R,24R,26S | 69.3 ± 8.0 | >33.3 | —e | |
| 50 | (−) | 4R,11R | 16R,18S,24S,26R | 69.3 ± 3.3 | 33.3 | 2.1 | |
| Ribavirinf | — | 8196.7 ± 45.6 | 1577.5 ± 84.4 | 5.2 | |||
| Pleconarilf | — | 40.5 ± 2.6 | 2.9 ± 0.5g | 14045.1 | |||
Discussion
As a rich source for bioactive natural products, a number of prenylated C6–C3 compounds with diverse structures and/or significant biological activity have been isolated from different parts of the pant I. oligandrum.17–20 Herein, spirooliganin (1), a prenylated C6–C3 compound, has been isolated from the plant stem bark and structurally characterized. It features an unprecedented linear tetracycle with six dense chiral centers and was characterized as the first example of a hybrid of spiro-type prenylated C6–C3 derivative and himachalane sesquiterpenoids (C16–C30) from I. oligandrum.For the SSAR study, we applied an effective total synthesis route that maximizes isomer formations to construct a library of stereochemically diverse isomers of spirooliganin. The 17-step total synthesis started from readily available materials and included five main transformations: (1) an intramolecular ring-closure reaction to furnish 2-cycloheptenone and a photo-induced Diels–Alder cycloaddition to assemble the trans-fused 6–7 ring system; (2) a three-component regioselective hetero-Diels–Alder cycloaddition reaction to build the 6–6–6–7 tetracyclic core; (3) aromatization of the α,β-unsaturated six-membered ketone ring; (4) a series of substitution reactions and aromatic Claisen rearrangements to introduce the side chain; and (5) tandem oxidative dearomatization/cyclization to produce the 5–6 oxaspiro moiety. X-ray crystallography studies confirmed the structures of the intermediates and one final diastereoisomer product. The photochemical Diels–Alder reaction was shown to be an unusual trans-selective cycloadditon, which broke the common Diels–Alder reaction rule and a 100% trans-fused product was obtained. This has been completely evidenced, by density functional theory calculations, to be due to the unreactivity of the cis-cycloheptenone in cycloadditon.
Conclusions
In summary, we obtained 16 diastereoisomers and 16 regioisomers of novel skeleton natural product spirooliganin in total with 2.23% overall yield, in which the isomerization derived from the non-selective UV-promoted Diels–Alder cycloaddition and the regioselective thermal hetero-Diels–Alder reaction. The structures of all the synthesized compounds were unambiguously assigned by comprehensive analysis of spectroscopic data, especially 2D NMR, ECD analysis and modified Mosher's method. By such a library construction of the diverse isomers of 1, we were able to investigate the effect of regio and stereo configuration on the bioactivity of the whole structure. Preliminary biological evaluation revealed the essential role of the D-ring 16R,18R,24R, and 26R configuration for their antiviral activity in both diastereoisomers and regioisomers. Spirooliganin 1 and regioisomer 39 have been screened as potent inhibitors against CVB3 for further mechanism study, which will be published in due course. This work not only gives a good example of how to construct a stereoisomers library for complex natural product containing multiple chiral centers for investigating their SSAR, but is also of great benefit to the construction of 6–6–6–7 skeleton compounds in organic synthesis. The unusual and confirmed photo-induced trans-Diels–Alder reaction in this study has certain implications for the synthesis of other analogs with a similar trans-cycloaddition moiety.Author contributions
S.-S. Y. initiated the project. S.-S. Y., S.-G. M., K.·N.·H., J. Q., and R.-B. W. designed the experiments. S.-G. M. isolated the new spirooliganin (1) and elucidated all compounds, and crystallized synthetic intermediate samples and final products. R.-B. W. synthesized all compounds. S.-G. M., R.-B. W., J. Q., Y.-B. L., Y. L., and X.-J. W. performed compound isolation and characterization. K. N. H. and C.·S. J. performed the computational analysis. R.-M. G. and Y.-H. L. performed in vitro experiments. All authors analysed and discussed the results. S.-G. M., R.-B. W., K.·N.·H., J. Q., and S.-S. Y. prepared the manuscript. R.-B. W., S.-G. M., and C.·S. J. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (no. 21732008, 21672261 and 82073739), the CAMS Innovation Fund for Medical Sciences (no. 2016-I2M-3-010), and the National Mega-project for Innovative Drugs (no. 2018ZX09711001-008-001). We are grateful to the Department of Instrumental Analysis of our institute for the acquisition of the IR, NMR, and MS spectra, and to the State Key Laboratory of Natural and Biomimetic Drugs of Peking University and the Instrumental Analysis Center of Beijing University of Chemical Technology for the X-ray crystallographic measurements and analyses.Notes and references
- D. P. Curran, Q. Zhang, C. Richard, H. Lu, V. Gudipati and C. S. Wilcox, J. Am. Chem. Soc., 2006, 128, 9561–9573 CrossRef CAS PubMed.
- S. C. Sinha, A. Sinha, A. Yazbak and E. Keinan, J. Org. Chem., 1996, 61, 7640–7641 CrossRef CAS PubMed.
- C. S. Wilcox, V. Gudipati, H. Lu, S. Turkyilmaz and D. P. Curran, Angew. Chem., Int. Ed., 2005, 44, 6938–6940 CrossRef CAS PubMed.
- S. Das, L.-S. Li, S. Abraham, Z. Chen and S. C. Sinha, J. Org. Chem., 2005, 70, 5922–5931 CrossRef CAS PubMed.
- P. Das, P. Babbar, N. Malhotra, M. Sharma, G. R. Jachak, G. Rajesh, R. G. Gonnade, D. Shanmugam, K. Harlos, M. Yogavel, A. Sharma and D. S. Reddy, J. Med. Chem., 2018, 61, 5664–5678 CrossRef CAS PubMed.
- S. D. Edmondson and S. J. Danishefsky, Angew. Chem., Int. Ed., 1998, 37, 1138–1140 CrossRef CAS PubMed.
- S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147–2155 CrossRef CAS.
- F. V. Nussbaum and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 2175–2178 CrossRef.
- S. Singh, Chem. Rev., 2000, 100, 925–1024 CrossRef CAS PubMed.
- J. D. Moretti, X. Wang and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 7963–7970 CrossRef CAS PubMed.
- D. P. Curran, M. K. Sinha, K. Zhang, J. J. Sabatini and D. H Cho, Nat. Chem., 2012, 4, 124–129 CrossRef CAS PubMed.
- J. Ding and D. G. Hall, Angew. Chem., Int. Ed., 2013, 52, 8069–8073 CrossRef CAS PubMed.
- M. T. Oliveira, M. Luparia, D. Audisio and N. Maulide, Angew. Chem., Int. Ed., 2013, 52, 13149–13152 CrossRef CAS PubMed.
- M. A. Schafroth, G. Zuccarello, S. Krautwald, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 13898–13901 CrossRef CAS PubMed.
- S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed.
- S. G. Ma, W. Z. Tang, Y. X. Liu, Y. C. Hu, S. S. Yu, Y. Zhang, X. G. Chen, J. Qu, J. H. Ren, Y. B. Liu, S. Xu, Y. Y. Liu, Y. Li, H. N. Lü and X. F. Wu, Phytochemistry, 2011, 72, 115–125 CrossRef CAS PubMed.
- W. Z. Tang, S. G. Ma, J. Qu, S. S. Yu, Y. B. Liu, D. M. Su and J. Liu, J. Nat. Prod., 2011, 74, 1268–1271 CrossRef CAS PubMed.
- S. G. Ma, Y. Gao, H. Q. Wang, L. Li, Y. B. Liu, J. Qu, Y. Li, S. Xu, H. N. Lv, Y. H. Li and S. S. Yu, Tetrahedron, 2016, 72, 3003–3013 CrossRef CAS.
- S. G. Ma, R. M. Gao, Y. H. Li, J. D. Jiang, N. B. Gong, L. Li, Y. Lü, W. Z. Tang, Y. B. Liu, J. Qu, H. N. Lü, Y. Li and S. S. Yu, Org. Lett., 2013, 15, 4450–4453 CrossRef CAS PubMed.
- S. K. Chen, H. Li and P. Y. Chen, Flora of China, Science Press, Beijing, 1996, vol. 30, p. 224 Search PubMed.
- L. Y. Song, H. L. Yao and R. B. Tong, Org. Lett., 2014, 16, 3740–3743 CrossRef CAS PubMed.
- K. Takao, S. Noguchi, S. Sakamoto, M. Kimura, K. Yoshida and K. Tadano, J. Am. Chem. Soc., 2015, 137, 15971–15977 CrossRef CAS PubMed.
- N. Zhao, X. D. Ren, J. H. Ren, H. N. Lü, S. G. Ma, R. M. Gao, Y. H. Li, S. Xu, L. Li and S. S. Yu, Org. Lett., 2015, 17, 3118–3121 CrossRef CAS PubMed.
- M. T. Allingham, E. L. Bennett, D. H. Davies, P. M. Harper, A. Howard-Jones, Y. T. H. Mehdar, P. J. Murphy, D. A. Thomas, P. W. R. Caulkett, D. Potter, C. M. Lam and A. C. O'Donoghue, Tetrahedron, 2016, 72, 496–503 CrossRef CAS.
- H. M. L. Davies, Ø. Loe and D. G. Stafford, Org. Lett., 2005, 7, 5561–5563 CrossRef CAS PubMed.
- J. Nikolai, Ø. Loe, P. M. Dominiak, O. O. Gerlitz, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 10763–10772 CrossRef CAS PubMed.
- L. Wei, M. X. Xiao and Z. X. Xie, Org. Lett., 2014, 16, 2784–2786 CrossRef CAS PubMed.
- J. W. Zhang, Q. Q. Jiang, D. J. Yang, X. M. Zhao, Y. L. Dong and R. H. Liu, Chem. Sci., 2015, 6, 4674–4680 RSC.
- Y. F. Liang, S. Song, L. S. Ai, X. W. Li and N. Jiao, Green Chem., 2016, 18, 6462–6467 RSC.
- F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS PubMed.
- J. A. Marshall, Acc. Chem. Res., 1969, 2, 33–40 CrossRef CAS.
- K. N. Houk, L. N. Domelsmith, R. W. Strozier and R. T. Patterson, J. Am. Chem. Soc., 1978, 100, 6531–6533 CrossRef CAS.
- R. W. Strozier, P. Caramella and K. N. Houk, J. Am. Chem. Soc., 1979, 101, 1340–1343 CrossRef CAS.
- Y. X. Wang, Y. H. Li, Y. H. Li, R. M. Gao, H. Q. Wang, Y. X. Liu, L. M. Gao, Q. N. Lu, J. D. Jiang and D. Q. Song, Bioorg. Med. Chem. Lett., 2011, 21, 5787–5790 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2009862, 2009865, 2009868, 2009871 and 2009873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01277k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |