
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the abolition of intersubunit salt bridges on allosteric protein structural dynamics†
Minseo
Choi‡
 abc,
Jong Goo
Kim‡
c,
Srinivasan
Muniyappan‡
c,
Hanui
Kim
abc,
Tae Wu
Kim
c,
Yunbeom
Lee
abc,
Sang Jin
Lee
abc,
Seong Ok
Kim
c and
Hyotcherl
Ihee
*abc
abc,
Jong Goo
Kim‡
c,
Srinivasan
Muniyappan‡
c,
Hanui
Kim
abc,
Tae Wu
Kim
c,
Yunbeom
Lee
abc,
Sang Jin
Lee
abc,
Seong Ok
Kim
c and
Hyotcherl
Ihee
*abc
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: hyotcherl.ihee@kaist.ac.kr
bKI for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
cCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 10th May 2021
Abstract
A salt bridge, one of the representative structural factors established by non-covalent interactions, plays a crucial role in stabilizing the structure and regulating the protein function, but its role in dynamic processes has been elusive. Here, to scrutinize the structural and functional roles of the salt bridge in the process of performing the protein function, we investigated the effects of salt bridges on the allosteric structural transition of homodimeric hemoglobin (HbI) by applying time-resolved X-ray solution scattering (TRXSS) to the K30D mutant, in which the interfacial salt bridges of the wild type (WT) are abolished. The TRXSS data of K30D are consistent with the kinetic model that requires one monomer intermediate in addition to three structurally distinct dimer intermediates (I1, I2, and I3) observed in WT and other mutants. The kinetic and structural analyses show that K30D has an accelerated biphasic transition from I2 to I3 by more than nine times compared to WT and lacks significant structural changes in the transition from R-like I2 to T-like I3 observed in WT, unveiling that the loss of the salt bridges interrupts the R–T allosteric transition of HbI. Besides, the correlation between the bimolecular CO recombination rates in K30D, WT, and other mutants reveals that the bimolecular CO recombination is abnormally decelerated in K30D, indicating that the salt bridges also affect the cooperative ligand binding in HbI. These comparisons of the structural dynamics and kinetics of K30D and WT show that the interfacial salt bridges not only assist the physical connection of two subunits but also play a critical role in the global structural signal transduction of one subunit to the other subunit via a series of well-organized structural transitions.
Introduction
A function of a protein has an intimate connection with its three-dimensional structure. Thus, understanding the interactions inside the protein that affect the structure provides key clues for uncovering the function of a biological system. The structure of a protein is determined by the covalent interactions between amino acid residues to establish the backbone of the protein as well as the non-covalent interactions such as hydrogen bonds, van der Waals interactions, ionic interactions, and hydrophobic interactions that determine how the protein folds. The salt bridge is one of the typical structural factors generated by non-covalent interactions found in many proteins and is formed by a combination of hydrogen bonds and ionic interactions. In a protein, a salt bridge is mainly constructed between a negatively charged carboxylate of aspartic acid or glutamic acid and cationic ammonium of lysine or guanidinium. Many studies have been conducted to unveil the structural and functional roles of salt bridges in proteins. The salt bridge in the structural aspect mainly plays a role in stabilizing entropically unfavorable conformations, enhancing the stabilities of secondary, tertiary, and quaternary structures, and having resistance to protein aggregation.1,2 Since the salt bridge contributes to stabilizing the protein structure, the protein activity is inevitably regulated by the salt bridge. Many cases have been reported where the proteins cannot perform their original functions when the salt bridges are abolished.2–4 Although much research has been conducted on salt bridges due to their importance in determining the structures and functions, most studies have been conducted only on static equilibrium states. In reality, however, performing a protein function is not a static process in which the protein stays in a specific structure but a dynamic process in which the structure changes over time. Therefore, it is important to identify how the salt bridge affects the structural dynamics of a protein, for example, the structures of reaction intermediates and the associated reaction rates.In this regard, here we investigated how the structural dynamics of a protein are altered in the presence or absence of salt bridges to improve the understanding of the structural and functional role of the salt bridges in the course of the dynamic biological process. Specifically, we investigated the role of the salt bridges during the allosteric structural transition of homodimeric hemoglobin (HbI) from Scapharca inaequivalvis. Allosteric regulation is one of the essential mechanisms regulating the function of proteins, in which the structural change at one site leads to significant feedback at a remote site of the same macromolecular entity. A well-known example is human tetrameric hemoglobin (Hb), where the binding of dioxygen or carbon monoxide is known to benefit from allosteric regulation.5–12 While the tetrameric structure of Hb makes detailed experimental studies difficult,13 HbI offers a greatly simplified model system, thanks to its homodimeric structure. Despite being only a dimer, the characteristics of cooperative ligand binding and allosteric structural transitions are clearly expressed between the liganded R state with high ligand affinity and the unliganded T state with low ligand affinity.14–26
HbI has two symmetrically linked salt bridges between Lys30 and Asp89 at the subunit interface. The salt bridges are one of the essential factors composing the intersubunit network and stabilizing the dimeric assembly with two subunits connected. By two symmetrically linked intersubunit salt bridges between Lys30 in one subunit and Asp89 in the other subunit, one subunit may communicate to the other subunit. The K30D mutation, where the Lys30 residues of the two subunits consisting of the salt bridges of the wild type (WT) are replaced with Asp30 residues, abolishes the only salt bridges at the subunit interface by introducing charge repulsion between Asp30 and Asp89 (Fig. 1) so that this mutant is a relevant system for identifying the effect of the salt bridges on the structural dynamics of HbI.
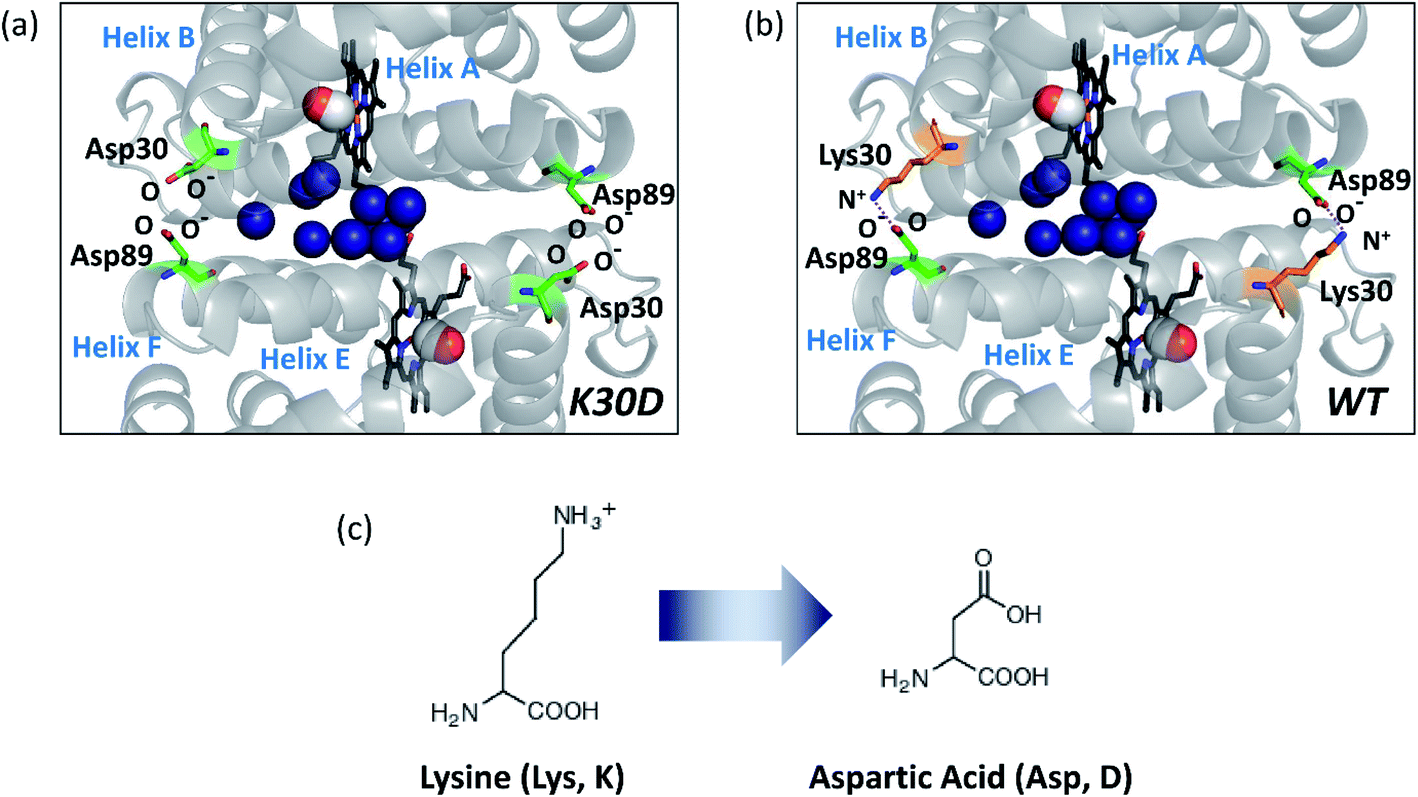 | ||
| Fig. 1 Subunit interface of HbI(CO)2. (a) Structure of K30D HbI(CO)2. (b) The crystal structure of WT HbI(CO)2 (PDB ID: 3SDH). The structure of K30D is based on the crystal structure of WT. In K30D, the Lys30 residue (shown in orange) in each subunit of WT is replaced by Asp30 (shown in green) as shown in (c). In (a) and (b), two carbon monoxide molecules are shown with connected red and white spheres. Eleven interfacial water molecules (shown in dark blue) are well organized and connected to two subunits and hemes. In WT, the network of the subunit interface is stabilized by symmetrical salt bridges between Lys30 and Asp89. In contrast, the interface is destabilized in K30D. | ||
A study using spectroscopic characterization on K30D reported the dimerization constants of unliganded and liganded derivatives, the oxygen equilibrium measurements, and the kinetics of oxygen release.27 It was reported that the cleavage of the salt bridges destabilizes the intersubunit network and dimeric assembly of HbI, resulting in a change in the mechanism of cooperativity.27 According to the reported dimerization constant,27 where WT mostly exists as the dimeric form in nature, the fraction of the monomer is not negligible in K30D due to the weak interaction at the interface. The local structural changes near the heme pocket and the kinetics upon oxygen release in K30D were investigated by spectroscopic studies and equilibrium oxygen-binding experiments.27 Still, more detailed kinetic and structural information, such as lifetimes and tertiary and quaternary structures of reaction intermediates, is unclear. In this regard, time-resolved X-ray solution scattering (TRXSS)28–33 can provide complementary dynamic information on the allosteric structural transition of K30D in solution for unveiling the effect of the salt bridges in HbI on the structural dynamics. TRXSS, also known as time-resolved X-ray liquidography (TRXL), is a useful technique that offers complementary advantages over conventional methods, as it allows for easy observation of the structural dynamics of proteins in physiological environments.
In this work, to elucidate how the interfacial salt bridges play a role in the allosteric structural transition in HbI, we performed the TRXSS experiment on K30D in solution and extracted the structures of the intermediates and determined kinetic parameters based on the modified kinetic model that considered the monomer fraction. In particular, we obtained the detailed structural parameters of the intermediates of K30D HbI, such as root-mean-square deviations (RMSDs), displacement plots, subunit rotation angles, heme–heme distances, and distances between Cα atoms of the key residues involved in the salt bridges in WT HbI, and quantitatively compared these structural parameters with those of WT HbI. We found that all the three dimer intermediates (I1, I2, and I3) of K30D have R-like structures, unlike WT involving a transition from R-like I1 and I2 to T-like I3. These observations indicate that the dramatic structural change associated with the R–T transition is remarkably suppressed by losing the salt bridges in K30D. Considering that the bimolecular CO recombination is decelerated in K30D although I3 of K30D has a structure more similar to the ground state compared to that of WT, the loss of the salt bridges may diminish the ligand-binding affinity and eventually make the CO recombination process slow.
Experimental
Sample preparation
The K30D mutation was introduced into the native recombinant HbI plasmid using the EZchange™ site-directed mutagenesis kit (Enzynomics) with the following oligonucleotide encoding K30D mutation: 5′-GGTTCGGACAAAGATGGTAACGG-3′ and 5′-CCGTTACCATCTTTGTCCGAACC (Genotech). The K30D mutant (K30D) was over-expressed in E. coli and purified according to the method34 described for WT. Carbonmonoxy derivatives of K30D solution for the photodissociation reaction were prepared as follows. A 2–4 mM deoxy K30D solution in 100 mM phosphate buffer (pH 7) was prepared in a rubber-topped air-tight vial. The protein concentration was determined from the absorbance at 578 nm using the absorption coefficient of heme-oxygenated derivatives (14.3 mM−1 cm−1). The deoxy K30D was reduced by adding 10 μL of 1 M sodium dithionite solution under a nitrogen atmosphere. The reduced samples were exposed to CO gas for 30 minutes to convert deoxy to CO-bound K30D. The sample solution was prepared just before the X-ray solution scattering measurement. An aliquot of the resulting CO-bound K30D solution was transferred into a quartz capillary with a 1 mm diameter (Hampton Research) and immediately sealed with epoxy to minimize gas exchange while CO gas was purged continuously into the capillary.Data acquisition
The TRXSS experiment of K30D was conducted at the BioCARS beamline 14IDB at the Advanced Photon Source using the experimental setup of a typical pump-probe method. Circularly polarized laser pulses generated from a picosecond laser system were sent to an optical parametric amplifier (TOPAS) to generate output pulses with a center wavelength of 532 nm and a temporal duration of ∼35 ps. The laser light was focused on 0.15 × 0.60 mm2 at the sample position and finally had an energy density of 1 mJ mm−2. The X-ray pulse peaked at 12 keV with a long wavelength tail of ∼4% and a photon flux of ∼109 photons/pulse. The size of the X-ray beam was adjusted to be 0.09 × 0.07 mm2 at the sample position, which was smaller than the laser spot size, to minimize the timing jitter caused by the spatial error between the laser and X-ray pulses. After a laser pulse passed through the sample to induce the reaction, an X-ray pulse arrived with a time delay, Δt, and time-resolved X-ray scattering data were measured at time delays in the range from 100 ps to 10 ms. The helium cone was utilized to reduce the air scattering by X-ray. Two-dimensional (2D) scattering patterns were collected using an area detector (MarCCD) located at a distance of 185 mm from the sample position. The capillary containing the protein sample was moved along the axis perpendicular to the pulse train of X-ray and the laser and was set to perform repetitive motion in the forward and backward directions to provide a fresh sample for each X-ray and laser pair. The obtained 2D X-ray scattering patterns have centrosymmetry due to the random orientation of the molecules in the solution phase, so that one-dimensional (1D) X-ray scattering curves can be obtained by azimuthally integrating the patterns as a function of the magnitude of the momentum transfer vector, q = (4π/λ)sin(2θ/2) where λ is the X-ray wavelength and 2θ is the scattering angle. The 1D X-ray scattering curves, S(q, Δt), contain information about the X-ray scattering occurring from the solvent pairs, solute pairs, and solvent–solute pairs at the corresponding time delay. The X-ray scattering signal from the solvent molecule was much larger than the signal of our interest resulting from the reaction of the solute molecules. The scattering curve at the negative time delay, −5 μs, containing the signal from the unreacted sample solution was subtracted from the scattering curve for each time delay, and the scattering signal generated by the bulk solvent was removed by this process. As a result, we obtained the time-resolved difference X-ray solution scattering curves, ΔS(q, Δt). The time delays used in the experiments are as follows:−5 μs, 100 ps, 178 ps, 316 ps, 562 ps, 1 ns, 1.78 ns, 3.16 ns, 5.62 ns, 10 ns, 17.8 ns, 31.6 ns, 56.2 ns, 100 ns, 178 ns, 316 ns, 562 ns, 1 μs, 1.78 μs, 3.16 μs, 5.62 μs, 10 μs, 17.8 μs, 31.6 μs, 56.2 μs, 100 μs, 178 μs, 316 μs, 562 μs, 1 ms, 1.78 ms, 3.16 ms, 5.62 ms, and 10 ms.
The measured difference X-ray scattering curves contained the signal from the thermal heating of the solvent. For the data analysis, we removed the thermal heating contribution using a well-established method.35–38 During the experiment, the temperature was maintained at 25 °C by a stream of cold nitrogen gas (Oxford Cryostream).
Results and discussion
Kinetic analysis of time-resolved difference solution scattering curves
Time-resolved difference X-ray solution scattering curves, ΔS(q, t), obtained for time delays (t) from 100 ps to 10 ms are shown in Fig. 2a. The data exhibit oscillatory features along the q-axis due to protein structural changes. To extract kinetic information of intermediates and their structures from ΔS(q, t), we followed the well-established procedure35–38 which had been applied in previous TRXSS studies on WT and various mutants of HbI, consisting of kinetic analysis using singular value decomposition (SVD) and principal component analysis (PCA). As detailed in the ESI,† SVD and the global fit of left singular vectors (lSVs) show that the kinetics involves four structurally distinct intermediates and five time constants of 4.6 (±0.7) ns, 47 (±13) ns, 588 (±81) ns, 616 (±109) μs, and 5.34 (±5.22) ms. The kinetics of all the HbI proteins so far studied with TRXSS, including WT, F97Y,37 T72V,36 and I114F35 can be explained with the same kinetic framework with only minor variations. This framework has three intermediates, a transition from the first intermediate to the second intermediate (I1 → I2), a biphasic transition from the second intermediate to the third intermediate (I2 → I3), and bimolecular recombination to recover CO-bound HbI(CO)2. Compared to the WT data, which exhibit three intermediates and seven time constants, K30D has one additional intermediate and a smaller number of kinetic components, requiring a more significant alteration to the known the kinetic framework.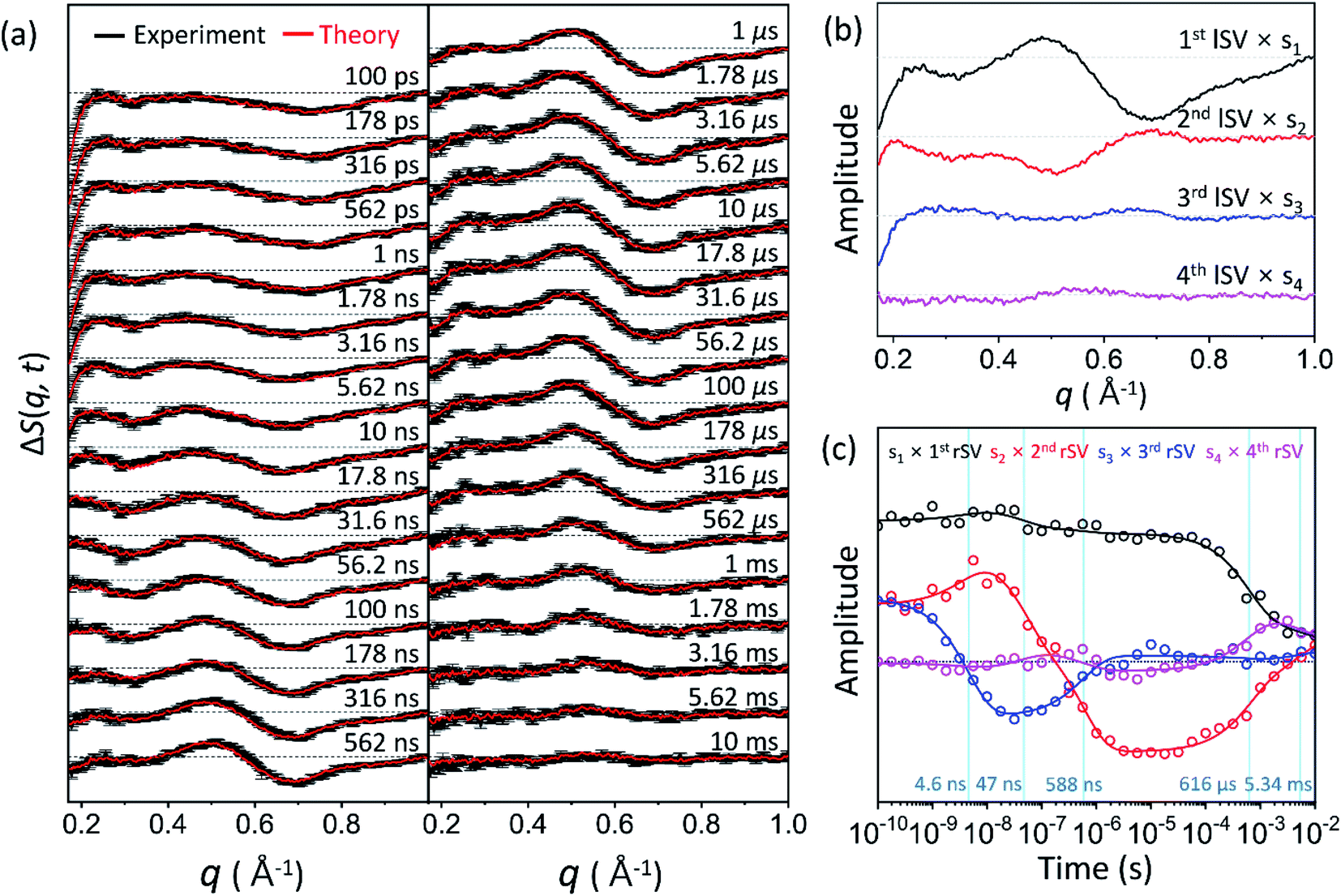 | ||
| Fig. 2 (a) Time-resolved X-ray solution scattering curves, ΔS(q, t), at time delays from 100 ps to 10 ms were obtained for the K30D mutant of HbI in solution. Experimental curves with standard deviation (shown in black) are compared with theoretical curves (shown in red) generated from a linear combination of left singular vectors (lSVs) extracted from the kinetic analysis. (b) The first four lSVs were multiplied by singular values (Fig. S9†) from singular value decomposition (SVD) analysis. (c) The first four right singular vectors (rSVs) were multiplied by singular values and fitted by the sum of exponential functions sharing five time constants of 4.6 ns, 47 ns, 588 ns, 616 μs, and 5.34 ms. | ||
To establish the kinetic framework (Fig. 3a), we used two effective methods that can greatly facilitate narrowing down the kinetic models compatible with the experimental data: (i) the number of species from the SVD results on various reduced time ranges39 in addition to the whole data and (ii) SVD-aided pseudo PCA analysis (SAPPA).39 As detailed in the ESI,† such analyses reveal that the first time constant of 4.6 ns can be attributed to the transition from I1 to I2, and the two time constants of 47 ns and 588 ns need to be assigned to the biphasic transition from I2 to I3. As a result, among five time constants, two (616 μs and 5.34 ms) remain to be assigned, and the identity of one intermediate among four remains to be determined.
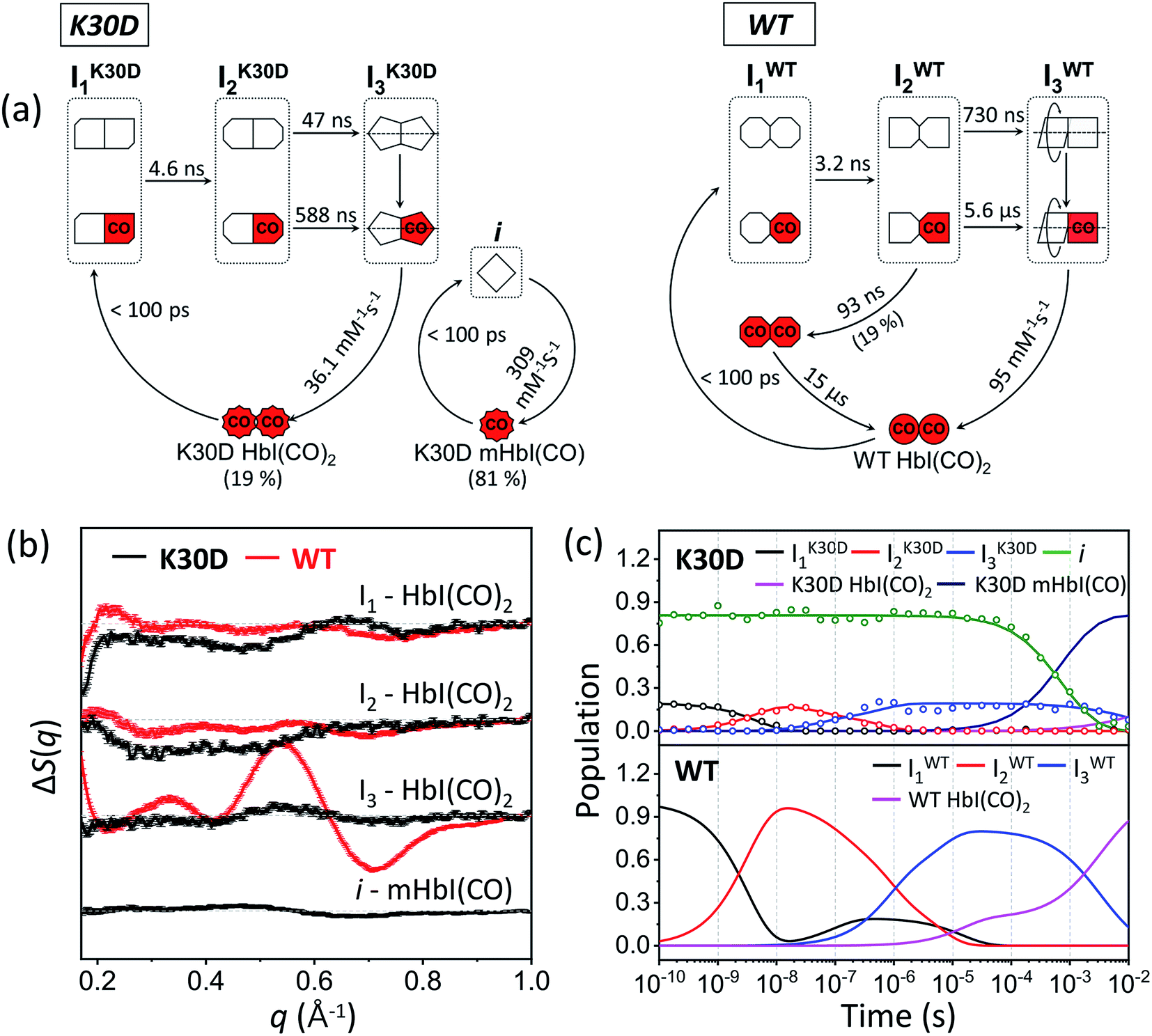 | ||
| Fig. 3 (a) Kinetic models for K30D (left) and WT (right). The red (with “CO”) and white symbols represent ligated and photolyzed subunits, respectively. The subunits of each intermediate are represented in different shapes to describe the change in the tertiary structure with the progress of transition from I1 to I2. To indicate quaternary structure change for the transition from I2 to I3, one subunit of I3 is described as rotation with respect to the other. To indicate the photolyzed monomer, i, the diamond-shaped symbols are used. For WT, two red octagons represent a ligated form of I1 formed by geminate recombination of CO with I2 and is structurally indistinguishable from the photolyzed forms of I1. (b) Species-associated difference scattering curves (SADSs) of the intermediates of K30D (black) and WT (red) HbI. (c) Population changes of the intermediates and initial HbI(CO)2 of K30D (upper graph) and WT (lower graph) extracted from the kinetic analysis. The open circles represent the optimized populations obtained by fitting the experimental curve at each time point with a linear combination of SADSs for the four intermediates. | ||
We considered the possibility that this remaining intermediate is either a monomer or a dimer and built the kinetic models for both scenarios (Fig. S2a and b,† respectively). The calculated curves from both kinetic models (Fig. S2c and d,† respectively) gave equally satisfactory agreements with the experimental data, meaning that the fit quality alone cannot discriminate whether the fourth species is a dimer or a monomer. Nevertheless, we favor the possibility of a “monomer” intermediate for the following reasons. (i) K30D has a weakened network of interactions between Asp30 and Asp89 located at the subunit interface. K30D has a dimerization constant of 1.2 × 103 M−1 at 10 °C under the equivalent conditions,27 and the ratio of the monomer is 41% in an aqueous solution. (ii) The monomeric unit of K30D can be regarded as a monomeric heme protein, whose photoinduced structural changes were observed in many time-resolved studies,28,40–55 and thus it is plausible that K30D monomer undergoes the structural change induced by photoexcitation. (iii) The simulated difference scattering intensity of the monomer shows a magnitude that cannot be neglected compared to that of the dimer (as detailed in the ESI and Fig. S3†). This result demonstrates that if the dimer intermediates can be distinguished in the TRXSS signals, the monomer intermediate should also be distinguished. Therefore, we considered the possibility of the monomer intermediate in the kinetic model. To avoid any confusion, from now on, intermediates refer to the dimer intermediates and i refers to the monomer intermediate. A more detailed explanation for the process of choosing the best kinetic model is described in the ESI.†
Finally, species-associated difference scattering curves (SADSs) and time-dependent concentrations of K30D are compared with those of WT in Fig. 3. To distinguish the intermediates of WT and K30D, we labeled I1, I2, and I3 of WT as IWT1, IWT2, and IWT3, respectively, and likewise labeled the intermediates of K30D as IK30D1, IK30D2, and IK30D3, respectively. For K30D, we labeled the monomeric form of the initial state as K30D mHbI(CO) and the dimeric form of the initial state as K30D HbI(CO)2. According to the determined kinetic model, K30D HbI(CO)2 is converted to the earliest intermediate, IK30D1, within the experimental time resolution (<100 ps), and IK30D1 transforms to IK30D2 with a time constant of 4.6 ns, which is slightly larger than that of WT (3.2 ns), indicating that the absence of salt bridges affects the earliest intermediates although the effect is not severe. K30D shows the biphasic IK30D2-to-IK30D3 transition, as in WT and other mutants, with time constants of 47 ns and 588 ns, which are smaller than those of WT (730 ns and 5.6 μs) by factors of 15.5 and 9.5, respectively, indicating acceleration with respect to WT. According to TRXSS studies on WT and mutants,35–38 this IWT2-to-IWT3 transition accounts for the quaternary structural change where both the rotation angle and the heme–heme distance significantly changes. Thus, the accelerated rates mean that either (i) these structural changes occur more rapidly or (ii) different structural changes are involved in K30D. This result suggests that the salt bridges between the two subunits may be involved in controlling the quaternary structural change. Then, IK30D3 returns to K30D HbI(CO)2 with a bimolecular rate constant of 36 (±3) mM−1 s−1, which is nearly three times smaller than that of WT (95 mM−1 s−1). For the monomeric form of K30D, initial K30D mHbI(CO) is converted to i, within the experimental time resolution (<100 ps), and returns to K30D mHbI(CO) with a bimolecular rate constant of 309 (±9) mM−1 s−1, which is more than eight times larger than that of IK30D3. The fit result shows 81 (±0.4) % for the fraction of monomers. The reported value at 10 °C is 41%.27 The fact that the TRXSS experiment was carried out at 25 °C can rationalize the higher monomer content. The estimated dimerization constant based on the monomer fractions at 25 °C is ∼77 (±1) M−1. We note that the relative portion of dimers in terms of the concentration that contributes to the difference scattering signal is ∼20% (and that of monomers is ∼80%), but this does not mean that the relative contribution to the difference scattering signal is 20%. One dimer contributes more strongly to the scattering signal than one monomer because a dimer is two times heavier than a monomer and a dimer generally has a larger structural change than a monomer. The relative contributions of dimers to the difference signal range from 25% to 88% depending on time, as shown in Fig. S12.†
We note that the intermediates of K30D have two substrates of the fully photolyzed and partially photolyzed forms, which are structurally indistinguishable from each other. The same was revealed from previous laser power dependency studies on WT and other mutants,35–38 which provided direct evidence that the partially photolyzed dimer subunit undergoes the same structural evolution as the fully photolyzed subunit, directly demonstrating the allosteric regulation of HbI.
Structural analysis of intermediates
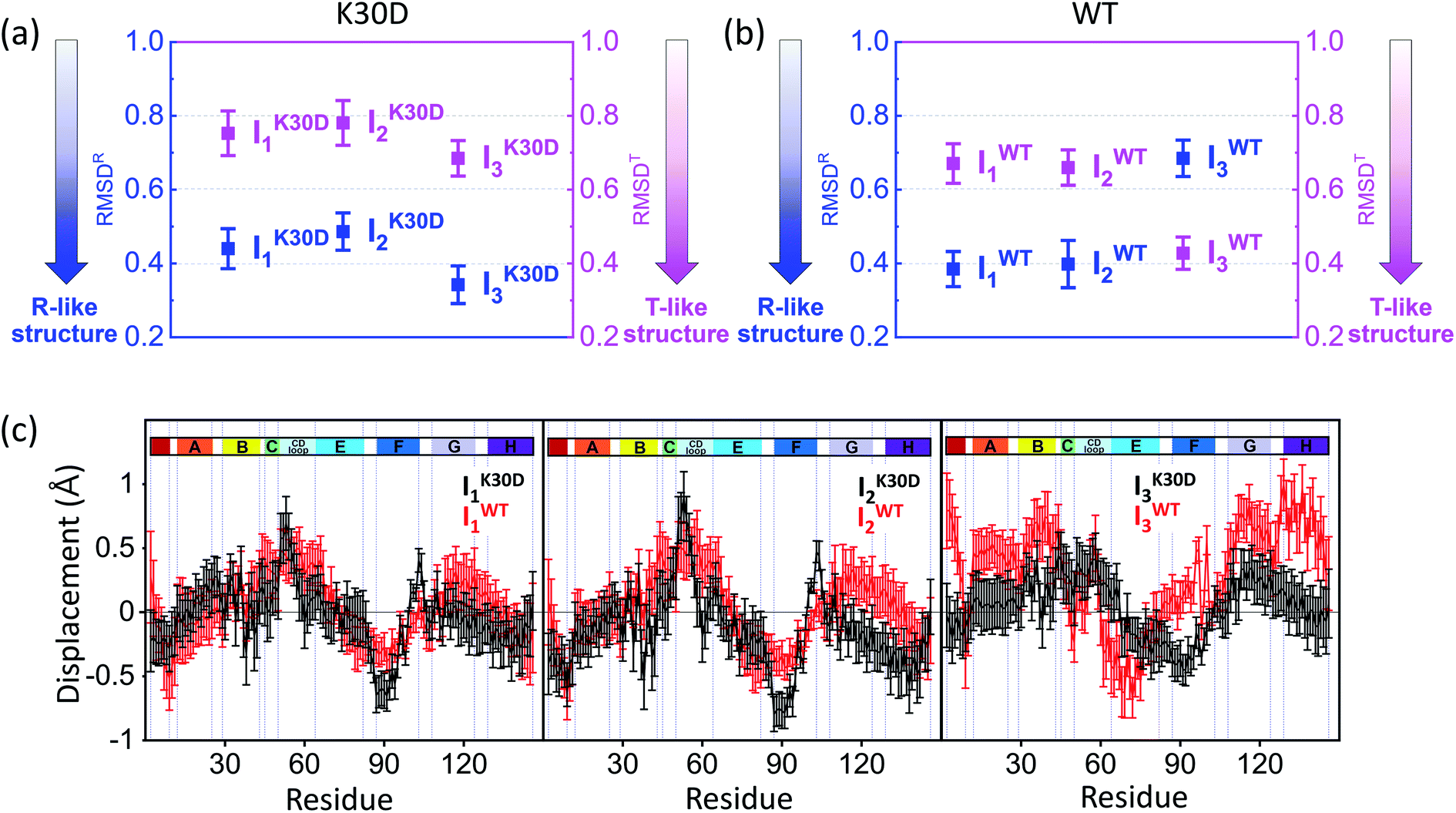 | ||
| Fig. 4 Root-mean-square deviations (RMSDs) were calculated from the candidate structures of the intermediates of (a) K30D and (b) WT. As described in the main text, RMSDR (blue) and RMSDT (magenta) were calculated with respect to the reference structures (for WT, the crystallographic structures and for K30D, modified structures based on these; see the ESI† for details) of the liganded state (R state) and the unliganded state (T state), respectively. The smaller RMSD indicates the structure closer to the corresponding reference structure. The plots display that all intermediates of K30D including I3 have the R-like structure rather than the T-like structure. In contrast, in WT, only I1 and I2 have the R-like structure, while I3 has the T-like structure. (c) Averaged displacement plots for K30D (black line) and WT (red line) and for I1 (left), I2 (middle), and I3 (right) were calculated with respect to K30D HbI(CO)2 (mutated from 3SDH) and HbI(CO)2 (3SDH), respectively. Error bars in (a)–(c) represent standard deviation values among various candidate structures of each intermediate. K30D and WT show similar displacement patterns for I1 and I2 whereas they show relatively more dissimilar patterns for I3. | ||
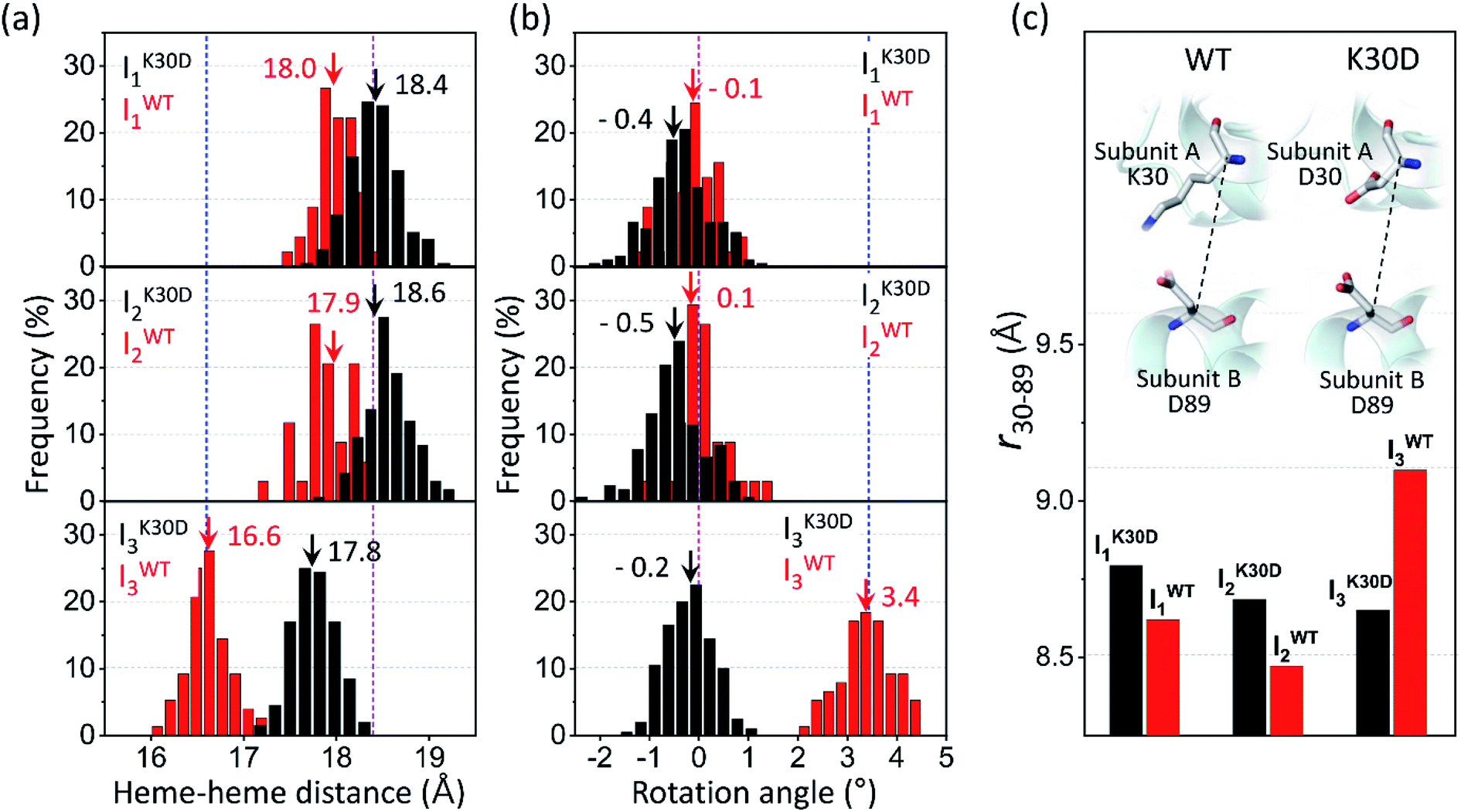 | ||
| Fig. 5 Occurrence distributions of (a) the heme–heme distance and (b) the rotation angle for the intermediates of K30D (black) and WT (red). The average values of K30D and WT are indicated with downward black and red arrows, respectively, for each intermediate. For comparison, the heme–heme distances and the rotation angles of the unliganded form (PDB ID: 4SDH) and the liganded form (PDB ID: 3SDH) of WT are indicated by vertical blue and magenta dotted lines, respectively. For all three intermediates, K30D undergoes smaller changes in the heme–heme distance compared to WT. In addition, the degree of rotation angles also shows that the I3 intermediate of K30D undergoes much less rotation than that of WT. (c) The average distance between Cα atoms in 30th and 89th residues, r30–89, for K30D (black) and WT (red). The 30th and 89th residues are involved in the interfacial salt bridges in WT. | ||
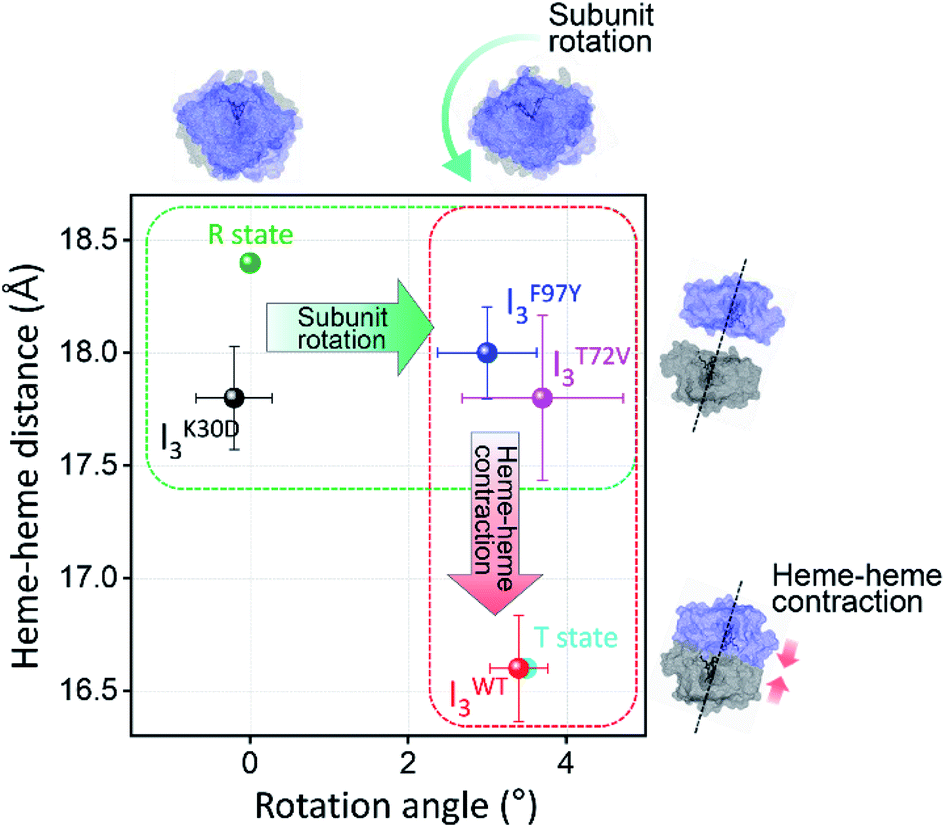 | ||
| Fig. 6 Heme–heme distances of the I3 intermediates of WT and various mutants including K30D are plotted as a function of the subunit rotation angle. For comparison, those of R and T states of WT, that is the crystallographic structures of HbI(CO)2 and unliganded HbI, respectively, are also shown as dots in green and cyan. The dots with errors in black, red, blue, magenta correspond to averaged candidate structures of IK30D3, IWT3, IF97Y3, and IT72V3, respectively. Error bars represent standard deviation values among various candidate structures of each intermediate. | ||
Conclusion
In this study, we applied TRXSS to the K30D mutant of HbI to investigate the structural and functional roles of the salt bridges located at the subunit interface in the allosteric structural transition of HbI. The kinetic analysis of the TRXSS data reveals that K30D has a kinetic framework with three structurally distinct intermediates of the dimer (I1, I2, and I3) as in WT. But, the kinetic model of K30D involves an additional intermediate (i) for the monomer, of which the fraction dramatically increases compared to WT due to the weak intersubunit interaction resulting from the loss of the salt bridges. A comparative study with WT reveals that the biphasic transition from I2 to I3, which involves allosteric structural R–T transition in WT, is accelerated in K30D. The reason for the acceleration of the process was scrutinized by subsequent structural analysis. Structural analysis using the SADSs of the three dimer intermediates shows that the tertiary and quaternary structural changes are suppressed in the IK30D2-to-IK30D3 transition compared to WT, in which the I2WT-to-IWT3 transition involves R–T structural transition with dramatic tertiary and quaternary structural changes. The structural similarity between R-like IK30D2 and IK30D3 can account for the acceleration of the biphasic IK30D2-to-IK30D3 transition. The observations from the kinetic and structural analyses indicate that the loss of salt bridges not only leads to the weakening of intersubunit interaction but also interrupts the R–T allosteric structural transition of HbI. Furthermore, the correlation between the bimolecular CO recombination rates and quaternary structures of I3 was inspected for K30D and WT, as well as other mutants studied by TRXSS such as F97Y and T72V. Thereby, it turned out that the bimolecular CO recombination of I3 is abnormally decelerated in K30D, even though I3 of K30D has a structure more similar to the ground state than I3 of WT, F97Y, and T72V. Considering that the bimolecular CO recombination process requires both the structural recovery to the ground state and the binding of CO ligands, the distinct correlation observed in K30D infers that the loss of the salt bridges degrades the ligand-binding affinity of K30D. Therefore, it can be considered that the salt bridges in HbI may play a functional role in ligand binding as well as the structural role contributing to the progression of R–T allosteric structural transition. These comparisons of the structural dynamics and kinetics of K30D and WT offer insights into the role of salt bridges. If interfacial salt bridges serve only as structural constraints to increase the rigidity between two subunits, one may expect that K30D, where the salt bridges are abolished, would have a more flexible intersubunit motion than WT. The data show the opposite; K30D has a suppressed intersubunit motion, suggesting a more sophisticated role of interfacial salt bridges beyond a simple structural glue between subunits. It should be noted that the structural changes of HbI are closely related to allosteric regulation. Once a ligand is attached to the heme of one subunit, this may trigger the change of the ligand-binding affinity of the other subunit via allosteric regulation. This consideration shows that the interfacial salt bridges not only assist the physical connection of two subunits but also play a critical role in the global structural signal transduction of one subunit to the other subunit via a series of well-organized structural transitions.Author contributions
H. I. directed the research. H. I. designed research. S. M. performed sample preparation. T. W. K., S. L., and S. K. performed the TRXSS experiments. M. C., J. G. K., H. K., and Y. L. analyzed the data. M. C., J. G. K., and H. I. wrote the paper, and all authors discussed the experimental results.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R004). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number P41GM118217. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The time-resolved setup at Sector 14 was funded in part through collaboration with P. Anfinrud (NIH/NIDDK).References
- D. Ferrari, D. Niks, L. H. Yang, E. W. Miles and M. F. Dunn, Biochemistry, 2003, 42, 7807–7818 CrossRef CAS PubMed
.
- M. Ghahramani, R. Yousefi, A. Krivandin, K. Muranov, B. Kurganov and A. A. Moosavi-Movahedi, Int. J. Biol. Macromol., 2020, 146, 1142–1160 CrossRef CAS PubMed
- J. M. Lv, J. Y. Chen, Z. P. Liu, Z. Y. Yao, Y. X. Wu, C. S. Tong and B. Cheng, Front. Immunol., 2020, 11, 583 CrossRef CAS PubMed
- M. Al-Haggar, A. Madej-Pilarczyk, L. Kozlowski, J. M. Bujnicki, S. Yahia, D. Abdel-Hadi, A. Shams, N. Ahmad, S. Hamed and M. Puzianowska-Kuznicka, Eur. J. Hum. Genet., 2012, 20, 1134–1140 CAS
- M. F. Perutz, Q. Rev. Biophys., 1989, 22, 139–237 CrossRef CAS PubMed
- S. Adachi, S. Y. Park, J. R. Tame, Y. Shiro and N. Shibayama, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7039–7044 CrossRef CAS PubMed
- S. Chang, M. Mizuno, H. Ishikawa and Y. Mizutani, Phys. Chem. Chem. Phys., 2018, 20, 3363–3372 RSC
- M. F. Perutz, Nature, 1970, 228, 726–739 CrossRef CAS PubMed
- W. A. Eaton, E. R. Henry, J. Hofrichter and A. Mozzarelli, Nat. Struct. Biol., 1999, 6, 351–358 CrossRef CAS PubMed
- B. Shaanan, J. Mol. Biol., 1983, 171, 31–59 CrossRef CAS PubMed
- G. Fermi, M. F. Perutz, B. Shaanan and R. Fourme, J. Mol. Biol., 1984, 175, 159–174 CrossRef CAS PubMed
- J. Baldwin and C. Chothia, J. Mol. Biol., 1979, 129, 175–220 CrossRef CAS PubMed
- J. Park, T. Lee and M. Lim, Photochem. Photobiol. Sci., 2013, 12, 1008–1015 CrossRef CAS PubMed
- W. E. Royer Jr., W. A. Hendrickson and E. Chiancone, Science, 1990, 249, 518–521 CrossRef CAS PubMed
- W. E. Royer Jr., A. Pardanani, Q. H. Gibson, E. S. Peterson and J. M. Friedman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14526–14531 CrossRef CAS PubMed
- J. E. Knapp, Q. H. Gibson, L. Cushing and W. E. Royer Jr., Biochemistry, 2001, 40, 14795–14805 CrossRef CAS PubMed
- J. E. Knapp and W. E. Royer Jr., Biochemistry, 2003, 42, 4640–4647 CrossRef CAS PubMed
- Y. Zhou, H. Zhou and M. Karplus, J. Mol. Biol., 2003, 326, 593–606 CrossRef CAS PubMed
- J. E. Knapp, M. A. Bonham, Q. H. Gibson, J. C. Nichols and W. E. Royer Jr., Biochemistry, 2005, 44, 14419–14430 CrossRef CAS PubMed
- Y. Seno, J. Comput. Chem., 2006, 27, 701–710 CrossRef CAS PubMed
- Y. Kiyota, R. Hiraoka, N. Yoshida, Y. Maruyama, T. Imai and F. Hirata, J. Am. Chem. Soc., 2009, 131, 3852–3853 CrossRef CAS PubMed
- J. E. Knapp, R. Pahl, J. Cohen, J. C. Nichols, K. Schulten, Q. H. Gibson, V. Srajer and W. E. Royer Jr., Structure, 2009, 17, 1494–1504 CrossRef CAS PubMed
- N. Yoshida, T. Imai, S. Phongphanphanee, A. Kovalenko and F. Hirata, J. Phys. Chem. B, 2009, 113, 873–886 CrossRef CAS PubMed
- J. S. Hub, M. B. Kubitzki and B. L. de Groot, PLoS Comput. Biol., 2010, 6, e1000774 CrossRef PubMed
- S. Fischer, K. W. Olsen, K. Nam and M. Karplus, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5608–5613 CrossRef CAS PubMed
- K. Nienhaus, J. E. Knapp, P. Palladino, W. E. Royer Jr. and G. U. Nienhaus, Biochemistry, 2007, 46, 14018–14031 CrossRef CAS PubMed
- P. Ceci, L. Giangiacomo, A. Boffi and E. Chiancone, J. Biol. Chem., 2002, 277, 6929–6933 CrossRef CAS PubMed
- H. S. Cho, N. Dashdorj, F. Schotte, T. Graber, R. Henning and P. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7281–7286 CrossRef CAS PubMed
- E. Malmerberg, Z. Omran, J. S. Hub, X. Li, G. Katona, S. Westenhoff, L. C. Johansson, M. Andersson, M. Cammarata, M. Wulff, D. van der Spoel, J. Davidsson, A. Specht and R. Neutze, Biophys. J., 2011, 101, 1345–1353 CrossRef CAS PubMed
- D. Arnlund, L. C. Johansson, C. Wickstrand, A. Barty, G. J. Williams, E. Malmerberg, J. Davidsson, D. Milathianaki, D. P. DePonte, R. L. Shoeman, D. Wang, D. James, G. Katona, S. Westenhoff, T. A. White, A. Aquila, S. Bari, P. Berntsen, M. Bogan, T. B. van Driel, R. B. Doak, K. S. Kjaer, M. Frank, R. Fromme, I. Grotjohann, R. Henning, M. S. Hunter, R. A. Kirian, I. Kosheleva, C. Kupitz, M. Liang, A. V. Martin, M. M. Nielsen, M. Messerschmidt, M. M. Seibert, J. Sjohamn, F. Stellato, U. Weierstall, N. A. Zatsepin, J. C. Spence, P. Fromme, I. Schlichting, S. Boutet, G. Groenhof, H. N. Chapman and R. Neutze, Nat. Methods, 2014, 11, 923–926 CrossRef CAS PubMed
- A. Bjorling, S. Niebling, M. Marcellini, D. van der Spoel and S. Westenhoff, J. Chem. Theory Comput., 2015, 11, 780–787 CrossRef PubMed
- T. Konuma, T. Kimura, S. Matsumoto, Y. Goto, T. Fujisawa, A. R. Fersht and S. Takahashi, J. Mol. Biol., 2011, 405, 1284–1294 CrossRef CAS PubMed
- P. L. Ramachandran, J. E. Lovett, P. J. Carl, M. Cammarata, J. H. Lee, Y. O. Jung, H. Ihee, C. R. Timmel and J. J. van Thor, J. Am. Chem. Soc., 2011, 133, 9395–9404 CrossRef CAS PubMed
- C. M. Summerford, A. Pardanani, A. H. Betts, A. R. Poteete, G. Colotti and W. E. Royer Jr., Protein Eng., Des. Sel., 1995, 8, 593–599 CrossRef CAS PubMed
- H. Kim, J. G. Kim, S. Muniyappan, T. W. Kim, S. J. Lee and H. Ihee, J. Phys. Chem. B, 2020, 124, 1550–1556 CrossRef CAS PubMed
- J. G. Kim, S. Muniyappan, K. Y. Oang, T. W. Kim, C. Yang, K. H. Kim, J. Kim and H. Ihee, Struct. Dyn., 2016, 3, 023610 CrossRef PubMed
- K. H. Kim, S. Muniyappan, K. Y. Oang, J. G. Kim, S. Nozawa, T. Sato, S. Y. Koshihara, R. Henning, I. Kosheleva, H. Ki, Y. Kim, T. W. Kim, J. Kim, S. Adachi and H. Ihee, J. Am. Chem. Soc., 2012, 134, 7001–7008 CrossRef CAS PubMed
- C. Yang, M. Choi, J. Kim, H. Kim, S. Muniyappan, S. Nozawa, S.-i. Adachi, R. Henning, I. Kosheleva and H. Ihee, Int. J. Mol. Sci., 2018, 19, 3633 CrossRef PubMed
- K. Y. Oang, C. Yang, S. Muniyappan, J. Kim and H. Ihee, Struct. Dyn., 2017, 4, 044013 CrossRef PubMed
- V. Srajer, T. Teng, T. Ursby, C. Pradervand, Z. Ren, S. Adachi, W. Schildkamp, D. Bourgeois, M. Wulff and K. Moffat, Science, 1996, 274, 1726–1729 CrossRef CAS PubMed
- E. Chen, M. J. Wood, A. L. Fink and D. S. Kliger, Biochemistry, 1998, 37, 5589–5598 CrossRef CAS PubMed
- V. Srajer, Z. Ren, T. Y. Teng, M. Schmidt, T. Ursby, D. Bourgeois, C. Pradervand, W. Schildkamp, M. Wulff and K. Moffat, Biochemistry, 2001, 40, 13802–13815 CrossRef CAS PubMed
- F. Schotte, M. Lim, T. A. Jackson, A. V. Smirnov, J. Soman, J. S. Olson, G. N. Phillips Jr., M. Wulff and P. A. Anfinrud, Science, 2003, 300, 1944–1947 CrossRef CAS PubMed
- M. Schmidt, K. Nienhaus, R. Pahl, A. Krasselt, S. Anderson, F. Parak, G. U. Nienhaus and V. Srajer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11704–11709 CrossRef CAS PubMed
- R. t. Aranda, E. J. Levin, F. Schotte, P. A. Anfinrud and G. N. Phillips Jr., Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 776–783 CrossRef PubMed
- M. Cammarata, M. Levantino, F. Schotte, P. A. Anfinrud, F. Ewald, J. Choi, A. Cupane, M. Wulff and H. Ihee, Nat. Methods, 2008, 5, 881–886 CrossRef CAS PubMed
- S. Ahn, K. H. Kim, Y. Kim, J. Kim and H. Ihee, J. Phys. Chem. B, 2009, 113, 13131–13133 CrossRef CAS PubMed
- S. Westenhoff, E. Nazarenko, E. Malmerberg, J. Davidsson, G. Katona and R. Neutze, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 207–219 CrossRef CAS PubMed
- K. H. Kim, K. Y. Oang, J. Kim, J. H. Lee, Y. Kim and H. Ihee, Chem. Commun., 2011, 47, 289–291 RSC
- K. Y. Oang, J. G. Kim, C. Yang, T. W. Kim, Y. Kim, K. H. Kim, J. Kim and H. Ihee, J. Phys. Chem. Lett., 2014, 5, 804–808 CrossRef CAS PubMed
- K. Y. Oang, K. H. Kim, J. Jo, Y. Kim, J. G. Kim, T. W. Kim, S. Jun, J. Kim and H. Ihee, Chem. Phys., 2014, 422, 137–142 CrossRef PubMed
- J. G. Kim, T. W. Kim, J. Kim and H. Ihee, Acc. Chem. Res., 2015, 48, 2200–2208 CrossRef CAS PubMed
- A. Shimada, M. Kubo, S. Baba, K. Yamashita, K. Hirata, G. Ueno, T. Nomura, T. Kimura, K. Shinzawa-Itoh, J. Baba, K. Hatano, Y. Eto, A. Miyamoto, H. Murakami, T. Kumasaka, S. Owada, K. Tono, M. Yabashi, Y. Yamaguchi, S. Yanagisawa, M. Sakaguchi, T. Ogura, R. Komiya, J. Yan, E. Yamashita, M. Yamamoto, H. Ago, S. Yoshikawa and T. Tsukihara, Sci. Adv., 2017, 3, e1603042 CrossRef PubMed
- T. W. Kim, S. J. Lee, J. Jo, J. G. Kim, H. Ki, C. W. Kim, K. H. Cho, J. Choi, J. H. Lee, M. Wulff, Y. M. Rhee and H. Ihee, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14996–15005 CrossRef PubMed
- C. Zang, J. A. Stevens, J. J. Link, L. Guo, L. Wang and D. Zhong, J. Am. Chem. Soc., 2009, 131, 2846–2852 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01207j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |