
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolding-controlled assembly of ortho-phenylene-based macrocycles†
Viraj C.
Kirinda
 and
C. Scott
Hartley
*
and
C. Scott
Hartley
*
Department of Chemistry & Biochemistry, Miami University, Oxford, OH 45056, USA. E-mail: scott.hartley@miamioh.edu
First published on 14th April 2021
Abstract
The self-assembly of foldamers into macrocycles is a simple approach to non-biological higher-order structure. Previous work on the co-assembly of ortho-phenylene foldamers with rod-shaped linkers has shown that folding and self-assembly affect each other; that is, the combination leads to new emergent behavior, such as access to otherwise unfavorable folding states. To this point this relationship has been passive. Here, we demonstrate control of self-assembly by manipulating the foldamers' conformational energy surfaces. A series of o-phenylene decamers and octamers have been assembled into macrocycles using imine condensation. Product distributions were analyzed by gel-permeation chromatography and molecular geometries extracted from a combination of NMR spectroscopy and computational chemistry. The assembly of o-phenylene decamers functionalized with alkoxy groups or hydrogens gives both [2 + 2] and [3 + 3] macrocycles. The mixture results from a subtle balance of entropic and enthalpic effects in these systems: the smaller [2 + 2] macrocycles are entropically favored but require the oligomer to misfold, whereas a perfectly folded decamer fits well within the larger [3 + 3] macrocycle that is entropically disfavored. Changing the substituents to fluoro groups, however, shifts assembly quantitatively to the [3 + 3] macrocycle products, even though the structural changes are well-removed from the functional groups directly participating in bond formation. The electron-withdrawing groups favor folding in these systems by strengthening arene–arene stacking interactions, increasing the enthalpic penalty to misfolding. The architectural changes are substantial even though the chemical perturbation is small: analogous o-phenylene octamers do not fit within macrocycles when perfectly folded, and quantitatively misfold to give small macrocycles regardless of substitution. Taken together, these results represent both a high level of structural control in structurally complex foldamer systems and the demonstration of large-amplitude structural changes as a consequence of a small structural effects.
Introduction
Nature's structurally complex, folded biomacromolecules have inspired decades of development of non-biological foldamers.1–7 This work has now yielded many elegant examples of functional systems with well-defined secondary structures.8–14 However, the added complexity of higher-order structure is key to biological function.3 Synthetic tertiary structure remains rare, especially in systems that are structurally dissimilar from biomacromolecules,15,16 and we presently lack design principles for even simple versions of higher-order structure. Beyond structure itself, control of dynamics is needed to replicate the many examples of biochemical machinery that undergo large-scale structural changes as part of their function.5The challenge then is to control the placement of discrete folded structures in space. Arguably the simplest approach to this problem is to confine folded segments within a ring, restricting their motion. This intersection of foldamer and macrocycle chemistry has been reasonably well-explored; for example, macrocycles have been reported that are themselves folded,17–23 as have macrocycles comprising a single foldamer moiety with its ends connected via unfolded segments.24,25 It is less common to construct rings from multiple discrete foldamer subunits, but there are examples: Huc has demonstrated homochiral self-sorting of folded segments within macrocycles,26 as has Jiang,27 and structurally intricate, foldamer-based coordination complexes have been reported by Sawada and Fujita.28,29
Recent work from our group30–32 has focused on the self-assembly of foldamer monomers into macrocycles via dynamic covalent chemistry,33,34 as shown in Fig. 1a. In particular, we are interested in how folding affects self-assembly and vice versa: confinement within a cyclic structure imposes conformational constraints on the foldamer subunits, affecting their geometries; at the same time, the (dynamic) geometric preferences of the foldamer ultimately dictate the size and shape of the product that is obtained. The interplay between these effects leads to new emergent behavior, such as new folding patterns and complex dynamics.22,30,31
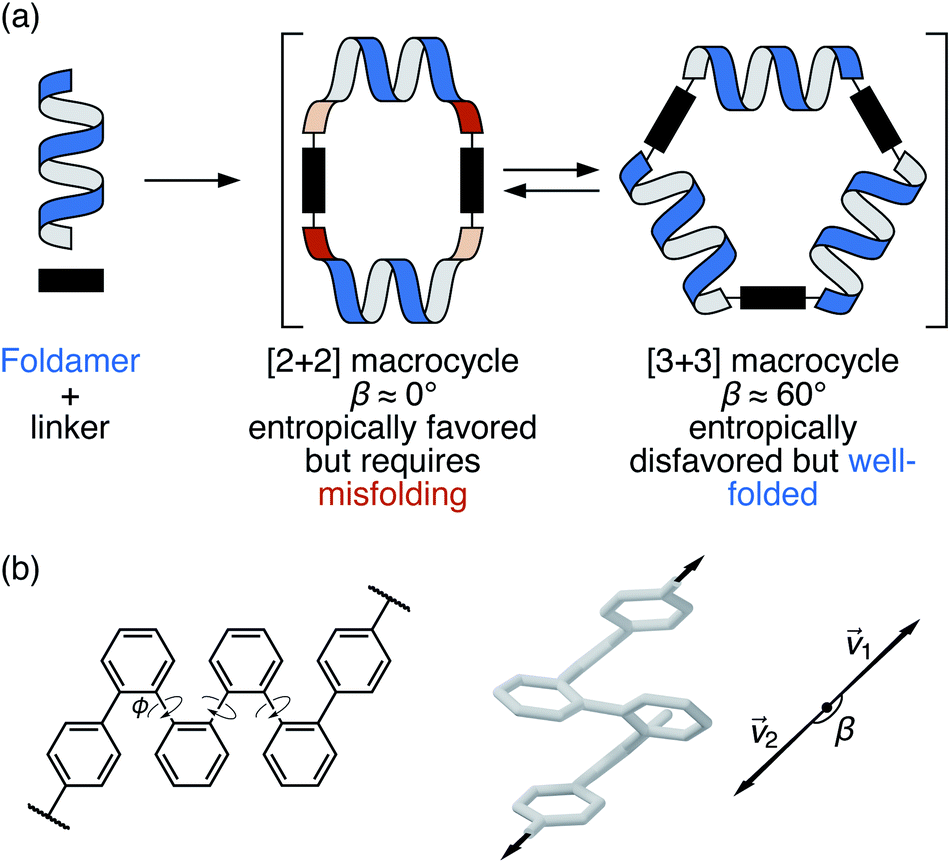 | ||
| Fig. 1 (a) Assembly of foldamers and linkers into [2 + 2] and [3 + 3] macrocycles. (b) Folding and bite angle (β) of a terminally functionalized o-phenylene hexamer. | ||
Our chosen foldamer system is the o-phenylenes, a simple class of helical aromatic foldamers.35–37 In solution, o-phenylenes fold into helical geometries (Fig. 1b) driven by arene–arene stacking between every third repeat unit. They are particularly well-suited to study within larger architectures: their conformational behavior is relatively simple and there is a close, predictable relationship between their NMR properties and their geometries, which allows their folding state to be determined in solution. Importantly, while o-phenylenes fold well, their folding propensity is tunable and small enough to be perturbed by external influences. This latter property is critical for achieving complex dynamic behavior: large-amplitude motions in biomolecules require distinct, energetically accessible folding states.38,39
Previous work has shown that amino-terminated o-phenylene tetramers, hexamers, and decamers can be assembled with dialdehyde linkers to give [2 + 2] and [3 + 3] macrocycles (Fig. 1a).30,31,40 For a particular length of o-phenylene, the size of the product macrocycle and the folding state are related via the foldamer's bite angle β, the angle made by the terminal connection points (β = arccos(![[small nu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0ea.gif) 1 × 2) , Fig. 1b).30 That is, only certain bite angles fit within certain macrocycle sizes. Quantifying the fit in this way is clearly a simplification, since the relationships between three-dimensional objects are being reduced to single numbers; nevertheless, the approximation works well for these systems, and is consistent with the observation of generally low (albeit nonzero) chiral “communication” between o-phenylene subunits.
1 × 2) , Fig. 1b).30 That is, only certain bite angles fit within certain macrocycle sizes. Quantifying the fit in this way is clearly a simplification, since the relationships between three-dimensional objects are being reduced to single numbers; nevertheless, the approximation works well for these systems, and is consistent with the observation of generally low (albeit nonzero) chiral “communication” between o-phenylene subunits.
The relationship between β and macrocycle architecture creates a tension: at equilibrium, entropy favors the formation of the smallest macrocycle,41 but for suitably chosen o-phenylenes there is an enthalpic penalty if the oligomer must unfold to fit. Up to now, this relationship has been passive: at certain oligomer lengths multiple products of assembly are obtained, showing that there is a delicate balance between entropic and enthalpic favorability, but there has been no element of control. Here, we show that modulating the folding propensity of the o-phenylene gives different outcomes of self-assembly. We tune the folding of the o-phenylenes through substituent effects,42 and, as a consequence, obtain differently sized macrocycles with very different folding patterns. While this system is not itself actively responsive, it demonstrates that small chemical perturbations can lead to dramatic structural changes in nonbiological foldamer systems.
Results and discussion
Oligomer design
We first briefly summarize the folding behavior of o-phenylenes.36 The folding state of an o-phenylene [n]-mer is defined by the n – 3 internal biaryl dihedral angles ϕ (Fig. 1b). “Perfect”37 folding corresponds to a compact helix that maximizes aromatic stacking. In this state, all the dihedral angles are ϕ ≈ −55° (left-handed helix) or ϕ ≈ +55° (right-handed helix), which we call the A or A′ states, respectively (e.g., a perfectly folded left-handed o-phenylene decamer is in the “AAAAAAA”, or “A7”, conformation). Misfolds correspond to dihedral angles of ϕ ≈ +135° or ϕ ≈ −135°, called the B or B′ states. Each misfold disrupts one arene–arene stacking interaction. Beyond this simple description, the folding is governed by two rules: within a single o-phenylene molecule, only A and B states can coexist (likewise A′ and B′), and “ABA” sequences are forbidden because of a steric clash.This model is idealized, but very useful for discussing and predicting the properties of o-phenylenes. To understand their fit within macrocycles, the relative energies ΔE of different folding states and their associated β can be computationally predicted for all possible conformers. The resulting plots are shown in Fig. 2 for (unsubstituted) octa(o-phenylene) and (previously reported30) deca(o-phenylene), which are of greatest relevance to this study. For example, a [3 + 3] macrocycle is quasi-triangular and requires approximately 60° angles at its corners.43 As shown in Fig. 2b, the AAAAAAA conformer of deca(o-phenylene) should be the best fit: it is both the most stable and has β = 69°, very close to the optimum value. Most of the other folding states are both less stable and have larger β and so would be disfavored under all conditions. However, some misfolded conformers (e.g., AABBAAB, BAAAAAB, AAABBBA, AAAABBB, ABBAAAB, etc.) have smaller β and can therefore potentially fit into [2 + 2] macrocycles, which require β ≈ 0°. In these cases, the entropic preference for the smaller macrocycles41,44 may be sufficient to compensate for the enthalpic penalty of misfolding so long as the differences in stability are not too large.
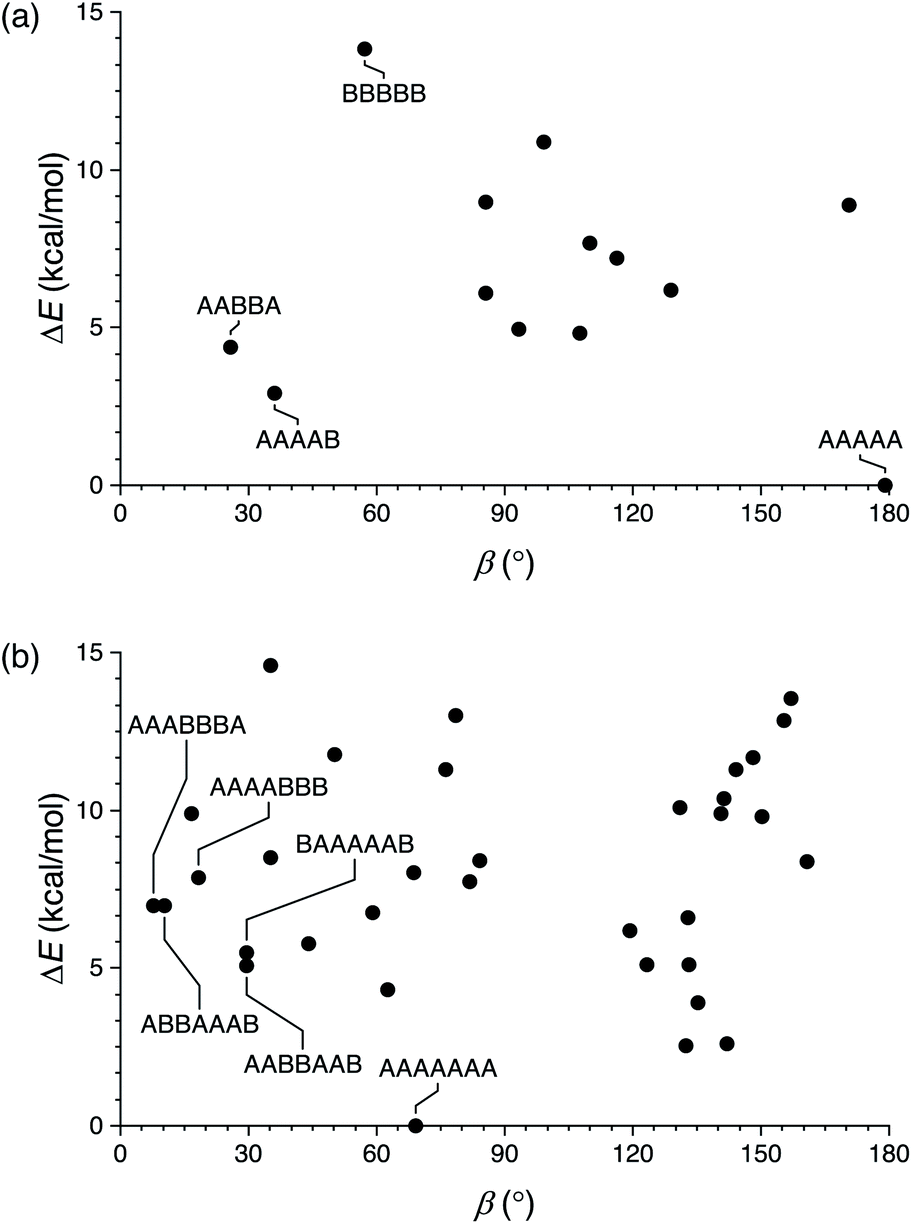 | ||
| Fig. 2 Calculated relative energy vs. bite angle for all conformers of (a) octa(o-phenylene) and (b) deca(o-phenylene) (B97-D/cc-pVDZ). | ||
The predictions of these energy surfaces are highly dependent on o-phenylene length. For octa(o-phenylene) (Fig. 2a), the perfectly folded conformer, although it is energetically favorable, has a bite angle of 179° and thus cannot form the corners of a reasonably sized macrocycle. The best fit to a [3 + 3] macrocycle is the completely unfolded BBBBB folding state (β = 57°), but it is also the least stable (ΔE ≈ 13 kcal mol−1). Some misfolded conformers, such as AABBA and AAAAB, are both good fits to smaller macrocycles and sufficiently stable that they should be energetically accessible.45
The shapes of these conformational energy profiles should be controllable through modulation of the aromatic stacking interactions within the foldamer which, to a first approximation, should affect ΔE without substantially changing ϕ. It is well-known, both experimentally and computationally, that arene–arene interactions are sensitive to substituent effects, as measured, for example, by Hammett substituent constants σ.46–49 Accordingly, the folding propensity of o-phenylenes shows a linear correlation with σm of the substituents.42
In this work, we examine the assembly and properties of macrocycles comprising the o-phenylene decamers and octamers in Chart 1, paired with linkers Phen and DPB. Compound oP10H(NH2) was previously used in assembly experiments and serves as a useful reference point.30 The substituents (H, F, and OMe) give reasonably good coverage of Hammett values while being synthetically accessible. Hexyloxy groups were included on most oligomers in order to ensure solubility; for oP10OMe(NH2), they complicated the synthesis (see ESI, Scheme S1†), and so the fully methoxylated oligomer was used instead.
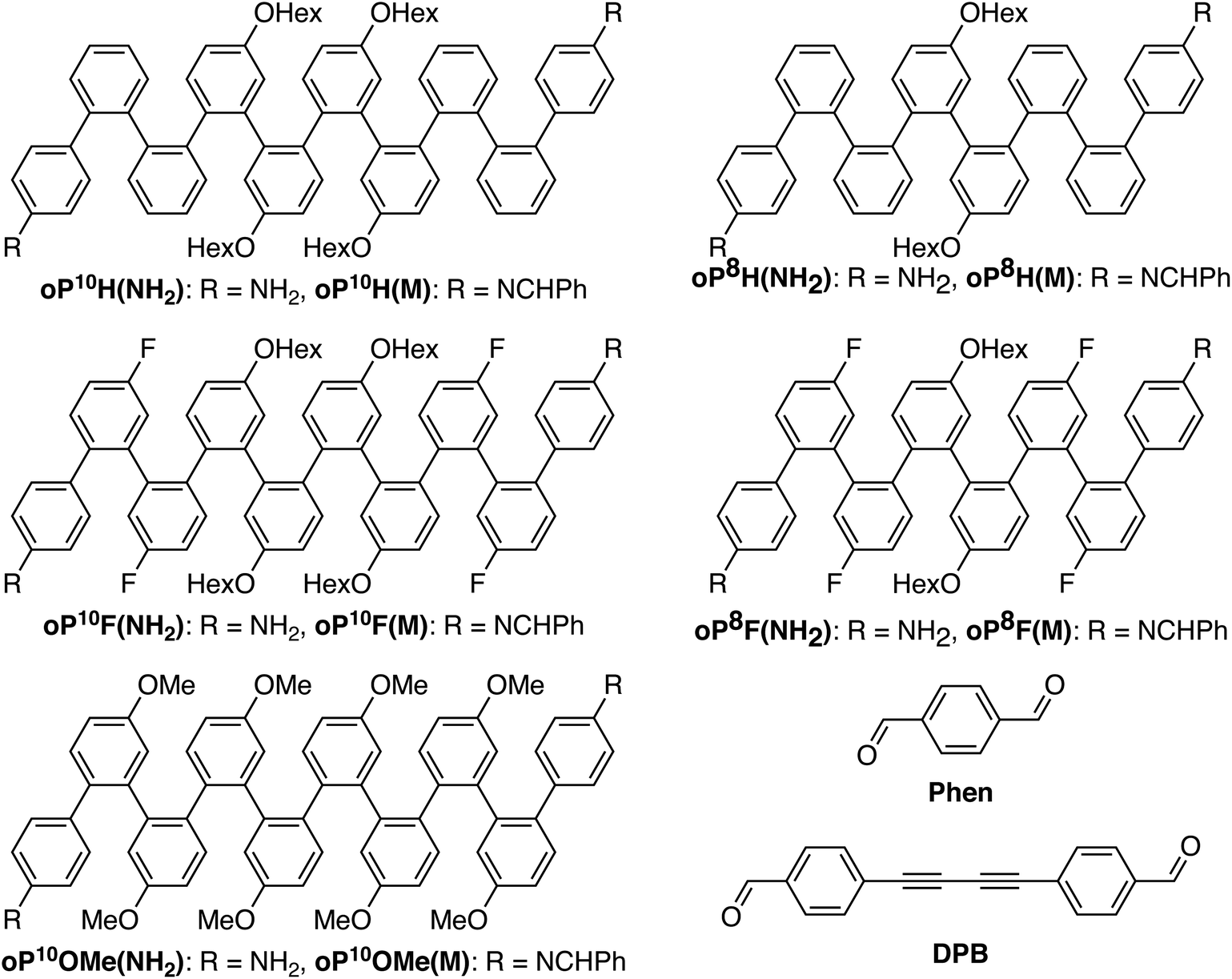 | ||
| Chart 1 o-Phenylene oligomers, acyclic model compounds, and linkers. | ||
Assembly of o-phenylene decamers
Conditions for assembly were based on the previous work with oP10H(NH2).30 Compounds oP10F(NH2) or oP10OMe(NH2) (1.1 equiv.) were mixed with Phen or DPB (1.0 equiv., 1.5 mM) in chloroform with TFA (0.1 equiv.) and 3 Å molecular sieves. The reactions were monitored by 1H NMR spectroscopy, reaching steady states after 6 d for oP10F(NH2) and 9 d for oP10OMe(NH2). For consistency with oP10H(NH2), the reactions were allowed to react for 11 d total before being quenched with triethylamine. Although self-catalysis of transimination is possible by the excess amines remaining after the quench,50,51 we have previously found this to be insignificant under these conditions.31 The quenched reaction mixtures were then analyzed by analytical GPC (see Fig. 3; the previously reported oP10H(NH2) system was retested to ensure accurate comparisons). Peaks corresponding to the [2 + 2] and [3 + 3] macrocycles were identified by subsequent isolation and characterization by mass spectrometry and NMR spectroscopy (see below).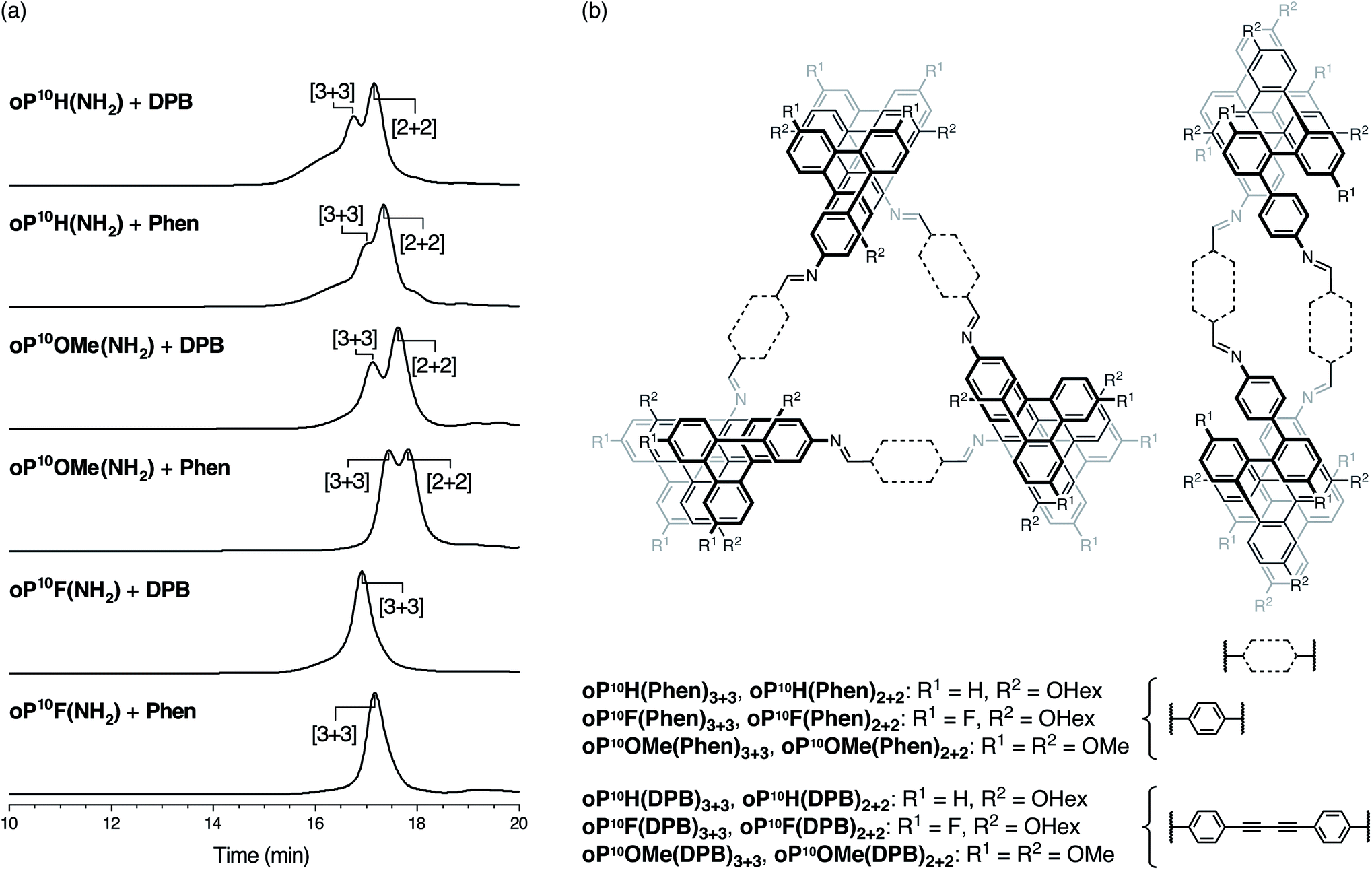 | ||
| Fig. 3 (a) GPC traces for the assembly of o-phenylene decamers with linkers DPB and Phen. (b) Structures of the [3 + 3] and [2 + 2] macrocycle products. | ||
The new, substituted o-phenylenes assemble cleanly into macrocycles. Close inspection of the GPC chromatograms in Fig. 3a reveals broad peaks at lower retention times for the previously reported assembly of oP10H(NH2) with both linkers, indicating a significant amount of higher-molecular-weight byproducts. These are reduced or absent for assembly of oP10OMe(NH2) and oP10F(NH2). With oP10OMe(NH2), two main peaks were observed in the GPC chromatograms of the crude products, corresponding to the [3 + 3] macrocycles oP10OMe(DPB)3+3 or oP10OMe(Phen)3+3 and [2 + 2] macrocycles oP10OMe(DPB)2+2 or oP10OMe(Phen)2+2. With the DPB-based linker, a molar ratio of oP10OMe(DPB)3+3 to oP10OMe(DPB)2+2 of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 was obtained (based on the deconvoluted refractive index detector response), similar to the oP10H(NH2) system. The behavior with the Phen-based linker is slightly different, with more oP10OMe(Phen)3+3 obtained relative to oP10OMe(Phen)2+2 (roughly 2:3 molar ratio).
:7 was obtained (based on the deconvoluted refractive index detector response), similar to the oP10H(NH2) system. The behavior with the Phen-based linker is slightly different, with more oP10OMe(Phen)3+3 obtained relative to oP10OMe(Phen)2+2 (roughly 2:3 molar ratio).
In contrast, the products of assembly of oP10F(NH2) with both linkers gave only single peaks in the chromatograms, corresponding to the macrocycles oP10F(DPB)3+3 and oP10F(Phen)3+3. No lower-molecular-weight species, including the [2 + 2] macrocycle, were detectable.
The methoxy-substituted macrocycles were isolated by multiple runs of semi-preparative GPC in 24% and 18% yields for oP10OMe(DPB)2+2 and oP10OMe(DPB)3+3, and 19% and 36% yields for oP10OMe(Phen)2+2 and oP10OMe(Phen)3+3. Comparatively easy purification resulted in high isolated yields of 92% and 98% for the fluoro-substituted macrocycles oP10F(DPB)3+3 and oP10F(Phen)3+3.
Structural analysis of deca(o-phenylenes)
Unfortunately, attempts to grow crystals suitable for crystallography have so far been unsuccessful in these systems; our analysis of the folding of the o-phenylenes within the macrocycles therefore relies on their useful NMR properties. o-Phenylene backbones are in slow conformational exchange on the NMR time scale at (or below) room temperature;36,52 that is, all backbone geometries are observed separately in the same 1H NMR spectrum, and the chemical shifts can be assigned using standard 2D NMR spectroscopy methods (COSY, TOCSY, HSQC, HMBC, NOESY/EXSY). The chemical shifts are highly geometry-sensitive and quantitatively predictable using computational methods, allowing detailed information about the folding state of the o-phenylenes to be extracted from the NMR data. In principle, the strategy is simple: isotropic shieldings (σ) are predicted for every possible o-phenylene backbone conformation (determined using the model described above) and then simply compared to the experimental chemical shifts. This approach works well for acyclic o-phenylenes in isolation,36 but for longer o-phenylenes embedded within larger architectures it quickly becomes impractical. For example, o-phenylene decamers have 37 possible backbone geometries that would have to be considered for every macrocycle configuration (e.g., homo-, heterochiral) and combination of substituents (including the possibility of multiple orientations of polyatomic substituents).To simplify the NMR analysis, we have found it useful to calculate chemical shift differences (Δδ) between the o-phenylenes within the macrocycles and acyclic model compounds for which the dominant conformation is easily established (and generally the perfectly folded AA…A conformer).30 Thus, for a proton Hn:
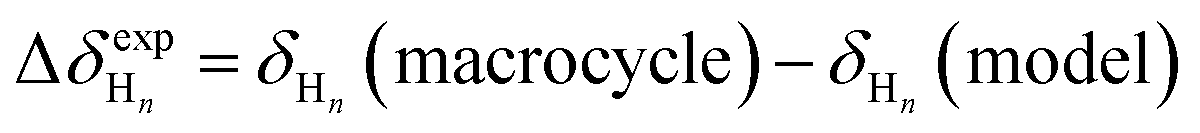 | (1) |
The resulting set of Δδ values is characteristic of changes in folding state as protons move in and out of the (de)shielding zones of nearby aromatic rings. Importantly, substituent effects on the chemical shifts approximately cancel out. This allows for comparison of experimental data to computational libraries of unsubstituted o-phenylenes. These calculations are as simple as possible and a new library does not need to be generated for every substitution pattern. For example, assuming the model compound was (experimentally) determined to be in the AA…A conformer, then for proton Hn in the conformer I:
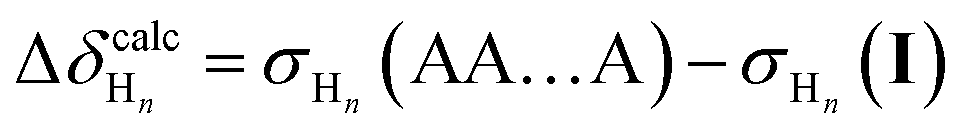 | (2) |
The pattern of Δδ is a fingerprint, characteristic of a folding state, so the set of Δδexp is then matched to Δδcalc. If the o-phenylene is in the same folding state in both the macrocycle and model, the similar proton environments will give all Δδexp close to 0 (and all Δδcalc exactly 0). If, however, the folding states are different, there will be significant chemical shift deviations (on the order of 1 ppm for key protons). For the o-phenylene decamer macrocycles considered here, we use a library of Δδcalc predicted for deca(o-phenylene) at the GIAO/PCM(CHCl3)/WP04/6-31G(d)//B97-D/cc-pVDZ level that was previously reported.30
Model compounds oP10F(M) and oP10OMe(M), shown in Chart 1, were prepared by reacting oP10F(NH2) and oP10OMe(NH2) with benzaldehyde. Their 1H NMR spectra are shown in Fig. 4 (CDCl3 at 0 °C (ref. 53)). The spectra are typical of o-phenylenes, with dominant signals from the major conformer complicated by small signals from misfolding.54 The signals corresponding to the major conformers are easily distinguished and were assigned using 2D NMR methods. Computational models for possible geometries of oP10F(M) and oP10OMe(M) were then optimized at the PCM(CHCl3)/B97-D/cc-pVDZ level and their corresponding isotropic magnetic shielding values were calculated at the PCM(CHCl3)/WP04/6-31G(d) level.55,56 Comparisons of the NMR predictions and the experimental data confirm that the major conformers adopted by both oP10F(M) and oP10OMe(M) are the perfectly folded AAAAAAA geometry (see ESI, Fig. S3†).
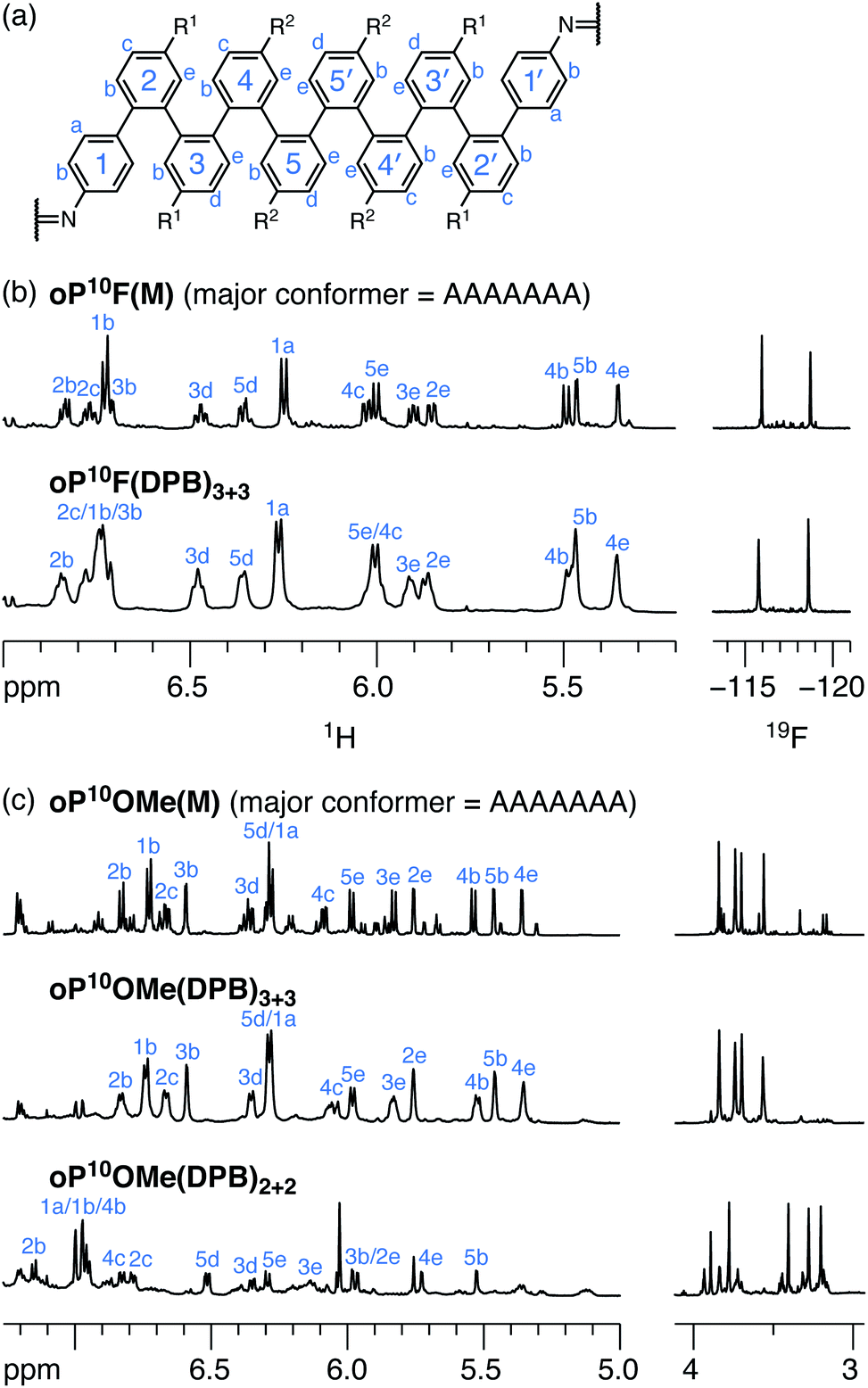 | ||
| Fig. 4 (a) Labeling scheme used for NMR assignments of o-phenylene decamers. (b) 1H NMR spectra (600 MHz, CDCl3, 0 °C) and 19F NMR spectra (188 MHz, CDCl3, rt) of oP10F(M) and oP10F(DPB)3+3. (c) 1H NMR spectra (600 MHz, CDCl3, 0 °C) of oP10OMe(M), oP10OMe(DPB)3+3, and oP10OMe(DPB)2+2. In all spectra, 1H assignments corresponding to the major conformation are labeled. | ||
As expected from the substituent effects on arene–arene stacking,42 the fluoro-substituted model oP10F(M) is well-folded into the AAAAAAA conformer (roughly 73% of the total population based on integration of the imine signals), with only small signals corresponding to misfolded states. The AAAAAAA state also predominates for oP10OMe(M) (roughly 53% the total intensity), but, as is clear from the eight distinguishable methoxy-group signals, a low-symmetry misfolded state is also significant, very likely the AAAAAAB state.
The 1H NMR spectra of macrocycles oP10F(DPB)3+3 and oP10OMe(DPB)3+3 are shown in Fig. 4 (CDCl3 at 0 °C). As previously observed,30,31 one of the advantages of the diarylbutadiyne linker is that it is sufficiently flexible57 that diastereomers resulting from different relative configurations of the o-phenylenes are indistinguishable by NMR spectroscopy, giving more easily interpreted spectra. That is, the spectra only provide information on the local folding of the o-phenylene moieties. Qualitatively, the spectra of the [3 + 3] macrocycles are very similar to those of the model compounds oP10F(M) and oP10OMe(M), suggesting that the o-phenylenes adopt the same folding state in both cases. Quantitatively, the Δδexp are very near 0 for all protons, as shown in Fig. 5a for oP10OMe(DPB)3+3. Unsurprisingly then, the sets of Δδexp values give the best match to Δδcalc for the AAAAAAA conformer from the deca(o-phenylene) library (where Δδcalc = 0 for all protons), with RMSDs of only 0.04 and 0.03 ppm for oP10F(DPB)3+3 and oP10OMe(DPB)3+3, respectively. As shown in Fig. 5b, these RMSDs are much lower than all other possible comparisons; the matches are significantly better than that to the next-best cases (AAAAAAB for both systems) at the >99% confidence level.
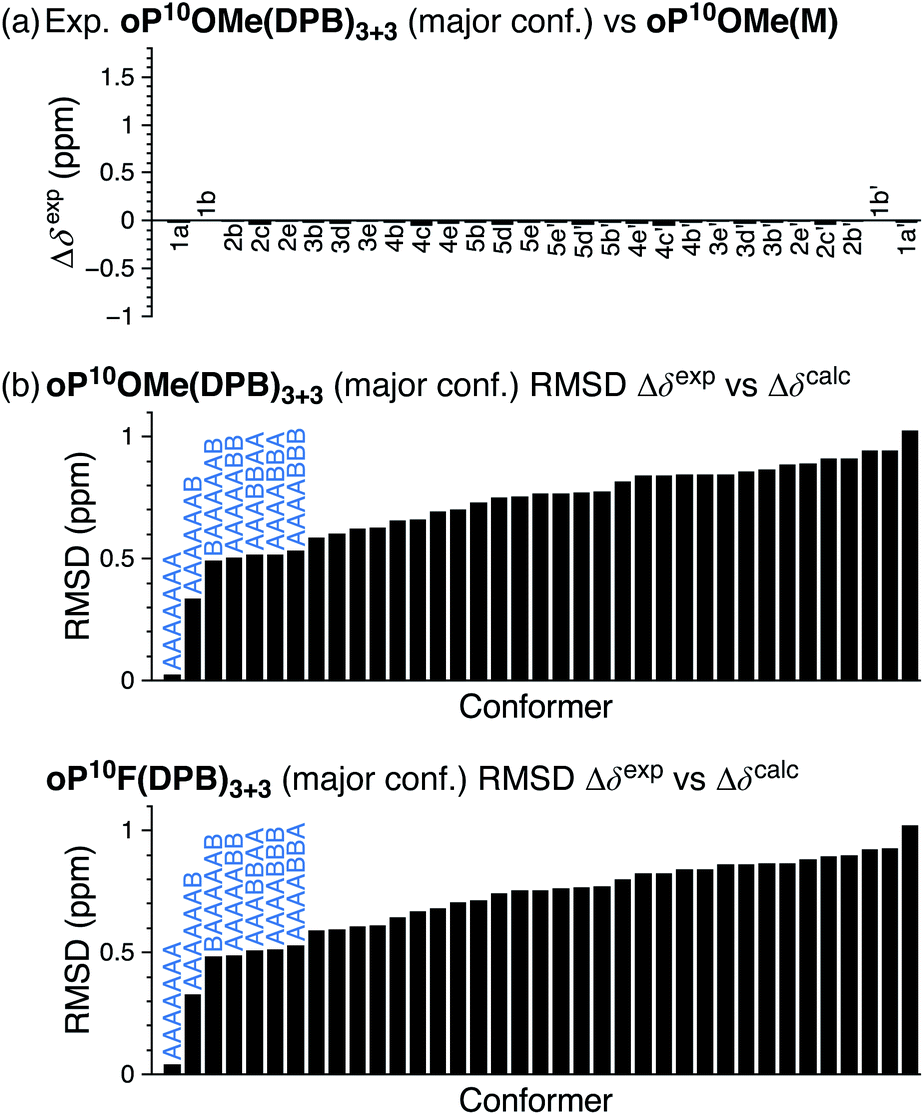 | ||
| Fig. 5 (a) Experimental Δδexp for oP10OMe(DPB)3+3 (major conformer) vs.oP10OMe(M) (AAAAAAA conformer); for comparision, the predicted Δδcalc are all exactly 0. (b) RMSD of Δδexpvs. Δδcalc for oP10OMe(DPB)3+3 and oP10F(DPB)3+3 relative to all possible deca(o-phenylene) conformers. | ||
We therefore conclude that the oligomers are folded in the AAAAAAA state within the [3 + 3] macrocycles. Additional signals in the 19F NMR spectrum of oP10F(DPB)3+3 and the methoxy region of the 1H NMR spectrum of oP10OMe(DPB)3+3 support this. The two fluorine peaks for oP10F(DPB)3+3 and four principal methoxy peaks for oP10OMe(DPB)3+3, in both cases with very similar chemical shifts as in the model compounds, indicate that the o-phenylenes are twofold symmetric and well-folded. We also note that the minor peaks resulting from misfolding are substantially suppressed compared to the models, particularly for oP10OMe(DPB)3+3, indicating that while the major conformers are the same, there are subtle differences in folding population.
As expected,30,31 macrocycles incorporating the relatively rigid Phen linker show distinct 1H NMR signals for different diastereomers (see ESI, Fig. S111 and S130†); that is, they capture the overall structure. This is most easily seen in the imine regions of the spectra, which show four prominent imine signals for both oP10F(Phen)3+3 and oP10OMe(Phen)3+3. These can be assigned to the homochiral D3-symmetric (1 signal) and heterochiral C2-symmetric (3 signals) diastereomers. The ratio between the D3 and C2 stereoisomers is approximately 1:2 for both macrocycles, implying little stereoselectivity (the statistical ratio based on symmetry would be 1:3).
The folding of the o-phenylenes within the [2 + 2] macrocycles is more complex. The 1H NMR spectrum of oP10OMe(DPB)2+2 is shown in Fig. 4c. There are now clear differences in the chemical shifts relative to those of oP10OMe(M), indicating a change in folding state. The difference is also obvious from the methoxy region of the spectrum, where four clear singlets are observed but with very different chemical shifts from those of the AAAAAAA conformer of oP10OMe(M) (or oP10OMe(DPB)3+3). The 1H NMR signals corresponding to the major conformer of oP10OMe(DPB)2+2 were assigned as before. As shown in Fig. 6a, the Δδexp values are no longer all close to 0, with some chemical shift variations in excess of 1 ppm. Comparison of the set of Δδexp to the Δδcalc from the computational library gave a clear match to the BAAAAAB conformation, with the RMSD values shown in Fig. 6b. The match is better than the next-best possibility (AAAAAAB) at the >99% confidence level.
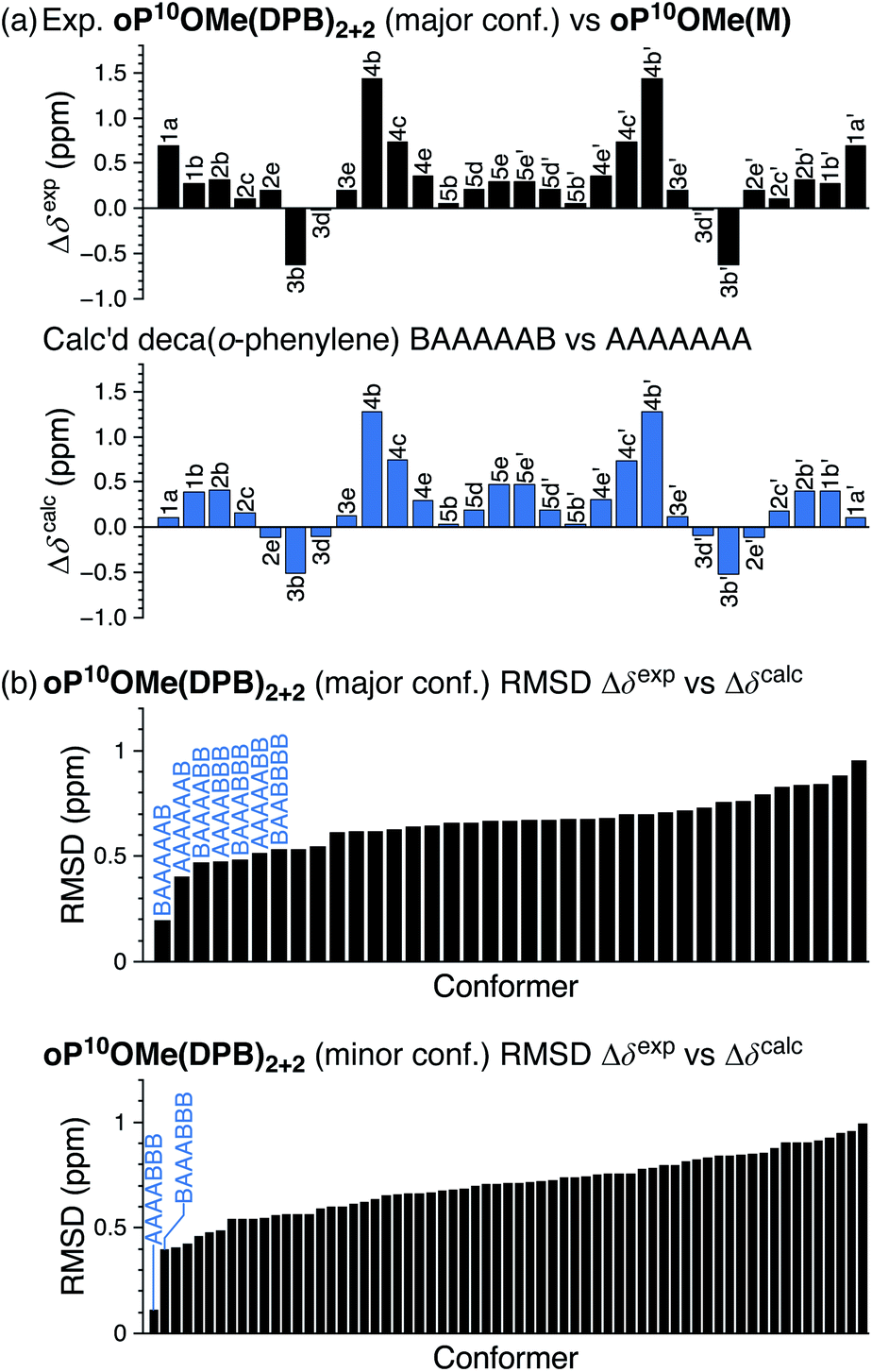 | ||
| Fig. 6 (a) Experimental Δδexp for oP10OMe(DPB)2+2 (major conformer) vs.oP10OMe(M) (AAAAAAA conformer) and predicted Δδcalc for the parent deca(o-phenylene) BAAAAAB vs. AAAAAAA. (b) RMSD of Δδexpvs. Δδcalc for oP10OMe(DPB)2+2 (major conformer) and oP10OMe(DPB)2+2 (minor conformer) relative to all possible deca(o-phenylene) conformers. | ||
Similarly, chemical shifts could also be assigned for second-most populated conformer of oP10OMe(DPB)2+2, which is unsymmetrical (e.g., eight distinct methoxy signals). Analysis of its Δδ values indicates that it corresponds to the AAAABBB geometry (Fig. 6b, significant at the >99% confidence level). This match relies on some assumptions,58 but is reasonable given that the AAAABBB conformation had been previously found to predominate for oP10H(DPB)2+2 (with the BAAAAAB conformer as the minor state).30
While linker effects are not the focus of this work, there is a notable difference in behavior between oP10OMe(Phen)2+2 and oP10OMe(DPB)2+2. In both cases, the BAAAAAB and AAAABBB states of the o-phenylenes are observed. However, the balance is different, favoring the BAAAAAB state for oP10OMe(DPB)2+2 but AAAABBB for oP10OMe(Phen)2+2 (see spectra in ESI†). It is not immediately obvious what is causing this effect, and the differences in stability between the two states must be very small. As was the case for the [3 + 3] macrocycles, the 1H NMR spectrum of oP10OMe(Phen)2+2 is complicated by chemical inequivalence related to the overall macrocyclic structure; in this case, the issue is likely the possibility of different stereoisomers and the presence of parallel/antiparallel configurations involving the unsymmetrical AAAABBB state.
To visualize possible geometries for the macrocycles, unsubstituted versions of the [3 + 3] macrocycle were optimized at the PCM(CHCl3)/B97-D/cc-pVDZ level. At this level of theory, the two stereoisomers are effectively isoenergetic (within roughly 0.1 kcal mol−1), consistent with the observation of both by NMR spectroscopy (with the Phen linker). The geometries are shown in Fig. 7; examples of the previously reported [2 + 2] macrocycle geometries30 are also included for reference. In all cases, the o-phenylene conformations identified experimentally are good fits for the corresponding macrocycle sizes, in good agreement with the predictions in Fig. 2b.
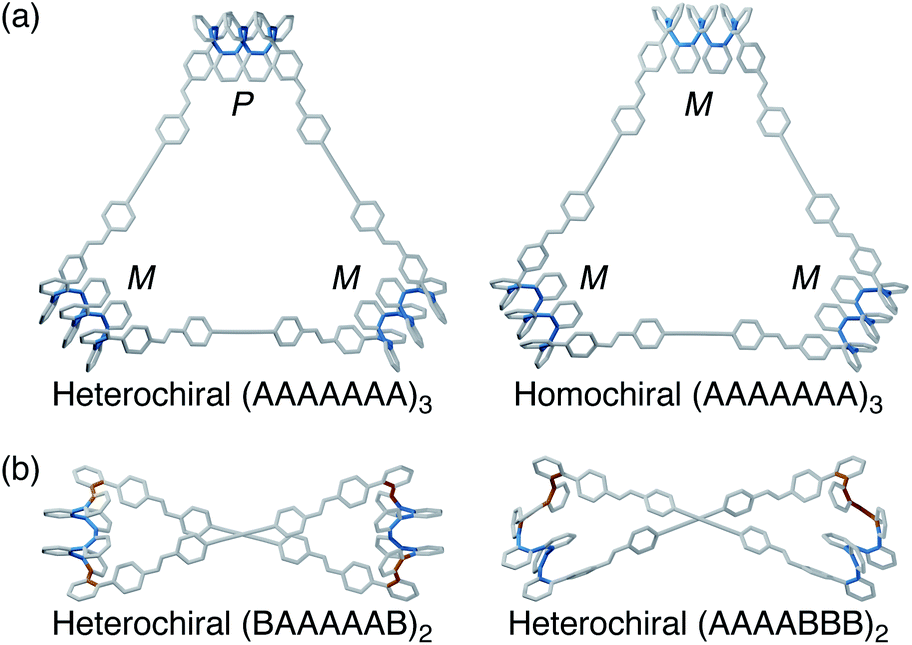 | ||
| Fig. 7 (a) Molecular geometries of heterochiral and homochiral o-phenylene decamer [3 + 3] macrocycles, optimized at the PCM(CHCl3)/B97-D/cc-pVDZ level. (b) Previously reported30 geometries of AAAABBB- and BAAAAAB-containing o-phenylene decamer [2 + 2] macrocycles (PCM(CHCl3)/B97-D/cc-pVDZ). Biaryl bonds in the A state are shown in blue, and those in the B state are shown in orange. | ||
Octa(o-phenylene) assembly
As for the o-phenylene decamers, oP8H(NH2) and oP8F(NH2) were co-assembled with rod-shaped linkers Phen and DPB. Monitoring by 1H NMR spectroscopy showed that the reactions reached a steady-state in approximately 2 d and were quenched after 5 d. The crude products were first analyzed by GPC, shown in Fig. 8a. The chromatograms indicate predominantly a single product in all cases, with no evidence for polymerization or multiple sizes of macrocycles. Analysis by MALDI MS showed that the major products are the [2 + 2] macrocycles (Fig. 8b) regardless of substituents or linker structure. The compounds could be isolated by semi-preparative GPC in 26% and 52% yields for oP8H(DPB)2+2 and oP8H(Phen)2+2, respectively, and 37% and 32% yields for oP8F(DPB)2+2 and oP8F(Phen)2+2 (these yields are lower than the actual yields of assembly because of loss during purification).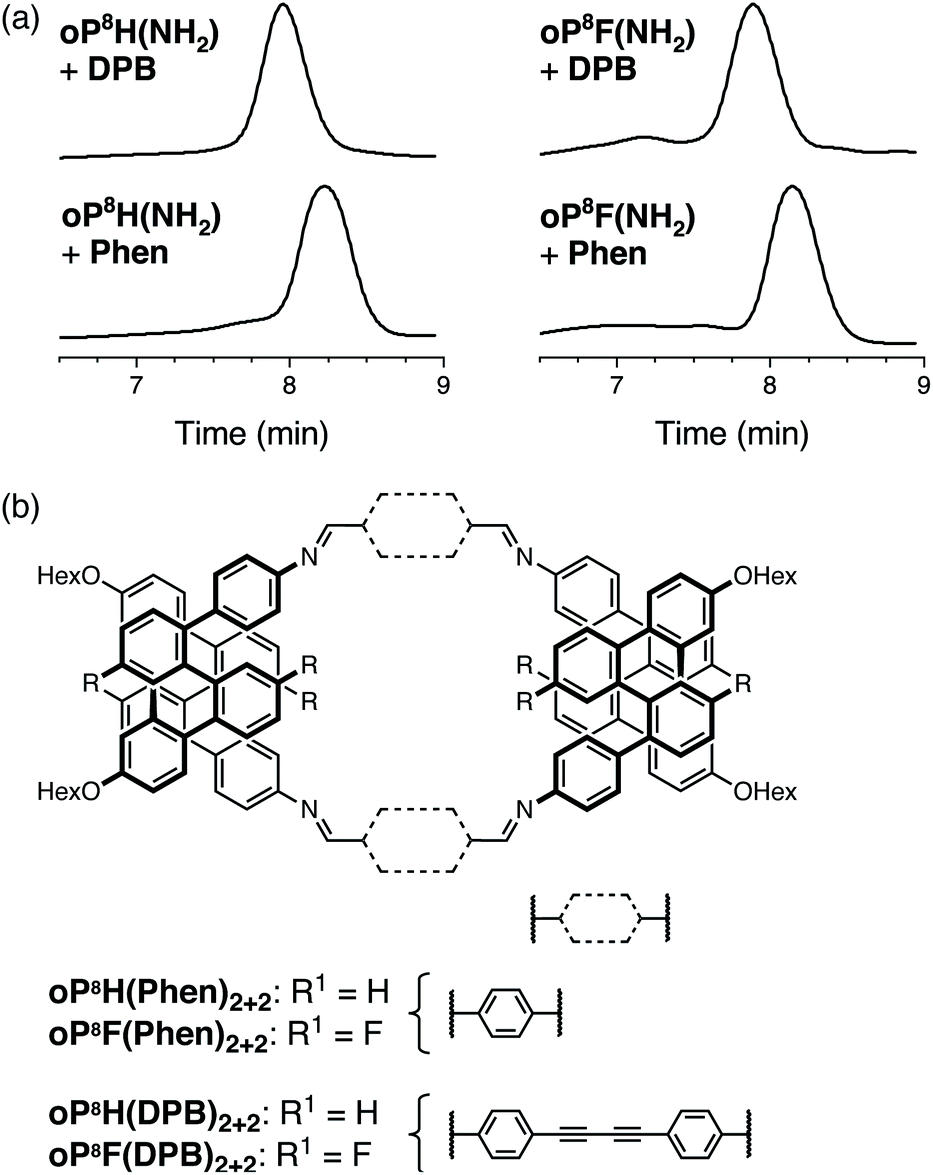 | ||
| Fig. 8 (a) GPC chromatograms of the assembly of o-phenylene octamers with linkers Phen and DPB. (b) Structures of the [2 + 2] macrocycle products. | ||
Acyclic models oP8F(M) and oP8H(M) (Chart 1) were synthesized as models from which to determine the folding of the o-phenylene octamers within the macrocycles. Their 1H NMR spectra are shown in Fig. 9 (CDCl3 at 0 °C). Geometry optimization (PCM(CHCl3)/B97-D/cc-pVDZ) and comparison of the calculated isotropic shieldings (PCM(CHCl3)/WP04/6-31G(d)) confirmed that, like the o-phenylene decamer models, both oP8F(M) and oP8H(M) adopt predominantly the perfectly folded AAAAA conformation. Better folding is observed for the fluoro-substituted system (85% well-folded for oP10F(M)vs. 49% for oP10H(M)).
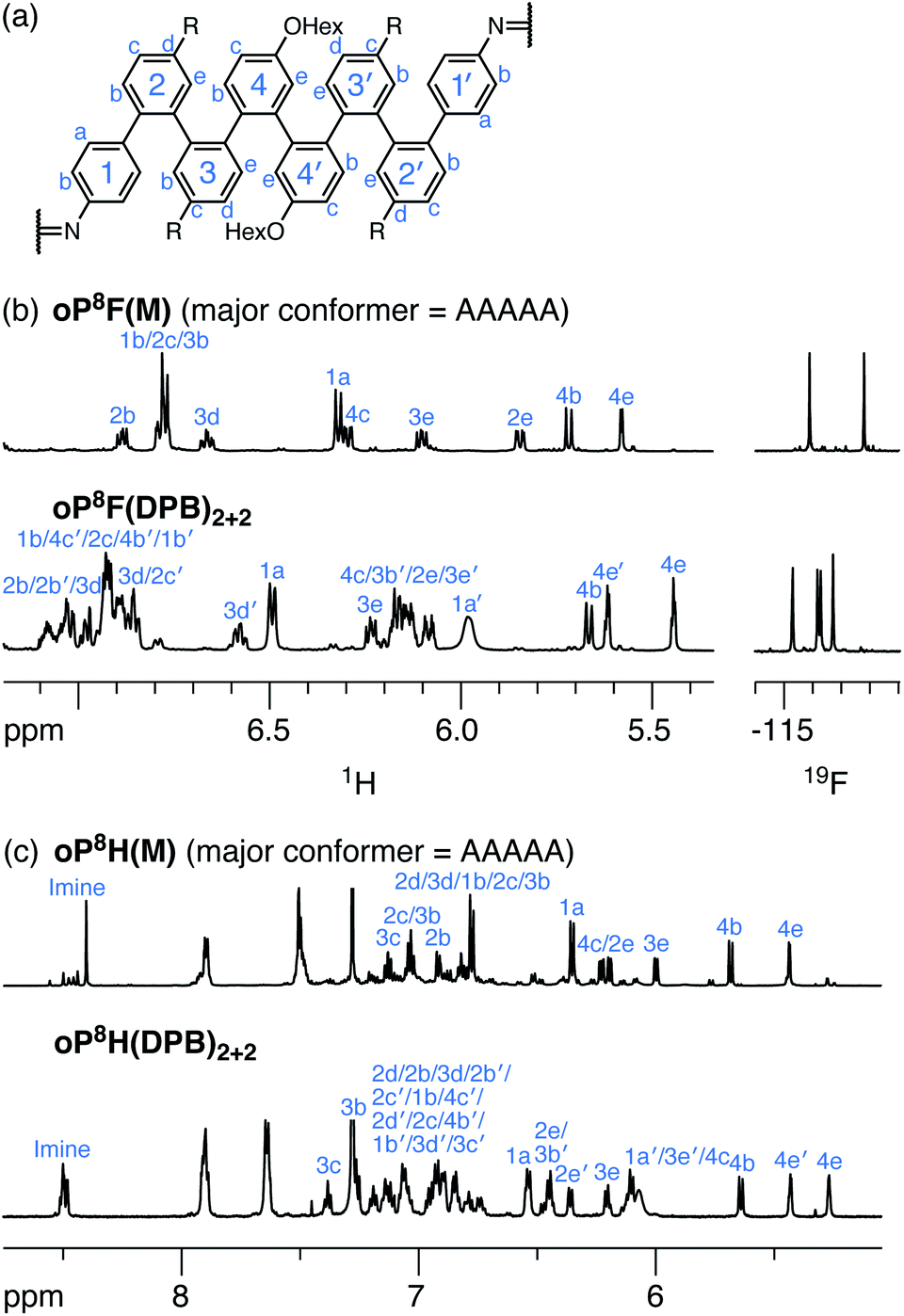 | ||
| Fig. 9 (a) Labeling scheme used for NMR assignments of o-phenylene octamers. (b) 1H NMR spectra (600 MHz, CDCl3, 0 °C) and 19F NMR spectra (188 MHz, CDCl3, rt) of oP8F(M) and oP8F(DPB)2+2. (c) 1H NMR spectra (600 MHz, CDCl3, 0 °C) of oP8H(M) and oP8H(DPB)2+2. 1H assignments corresponding to the major conformation are labeled in each case. | ||
As before, the NMR spectra of macrocycles with DPB-based linkers allow us to focus on the local folding behavior of the o-phenylenes. 1H NMR spectra of macrocycles oP8F(DPB)2+2 and oP8H(DPB)2+2 (CDCl3 at 0 °C) are also shown in Fig. 9. The spectra are significantly different from those of oP8F(M) and oP8H(M), indicating that the o-phenylenes have changed folding states. Analysis shows that the o-phenylenes are primarily in a single unsymmetrical conformation (e.g., the four distinct 19F singlets), with very little evidence of other folding states. Plots of Δδexp were then generated relative to the major conformers of the acyclic models; that of oP8F(DPB)2+2 is shown in Fig. 10. The plots are very similar for both oP8F(DPB)2+2 and oP8H(DPB)2+2, indicating similar behavior in both systems.
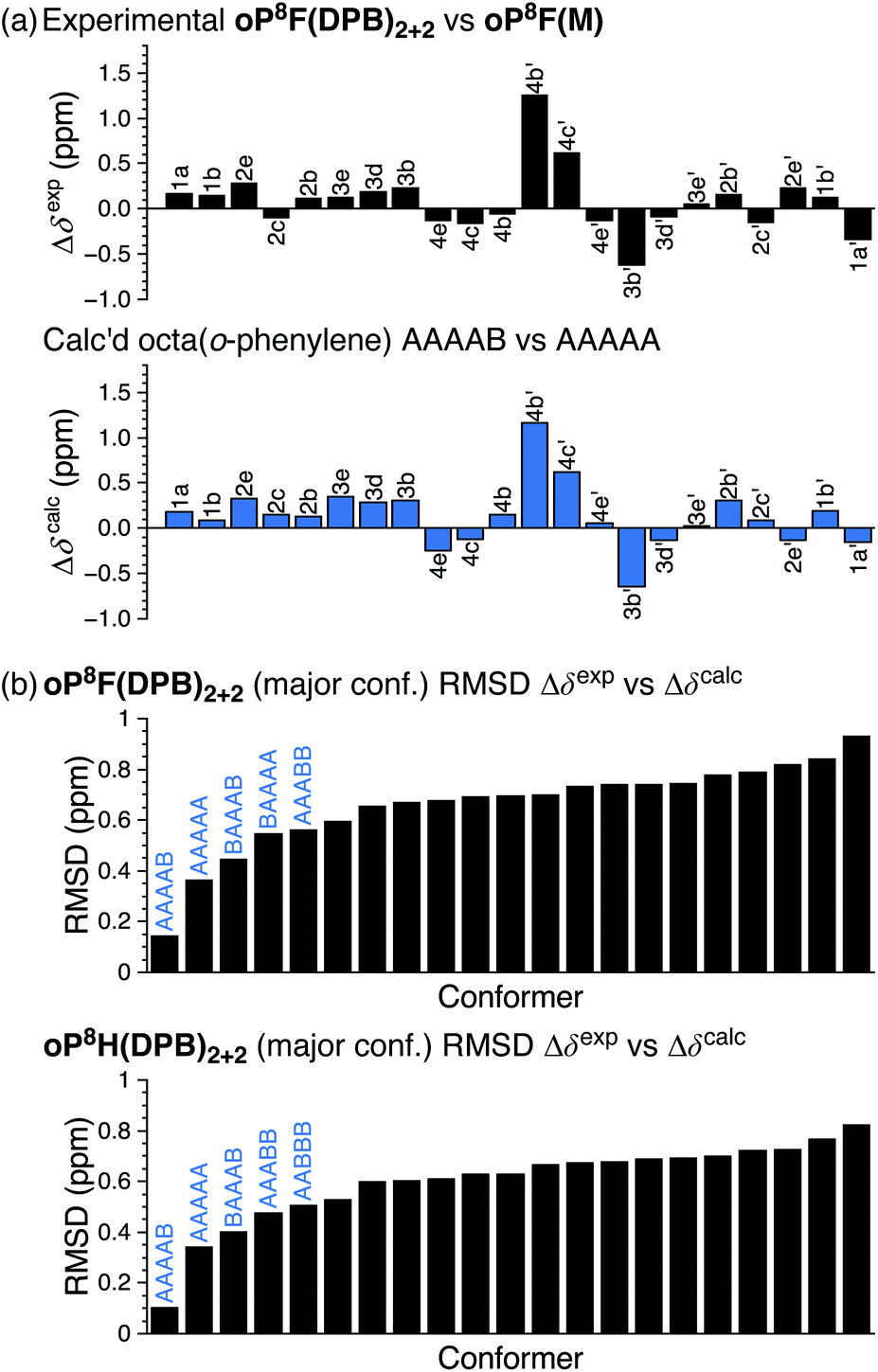 | ||
| Fig. 10 (a) Experimental Δδexp for oP8F(DPB)2+2 (major conformer) vs.oP8F(M) (AAAAA conformer) and predicted Δδcalc for the parent octa(o-phenylene) AAAAB vs. AAAAA. (b) RMSD of Δδexpvs. Δδcalc for oP8F(DPB)2+2 (major conformer) and oP8H(DPB)2+2 (major conformer) relative to all possible octa(o-phenylene) conformers. | ||
A library of all possible conformers of unsubstituted octa(o-phenylene) was generated with NMR properties calculated at the GIAO/PCM(CHCl3)/WP04/6-31G(d)//B97-D/cc-pVDZ level (see ESI, Tables S28–S30†). Comparison between the sets of Δδexp and Δδcalc indicates that the o-phenylenes in both oP8F(DPB)2+2 and oP8H(DPB)2+2 are folded into the AAAAB state. The RMSD values are shown in Fig. 10b; the matches can be made at a confidence level of >96% relative to the next-best possibility (AAAAA in both cases).
The spectra of oP8H(Phen)2+2 and oP8F(Phen)2+2 are similar to those of the DPB-based macrocycles, but more complex as they capture the overall structure (see ESI, Fig. S55 and S66†). In both cases, there are four distinct imine singlets; for oP8F(Phen)2+2, there are eight distinct 19F signals. The simplicity of the spectra suggests that there is a single explanation for this behavior; either a roughly 1:1 mixture of homo- and heterochiral macrocycles, a 1:1 mixture of macrocycles with parallel and antiparallel oligomer orientations, or a single geometry that is distorted from twofold symmetry. At this point, we cannot distinguish these possibilities.
The AAAAB conformation of an o-phenylene octamer has a small bite angle (β = 36°), and therefore should indeed be a good fit for a [2 + 2] macrocycle (Fig. 2). To visualize the structures, geometry optimizations of candidate macrocycles were performed at the PCM(CHCl3)/B97-D/cc-pVDZ level, explicitly considering the relative stereochemistry and alignment of the o-phenylenes. As shown in Fig. 11, the best fit (by 2 kcal mol−1) was found to be the homochiral-antiparallel geometry. The structure is reminiscent of the [2 + 2] macrocycles of o-phenylene decamers (Fig. 7), with crossed linkers to accommodate the nonzero β.
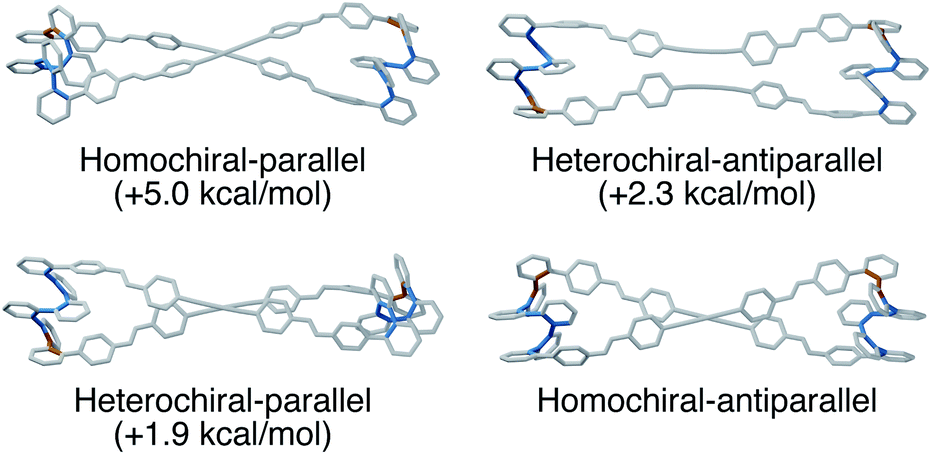 | ||
| Fig. 11 Molecular geometries of all [2 + 2] macrocycles of o-phenylene octamers in the AAAAB conformation co-assembled with DPB, optimized at the PCM(CHCl3)/B97-D/cc-pVDZ level. Biaryl bonds in the A state are shown in blue, and those in the B state are shown in orange. | ||
Controlled self-assembly of foldamer macrocycles
As discussed above, in these systems there is a trade-off between the enthalpic and entropic contributions to self-assembly: for an equilibrium between macrocycles of different sizes, in the simplest possible model, the enthalpic contribution is governed by the (mis)folding of the foldamer and the entropic contribution comes from the increased translational entropy of a larger number of smaller macrocycles. Thus, while all of the o-phenylenes discussed here prefer the perfectly folded conformation when free of restrictions (i.e., as measured for the acyclic model compounds), the net proportion of o-phenylenes that are misfolded increases when they are made to self-assemble (e.g., the roughly 3:7 ratio of oP10OMe(DPB)3+3:oP10OMe(DPB)2+2 requires that far more o-phenylenes misfold compared to the innate folding propensity of oP10OMe(M)). That is, as we have noted before,30 the entropic bias toward smaller products is sufficient to override the relatively weak folding preferences of the oligomers in these systems.
The o-phenylene octamer systems represent an extreme version of this principle. The perfectly folded conformer, while favored in the acyclic models (Fig. 9), cannot be incorporated into a macrocycle because of its large bite angle (179°). Instead, it quantitatively misfolds into the AAAAB conformer on assembly. This is easily rationalized by considering Fig. 2, which shows that the AAAAB state is both the second-most stable and a reasonable match for the smallest possible macrocycle ([1 + 1] macrocycles would be too strained to exist in these systems). The only conformer that is a good match for the [3 + 3] macrocycle is the BBBBB state, which is simply too unstable.
The key result here is that this balance can be controlled by adjusting the folding propensities of the o-phenylenes. The chemical structures of the macrocycles in Fig. 3b are identical except for substitution quite remote (≥8 bonds) from the bonding sites for assembly. Nevertheless, replacement of even single atoms (i.e., H → F) has profound effects on the obtained products. In the oP10H(NH2) and oP10OMe(NH2) systems, both the [2 + 2] and the [3 + 3] macrocycles are observed, with a slight preference for the [2 + 2] macrocycle. The hydrogen and methoxy substituents are not particularly good at inducing the folding of o-phenylenes,42,59 consistent with their well-known substituent effects on arene–arene stacking interactions.46–49 Consequently, even though the acyclic o-phenylenes are well-folded, there is relatively little penalty to misfolding, and two (BAAAAAB) or three (AAAABBB) stacking interactions can be sacrificed in order to adopt conformations that better fit into the entropically favored [2 + 2] macrocycle. In contrast, the fluoro substituents in the oP10F(NH2) systems strengthen arene–arene stacking. The difference in stability is not large (on the order of 0.3 kcal mol−1 per interaction on the basis of the behavior of the model compounds), yet it is enough shift the assembly fully toward the [3 + 3] products oP10F(DPB)3+3 and oP10F(Phen)3+3. Of course, this effect has limits. The observation of exclusively [2 + 2] macrocycles in the fluorinated octamer systems shows that the fluorinated systems are still able to misfold under the right circumstances and confirms that the overall change in folding stability is small.
Thus, a small substitution drives a substantial change in both local folding state and overall product geometry for o-phenylene macrocycles via a small perturbation of ΔH° that tips the balance with TΔS° to favor one macrocycle size. These results relate to an important concept from biological systems, where achieving large-amplitude motions in response to simple stimuli requires systems that are well-folded but in relatively shallow free energy wells that are easily perturbed.38,39 Here this is essentially a sequence effect: the o-phenylene system is not responsive,60 in the sense that it does not switch in response to changes in its environment.61 Nevertheless, the results represent a simple demonstration of large-scale structural changes in response to small chemical perturbations in an wholly abiotic foldamer system.
Although these systems are clearly not designed for post-assembly function, it is noteworthy that the control demonstrated here, providing near quantitative yields of the [3 + 3] macrocycle instead of mixtures, is in the direction with the most potential utility. That is, by switching to o-phenylenes functionalized with moderately electron-withdrawing substituents, we can fully bias the system towards larger macrocycles that have large central cavities with exposed folded surfaces that could be used to interact with other species. Of course, as is clear from the NMR spectra of the systems with Phen-based linkers, we still do not yet have full control over these structures: in particular, we must learn to control the twist-senses of the o-phenylenes in order to generate species that are truly structurally well-defined.
Conclusions
Differently substituted o-phenylene decamers octamers have been co-assembled with rod-shaped linkers via dynamic covalent chemistry. There is a delicate balance between enthalpic and entropic effects in these systems. In the o-phenylene decamer series, larger macrocycles containing perfectly folded o-phenylenes and smaller macrocycles containing misfolded o-phenylenes are possible. Whereas methoxy- and unsubstituted oligomers give mixtures of macrocycles, the fluorinated oligomer gives exclusive assembly into [3 + 3] macrocycles with perfectly folded o-phenylene segments. That is, substitution at a site remote from the site of bond formation controls the architecture of the product that is obtained, likely via the increased strength of arene–arene stacking interactions. o-Phenylene octamers, which cannot assemble into macrocycles when perfectly folded, gave only [2 + 2] macrocycles regardless of substitution or linker structure, confirming that the fluorinated system remains capable of misfolding under the appropriate conditions. This represents a demonstration of an abiotic system that undergoes large changes in folding and overall structure as a consequence of relatively small changes in energetics. The control of product distribution via folding represents a significant step towards the generation of complex, structurally well-defined, non-biological macromolecules.Author contributions
The study was conceived and planned by CSH and VCK. Experimental work was executed by VCK. DFT calculations were performed by CSH. Both VCK and CSH wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1904236) and the Volwiler Distinguished Research Professorship for support of this work.Notes and references
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852–4863 RSC.
- T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- Y. Zhang, Y. Zhong, A. L. Connor, D. P. Miller, R. Cao, J. Shen, B. Song, E. S. Baker, Q. Tang, S. V. S. R. K. Pulavarti, R. Liu, Q. Wang, Z.-l. Lu, T. Szyperski, H. Zeng, X. Li, R. D. Smith, E. Zurek, J. Zhu and B. Gong, J. Am. Chem. Soc., 2019, 141, 14239–14248 CrossRef CAS PubMed.
- Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477–15486 CrossRef CAS PubMed.
- P. Mateus, N. Chandramouli, C. D. Mackereth, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 5797–5805 CrossRef CAS PubMed.
- S. B. Seo, S. Lee, H.-G. Jeon and K.-S. Jeong, Angew. Chem., Int. Ed., 2020, 59, 10441–10445 CrossRef CAS PubMed.
- C.-B. Huang, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2020, 59, 7319–7330 CrossRef CAS PubMed.
- F. Chen, J. Shen, N. Li, A. Roy, R. Ye, C. Ren and H. Zeng, Angew. Chem., Int. Ed., 2020, 59, 1440–1444 CrossRef CAS PubMed.
- A. D. Peters, S. Borsley, F. della Sala, D. F. Cairns-Gibson, M. Leonidou, J. Clayden, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, E. Takano, J. Burthem, S. L. Cockroft and S. J. Webb, Chem. Sci., 2020, 11, 7023–7030 RSC.
- Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellman, Science, 2019, 366, 1528–1531 CrossRef CAS PubMed.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed.
- P. Reiné, J. Justicia, S. P. Morcillo, S. Abbate, B. Vaz, M. Ribagorda, Á. Orte, L. Álvarez de Cienfuegos, G. Longhi, A. G. Campaña, D. Miguel and J. M. Cuerva, J. Org. Chem., 2018, 83, 4455–4463 CrossRef PubMed.
- R. Katoono, K. Kusaka, K. Fujiwara and T. Suzuki, Chem.–Asian J., 2014, 9, 3182–3187 CrossRef CAS PubMed.
- R. Katoono, Y. Tanaka, K. Fujiwara and T. Suzuki, J. Org. Chem., 2014, 79, 10218–10225 CrossRef CAS PubMed.
- F. Chen, T. Tanaka, Y. Hong, W. Kim, D. Kim and A. Osuka, Chem.–Eur. J., 2016, 22, 10597–10606 CrossRef CAS PubMed.
- T. Hjelmgaard, L. Nauton, F. De Riccardis, L. Jouffret and S. Faure, Org. Lett., 2018, 20, 268–271 CrossRef CAS PubMed.
- B. Liu, C. G. Pappas, E. Zangrando, N. Demitri, P. J. Chmielewski and S. Otto, J. Am. Chem. Soc., 2019, 141, 1685–1689 CrossRef CAS PubMed.
- R. Katoono, K. Kusaka, Y. Saito, K. Sakamoto and T. Suzuki, Chem. Sci., 2019, 10, 4782–4791 RSC.
- N. Fuentes, A. Martin-Lasanta, L. Alvarez de Cienfuegos, R. Robles, D. Choquesillo-Lazarte, J. M. García-Ruiz, L. Martínez-Fernández, I. Corral, M. Ribagorda, A. J. Mota, D. J. Cárdenas, M. C. Carreño and J. M. Cuerva, Angew. Chem., Int. Ed., 2012, 51, 13036–13040 CrossRef CAS PubMed.
- H. Wu, A. Acharyya, Y. Wu, L. Liu, H. Jo, F. Gai and W. F. DeGrado, ChemBioChem, 2018, 19, 902–906 CrossRef CAS PubMed.
- K. Urushibara, Y. Ferrand, Z. Liu, H. Masu, V. Pophristic, A. Tanatani and I. Huc, Angew. Chem., Int. Ed., 2018, 57, 7888–7892 CrossRef CAS PubMed.
- L. Zheng, S. Guo, C. Wang, Y. Wang, Y. Fan, X. Chen, K. Zhang and H. Jiang, ChemPlusChem, 2021, 86, 340–346 CrossRef CAS PubMed.
- T. Sawada, M. Yamagami, K. Ohara, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 4519–4522 CrossRef CAS PubMed.
- T. Sawada and M. Fujita, Chem, 2020, 6, 1861–1876 CAS.
- Z. J. Kinney, V. C. Kirinda and C. S. Hartley, Chem. Sci., 2019, 10, 9057–9068 RSC.
- Z. J. Kinney and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 4821–4827 CrossRef CAS PubMed.
- V. C. Kirinda, B. R. Schrage, C. J. Ziegler and C. S. Hartley, Eur. J. Org. Chem., 2020, 5620–5625 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68–73 CrossRef CAS PubMed.
- C. S. Hartley, Acc. Chem. Res., 2016, 49, 646–654 CrossRef CAS PubMed.
- S. Ando, E. Ohta, A. Kosaka, D. Hashizume, H. Koshino, T. Fukushima and T. Aida, J. Am. Chem. Soc., 2012, 134, 11084–11087 CrossRef CAS PubMed.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS PubMed.
- O. Miyashita, J. N. Onuchic and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12570–12575 CrossRef CAS PubMed.
- Z. J. Kinney and C. S. Hartley, Org. Lett., 2018, 20, 3327–3331 CrossRef CAS PubMed.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed.
- S. Mathew, L. A. Crandall, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2014, 136, 16666–16675 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS PubMed.
- Note that the calculated ΔE values are typically found to overestimate the actual relative stabilities in solution.
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS PubMed.
- F. Cozzi and J. S. Siegel, Pure Appl. Chem., 1995, 67, 683–689 CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- B. W. Gung, X. Xue and H. J. Reich, J. Org. Chem., 2005, 70, 3641–3644 CrossRef CAS PubMed.
- M. Ciaccia, S. Pilati, R. Cacciapaglia, L. Mandolini and S. Di Stefano, Org. Biomol. Chem., 2014, 12, 3282–3287 RSC.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. Di Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC.
- C. S. Hartley and J. He, J. Org. Chem., 2010, 75, 8627–8636 CrossRef CAS PubMed.
- NMR experiments were carried out at low temperature to slow conformational exchange, which gives a slight sharpening of the signals.
- The small signals were confirmed to arise from misfolding, as opposed to impurities, by EXSY spectroscopy.
- R. Jain, T. Bally and P. R. Rablen, J. Org. Chem., 2009, 74, 4017–4023 CrossRef CAS PubMed.
- K. W. Wiitala, T. R. Hoye and C. J. Cramer, J. Chem. Theory Comput., 2006, 2, 1085–1092 CrossRef CAS PubMed.
- S. Toyota, Chem. Rev., 2010, 110, 5398–5424 CrossRef CAS PubMed.
- HMBC correlations for the minor conformer of oP10OMe(DPB)2+2 were too weak to unambiguously determine the order of the repeat units.
- S. M. Mathew, J. T. Engle, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2013, 135, 6714–6722 CrossRef CAS PubMed.
- NMR spectra were run in other solvents (toluene and acetone), but with no significant effects.
- T. Sawada, Y. Inomata, M. Yamagami and M. Fujita, Chem. Lett., 2017, 46, 1119–1121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01270c |
| This journal is © The Royal Society of Chemistry 2021 |