
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ reversible tuning of chemical interface damping in single gold nanorod-based recyclable platforms through manipulation of supramolecular host–guest interactions†
Hui Bin
Jeon
a,
Sehoon
Park
a,
Kyeong Rim
Ryu
a,
Suman Kr
Ghosh
b,
Jaehoon
Jung
 a,
Kyeng Min
Park
c and
Ji Won
Ha
*ad
a,
Kyeng Min
Park
c and
Ji Won
Ha
*ad
aDepartment of Chemistry, University of Ulsan, 93 Daehak-ro, Nam-gu, Ulsan 44610, Republic of Korea. E-mail: jwha77@ulsan.ac.kr; Fax: +82 52 712 8002; Tel: +82 52 712 8012
bCenter for Self-Assembly and Complexity (CSC), Institute for Basic Science (IBS), Pohang, Gyeongsangbuk-do 37673, Republic of Korea
cDepartment of Biochemistry, School of Medicine, Daegu Catholic University, 33, 17-gil, Duryugongwon-ro, Nam-gu, Daegu 42472, Republic of Korea
dEnergy Harvest-Storage Research Center (EHSRC), University of Ulsan, 93 Daehak-ro, Nam-gu, Ulsan 44610, Republic of Korea
First published on 15th April 2021
Abstract
Recently, chemical interface damping (CID) has been proposed as a new plasmon damping pathway based on interfacial hot-electron transfer from metal to adsorbate molecules. It has been considered essential, owing to its potential implications in efficient photochemical processes and sensing experiments. However, thus far, studies focusing on controlling CID in single gold nanoparticles have been very limited, and in situ reversible tuning has remained a considerable challenge. In these scanning electron microscopy-correlated dark-field spectroscopic measurements and density functional theory calculations, cucurbit[7]uril (CB[7])-based host–guest supramolecular interactions were employed to examine and control the CID process using monoamine-functionalized CB[7] (CB[7]-NH2) attached to single gold nanorods (AuNRs). In situ tuning of CID through the CB[7]–oxaliplatin complexation, which can result in the variation of the chemical nature and electronic properties of adsorbates, was presented. In addition, in situ tuning of CID was demonstrated through the competitive release of the oxaliplatin guest from the oxaliplatin@CB[7] complex, which was then replaced by a competitor guest of spermine in sufficient amounts. Furthermore, nuclear magnetic resonance experiments confirmed that the release of the guest is the consequence of adding salt (NaCl). Thus, in situ reversible tuning of CID in single AuNRs was achieved through successive steps of encapsulation and release of the guest on the same AuNR in a flow cell. Finally, single CB[7]-NH2@AuNRs were presented as a recyclable platform for CID investigations after the complete release of guest molecules from their host–guest inclusion complexes. Therefore, this study has paved a new route to achieve in situ reversible tuning of CID in the same AuNR and to investigate the CID process using CB-based host–guest chemistry with various guest molecules in single AuNRs for efficient hot-electron photochemistry and biosensing applications.
Introduction
Plasmon damping pathways have been attracting considerable interest due to their potential implications in photochemical processes and sensing experiments.1,2 The localized surface plasmon resonance (LSPR) total damping channel has been regarded as the sum of the following: (1) bulk damping (Γbulk), (2) radiation damping (Γrad), (3) electron-surface scattering (Γsurf), and (4) chemical interface damping (CID, ΓCID).2–4 Among them, CID is the most recently proposed damping pathway, which is based on the interfacial hot-electron transfer from metal to adsorbate molecules, and its efficient use in various photochemical and photocatalytic reactions has been considered essential.1,5,6 Therefore, mastering CID and its optimization will aid in controlling the efficiency of hot-electron generation and transfer in artificial systems.7–9Single gold nanorods (AuNRs) are often the targeted metal due to their shape-dependent unique optical properties, easy surface modification, photostability, and biocompatibility.10–12 Binding between sulfur or nitrogen atoms and AuNRs, with their preferred and strong covalent soft–soft bonding, is often used in CID studies.13–19 In these chemical processes, the presence of closely or strongly interacting adsorbate molecules induces the direct generation of hot electrons in the empty lowest unoccupied molecular orbital (LUMO) of the adsorbate (Scheme 1A).1,20–23 In addition to the direct electron transfer, CID has a component of scattering of the electronic waves by a disordered potential at the surface. Accordingly, the LSPR spectral frequency is red-shifted and suffers a loss in intensity and a broadening of the full linewidth at half maximum (FWHM).4,24–26 Therefore, AuNRs that are attached to thiolated or amine-functionalized adsorbates are identified as good candidates for the effective investigation of CID in the total LSPR damping channel.
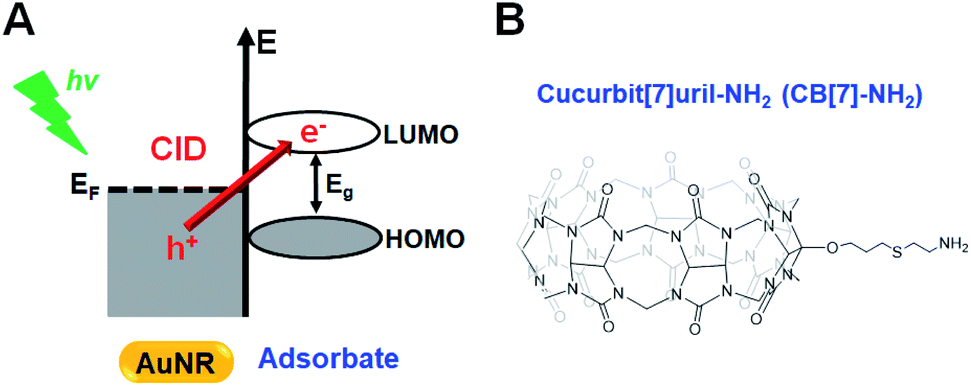 | ||
| Scheme 1 (A) Schematic diagram illustrating the direct mechanism (or CID). Hot electrons are directly generated in the LUMO, which leaves hot holes in the occupied state of the metal. (B) Chemical structure of monoamine-cucurbit[7]uril (CB[7]-NH2). | ||
Given the proportional relationship between the LSPR linewidth and damping rate, the CID relaxation pathway can be effectively examined using a homogeneous LSPR spectral linewidth by far-field single-particle spectroscopic techniques, overcoming the time scale limitation of ultrafast spectroscopic techniques.27–30 Recently, a homogeneous LSPR linewidth has been employed by several groups in studies examining the CID of single AuNRs with 1-alkanethiols or pyridines.4,15,18,23 For example, Foerster et al. reported the time evolution of the Langmuir adsorption model in surface chemistry and further confirmed that CID is dependent on the electrons reaching the surface.4 Meanwhile, Ryu et al. examined the effect of the preferred orientation of pyridine derivatives on CID in silver-coated AuNRs.31 Furthermore, Lee et al. were the first to investigate the tuning of homogeneous LSPR linewidths through interfacial electronic effects of thiolated aromatic benzene rings with additional functional groups.2 However, despite these recent contributions, thus far, research on the ability to tune CID in single AuNRs has been very limited, and more notably, in situ reversible tuning of CID in single AuNRs has remained a considerable challenge.
Cucurbituril (CB) is a family of homologues identified as the most favored cavitands for host–guest complex formation.32,33 The CB cavity provides a hydrophobic void for the binding of neutral hydrophobic molecules, while the two identical carbonyl portals represent docking sites for positively charged groups.34,35 Among the CB homologues, cucurbit[7]uril (CB[7]) has drawn particular attention due to its high biocompatibility, remarkable solubility in aqueous solution, and strong affinity with many kinds of guest molecules.34,36 In recent years, CB[7]-based host–guest systems have been employed in the control of target structure and function.37–41 Therefore, CB[7]-based supramolecular chemistry has the potential to be used for the reversible tuning of CID in single AuNRs with various guest molecules.
In this study, scanning electron microscopy (SEM)-correlated dark-field (DF) microscopy and spectroscopic measurements of single AuNRs were performed to examine their CID process in combination with density functional theory (DFT) calculations and nuclear magnetic resonance (NMR) measurements. This study employed host–guest supramolecular chemistry using monoamine-functionalized CB[7] (CB[7]-NH2, Scheme 1B) attached to single AuNRs to investigate and control the CID process. This investigation presents in situ reversible tuning of CID in single AuNRs by manipulating host–guest supramolecular interactions in a flow cell. Furthermore, single CB[7]-NH2@AuNRs are presented as a new recyclable platform for CID investigations after the release of guest molecules from their host–guest inclusion complexes, which is a result of the addition of salt.
Results and discussion
Single–particle correlation study on CID induced by CB[7]-NH2 in AuNRs
The AuNRs used in this study were purchased from Nanopartz (Loveland, CO, USA). Cetyltrimethylammonium bromide (CTAB)-stabilized AuNRs with a rod-like anisotropic shape were observed clearly via SEM and transmission electron microscopy (TEM) (Fig. S1†). Their mean size was determined to be 25.1(±2.78) nm × 75.4(±3.53) nm (Fig. S2†). Fig. S3† shows the ensemble extinction spectrum of AuNRs in water and further shows a high-energy transverse peak (522 nm) and a low-energy longitudinal peak (713 nm). However, ensemble studies remain limited by the slight heterogeneity (distribution of the size or shape) of individual particles, and hence, single-particle studies are needed to better understand the optical properties of AuNRs without ensemble averaging.In this study, SEM-correlated single-particle DF spectroscopy was performed to eliminate ensemble averaging effects. The experimental setup for single-particle DF microscopy and spectroscopy is shown in Fig. S4,† and scattering-based DF microscopy has been considered as a powerful technique in visualizing and characterizing single Au nanoparticles (Fig. S5†). As shown in Fig. 1, correlated SEM and optical imaging confirmed that single AuNRs were measured within the diffraction-limited optical detection area. The remaining results show only the widths from homogeneous spectral characterization.2 In this correlation study, a gold pattern generated using a TEM grid (Ted Pella) enabled the same AuNRs to be observed (Fig. S6, full details in the ESI†). Fig. 1B shows a correlated DF scattering image of single AuNRs excited by white light illumination. Furthermore, the amine group in CB[7]-NH2 have the preferred and strong soft–soft interactions (or dative covalent bonds) between nitrogen atoms and gold surfaces.18,31,42,43 However, to further facilitate the adsorption of CB[7]-NH2 onto AuNR surfaces, the capping material, CTAB, on the nanoparticle surfaces was removed by oxygen plasma treatment.31,44Fig. 1C shows the single-particle scattering spectra of AuNR1, highlighted in Fig. 1B, 60 min after the adsorption of CB[7]-NH2 molecules on the AuNR surface in water. Moreover, the scattering spectrum of single AuNR1 was well fitted with a Lorentzian function, which was used to obtain its homogeneous LSPR linewidth (or FWHM) and LSPR wavelength (Fig. 1C). After the adsorption of CB[7]-NH2 on the particle surface, the LSPR scattering peak of AuNR1 was red-shifted due to multiple factors, including an increase in the local dielectric constant surrounding the nanoparticle, a decrease in the electron density in the metal due to electron transfer to adsorbates, etc. (Fig. 1C and S7†). Furthermore, the LSPR linewidth considerably increased, together with a strongly decreased scattering intensity (Fig. 1C and S7†). The single-particle scattering spectra of AuNR2 shown in Fig. 1B are provided in the ESI (Fig. S8†). It should be noted that CID reduces the scattering intensity of AuNRs because a fraction of the plasmon energy is lost nonradiatively.4 Therefore, the LSPR broadening (or the loss in intensity) can be clearly attributed to CID induced by the adsorption of CB[7]-NH2 on the AuNR surface through the strong nitrogen–Au interaction.4,18,24,31,43
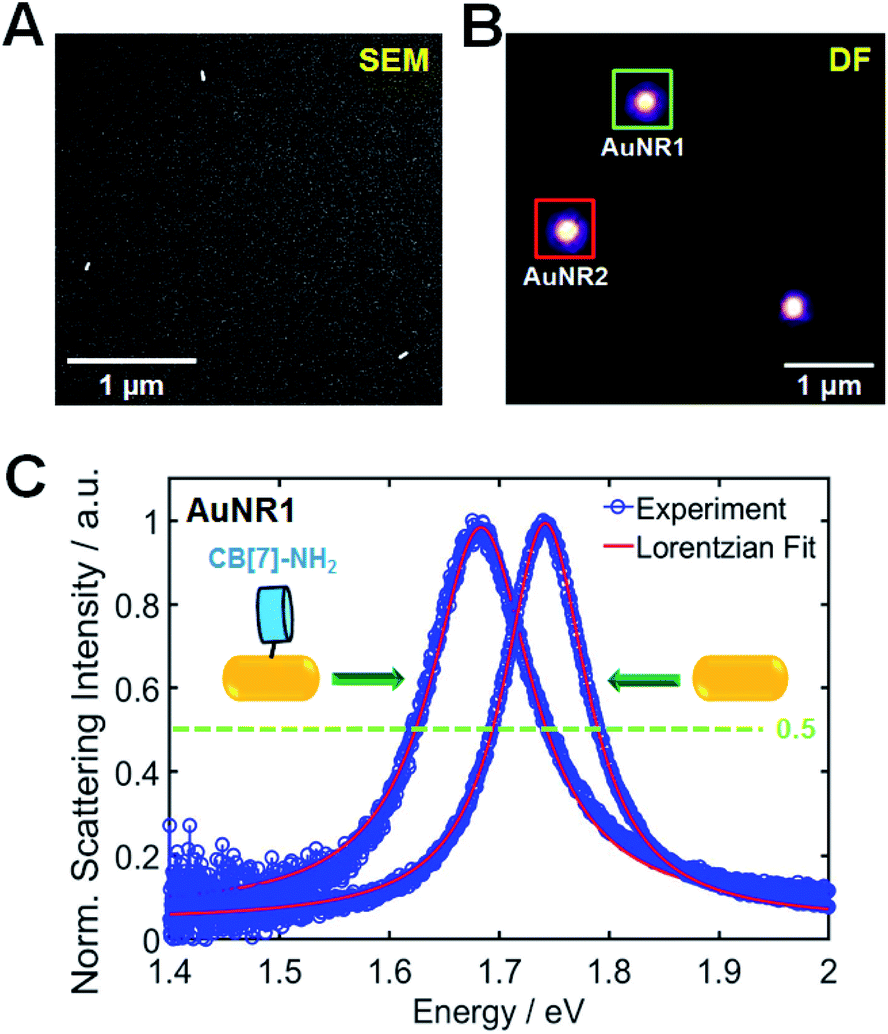 | ||
| Fig. 1 Single-particle correlation study. (A) SEM image of single AuNRs. (B) DF scattering images of single AuNRs (A) fixed on a glass slide in water and excited by white light illumination. (C) Overlaid DF scattering spectra of a single AuNR1 before and after the adsorption of CB[7]-NH2. Single-particle scattering spectra of AuNR1 are well fitted using a Lorentzian function to extract the homogeneous LSPR linewidth. | ||
Host–guest supramolecular approach for examining CID of single AuNRs
To obtain a new insight into the effect of host–guest supramolecular interactions on the LSPR linewidth broadening in single AuNRs, SEM-correlated single-particle DF spectroscopic measurements were further examined using a homemade flow cell with a syringe pump (Fig. S9†). As shown in Fig. 2A, CB[7]-NH2 was utilized as a starting material (step 1) to modify the chemical interface on the surface of single AuNRs in a flow cell. In our CID studies using single AuNRs, their orientations on the glass slide could affect the experimental results because CID is a function of the adsorbate coverage. Therefore, it was confirmed by a defocused orientation imaging technique45–47 that single AuNRs had an almost equal orientation (i.e., laid flat on the glass surface) on the glass slide. As shown in Fig. S10–S12,† we provided the measured and best-matched simulation patterns for single AuNRs. No orientation effects in CID experiments on AuNRs were observed, meaning that only chemical effects were focused on. Full details including the simulation are provided in the ESI.†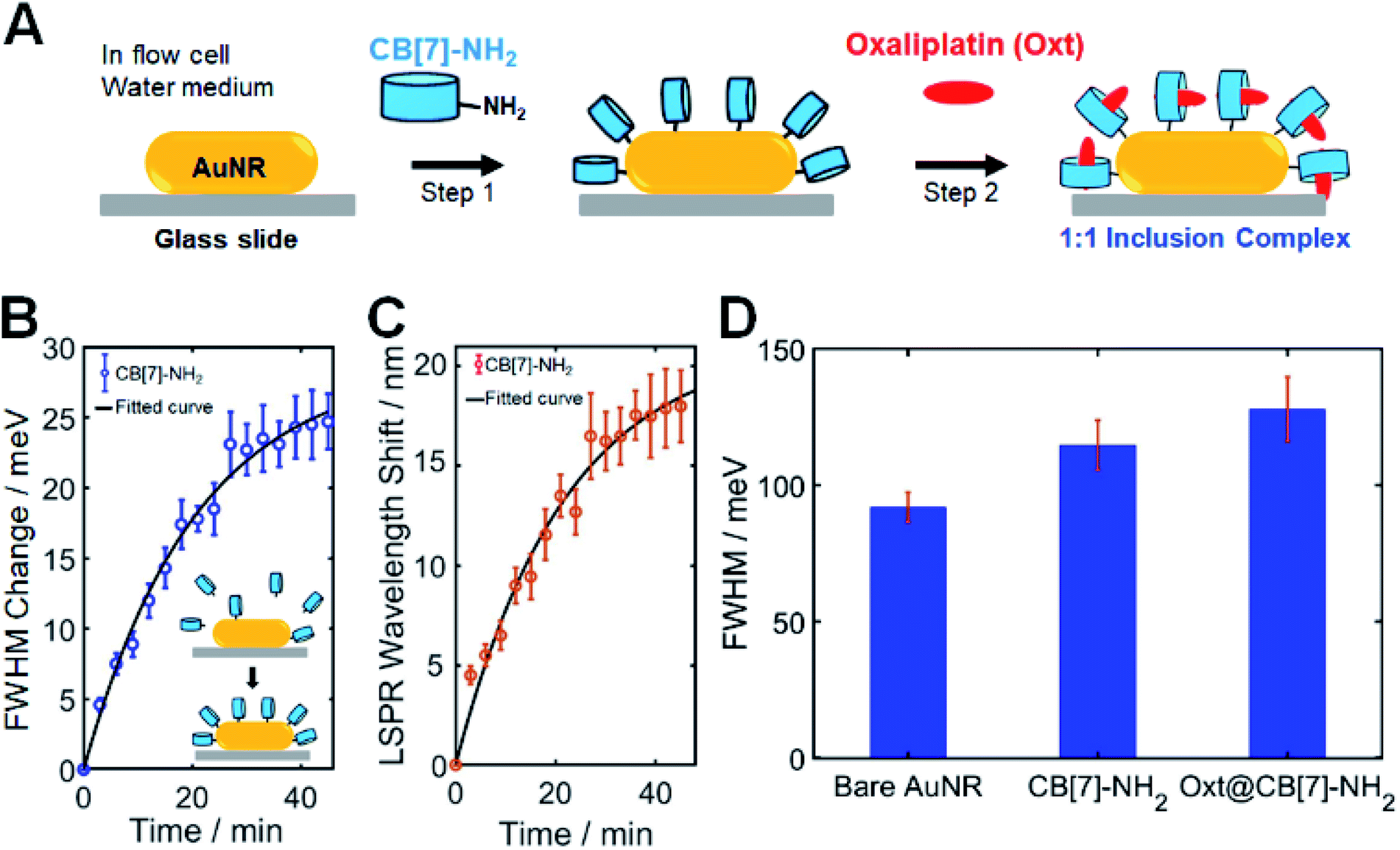 | ||
| Fig. 2 Host–guest supramolecular chemistry in investigating CID of single AuNRs in a flow cell. (A) Schematic to show CID induced by CB[7]-NH2 and its inclusion complex formed with CB[7] and the oxaliplatin guest. Using CB[7], oxaliplatin can be incorporated into its hydrophobic cavity. (B) Time dependence of CB[7]-NH2 adsorption observed via FWHM broadening of single AuNRs. The scattering spectra of 50 single AuNRs were repeatedly obtained for a period of approximately 60 min, and the LSPR linewidth, ΔΓ, was extracted with respect to the starting value. The fitted curves are shown in black. (C) Corresponding LSPR wavelength shift of single AuNRs shown in B. (D) Bar graphs to show the comparison of FHWM induced by bare AuNRs without the capping material as the control, AuNRs with CB[7]-NH2, and AuNRs with oxt@CB[7]-NH2. | ||
In order to quantitatively compare the amount of CID when using different adsorbate molecules, it was necessary to establish whether comparable, if not complete, coverage of adsorbate molecules was achieved. Therefore, a time evolution typical for a Langmuir adsorption isotherm was employed, i.e., θ = 1 − exp(−kLct), where the coverage θ depends on the time t, thiol concentration c, and Langmuir adsorption constant kL.2 Detailed information is provided in the ESI.†Fig. 2B shows the time dependence of CB[7]-NH2 adsorption observed via LSPR linewidth broadening of single AuNRs. The scattering spectra of 50 single AuNRs were repeatedly obtained over a period of approximately 60 min, and the LSPR linewidth, ΔΓ, was extracted with respect to the starting value (Fig. 2B). Fig. 2C shows the corresponding LSPR shifts induced by CB[7]-NH2. The presented kinetics of the FWHM change (or CID) and LSPR shifts (Fig. 2B and C) are averages of 50 single AuNRs and the error bars are their standard deviations. As clearly shown in Fig. 2B, the FWHM change (or broadening) increased after introducing a CB[7]-NH2 solution (1 mM) through a flow cell (at a flow rate of 1 μL min−1), showing saturation after 60 min, which was consistent with the linewidth broadening shown in Fig. 1C. Furthermore, a red shift of the LSPR wavelength was simultaneously observed over time due to the adsorption of CB[7]-NH2 on AuNR surfaces (Fig. 2C).
The second step (step 2) in Fig. 2A shows that oxaliplatin (Fig. S13†), which was used as a guest for CB[7], was flowed into CB[7]-NH2@AuNRs through a flow cell. Jeon et al. demonstrated that oxaliplatin forms a stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex with CB[7], as confirmed by NMR and X-ray crystallographic studies, and the 1,2-diaminocyclohexane ligand of the oxaliplatin guest is embedded in the CB[7] host cavity.48 They demonstrated that the amine nitrogen atoms of the guest were on the plane formed by seven oxygen atoms of CB[7] portals while forming hydrogen bonds with the oxygen atoms. In addition, they calculated the binding constant (Ka) measured in a TRIS buffer to be 2.3 × 105 M−1 in order for the guest oxaliplatin to bind to CB[7]. Using 1H NMR spectroscopy, this study confirmed that the addition of oxaliplatin to CB[7] in water resulted in the formation of an inclusion complex (Fig. S15†). Fig. S14† shows that all peaks (a–e) for the cyclohexyl ring protons of the guest are shifted upfield relative to those of the free guest (oxaliplatin), which indicates that the cyclohexyl ring is now located inside the cavity of CB[7]. Our NMR results are consistent with the previous study conducted by Jeon et al.48 Therefore, when introducing the oxaliplatin solution through a flow cell in this study, the guest was encapsulated by CB[7]-NH2 attached to the AuNR surface in order to form a stable 1:1 host–guest inclusion complex of oxt@CB[7]-NH2.36,48 Furthermore, Fig. 2D shows the equilibrium ΔΓCID values obtained by fitting a Langmuir adsorption process using a bar graph for each sample with error bars, which allowed a quantitative comparison of the amount of CID induced by CB[7]-NH2 without guest molecules and oxt@CB[7]-NH2 under the same experimental conditions. In Fig. 2D, the FWHM value of oxt@CB[7]-NH2 was obtained 60 min after the CB[7]-NH2 molecules were adsorbed on AuNR surfaces in a cell. This result was further confirmed by the equilibrium ΔΓCID value obtained by fitting a time-dependent Langmuir adsorption process for 50 single AuNRs after making a solution of the 1:1 inclusion complex and introducing the pre-made oxt@CB[7]-NH2 solution through a flow cell (Fig. S15†). Interestingly, compared to CB[7]-NH2 without guest molecules, additional LSPR linewidth broadening was observed for the oxt@CB[7]-NH2 complex, and the FWHM value increased further to ∼134 meV. Therefore, in Fig. 2D and S15,† the increased LSPR broadening for oxt@CB[7]-NH2 could be ascribed to more enhanced CID (or direct electron transfer to the empty LUMO of the adsorbate) in single AuNRs.
:1 inclusion complex with CB[7], as confirmed by NMR and X-ray crystallographic studies, and the 1,2-diaminocyclohexane ligand of the oxaliplatin guest is embedded in the CB[7] host cavity.48 They demonstrated that the amine nitrogen atoms of the guest were on the plane formed by seven oxygen atoms of CB[7] portals while forming hydrogen bonds with the oxygen atoms. In addition, they calculated the binding constant (Ka) measured in a TRIS buffer to be 2.3 × 105 M−1 in order for the guest oxaliplatin to bind to CB[7]. Using 1H NMR spectroscopy, this study confirmed that the addition of oxaliplatin to CB[7] in water resulted in the formation of an inclusion complex (Fig. S15†). Fig. S14† shows that all peaks (a–e) for the cyclohexyl ring protons of the guest are shifted upfield relative to those of the free guest (oxaliplatin), which indicates that the cyclohexyl ring is now located inside the cavity of CB[7]. Our NMR results are consistent with the previous study conducted by Jeon et al.48 Therefore, when introducing the oxaliplatin solution through a flow cell in this study, the guest was encapsulated by CB[7]-NH2 attached to the AuNR surface in order to form a stable 1:1 host–guest inclusion complex of oxt@CB[7]-NH2.36,48 Furthermore, Fig. 2D shows the equilibrium ΔΓCID values obtained by fitting a Langmuir adsorption process using a bar graph for each sample with error bars, which allowed a quantitative comparison of the amount of CID induced by CB[7]-NH2 without guest molecules and oxt@CB[7]-NH2 under the same experimental conditions. In Fig. 2D, the FWHM value of oxt@CB[7]-NH2 was obtained 60 min after the CB[7]-NH2 molecules were adsorbed on AuNR surfaces in a cell. This result was further confirmed by the equilibrium ΔΓCID value obtained by fitting a time-dependent Langmuir adsorption process for 50 single AuNRs after making a solution of the 1:1 inclusion complex and introducing the pre-made oxt@CB[7]-NH2 solution through a flow cell (Fig. S15†). Interestingly, compared to CB[7]-NH2 without guest molecules, additional LSPR linewidth broadening was observed for the oxt@CB[7]-NH2 complex, and the FWHM value increased further to ∼134 meV. Therefore, in Fig. 2D and S15,† the increased LSPR broadening for oxt@CB[7]-NH2 could be ascribed to more enhanced CID (or direct electron transfer to the empty LUMO of the adsorbate) in single AuNRs.
Nonspecific binding of the guest (oxaliplatin) is an important point that needs to be discussed regarding Fig. 2. Thus, to verify the effect of nonspecific adsorption of the guest, we performed single particle DF experiments and added the guest only to the AuNRs without prior CB[7]-NH2 adsorption. As shown in Fig. S16,† the FWHM value induced by the guest was comparable to that of the bare AuNR. In contrast, there was a significant change in the FWHM value induced by specific adsorption of CB[7]-NH2 on the AuNR surfaces (Fig. 2D and S16†).
An important point regarding Fig. 2 warrants further discussion. To date, CID investigations have been performed using adsorbate molecules containing thiol or amine groups that can strongly bind to AuNR surfaces.2,4,15,18,31,49 However, it has been a challenge to achieve the in situ change and tuning of CID after their strong adsorption on the AuNR surface. To overcome this challenge, the present study introduced CB[7]-based supramolecular host–guest chemistry into CID investigations using single AuNRs. As demonstrated by single-particle DF measurements in Fig. 2, we were able to use the complexation of a guest with the CB[7] host as a means to change the chemical nature and electronic structures of the adsorbate attached to AuNR surfaces.
In situ tuning of CID by competitive release and replacement in single AuNRs
In the previous section, we showed a new approach for in situ tuning of CID in single AuNRs through the encapsulation of various guest molecules into CB[7] cavities. Another way of achieving in situ tuning of CID in single AuNRs is through the competitive release and replacement by using a competitor guest. For example, after the complex formation of oxt@CB[7]-NH2 on the AuNR surface, we can further perform the in situ change and control of CID by inducing the competitive release and replacement using a competitor guest. To demonstrate this, we conducted single-particle DF spectroscopic measurements by adding spermine in water, as a competitor guest with positive charges and considerable binding affinity (Ka = 2.9 × 106 M−1)36 for CB[7], to oxt@CB[7]-NH2 bound to the AuNR surface (Fig. 3). Fig. 3A shows that by adding spermine at high concentration in water through a flow cell, the encapsulated oxt@CB[7] complex is destroyed, releasing the oxaliplatin guest from the CB[7] cavity.34,36 Similar to oxaliplatin, spermine forms a stable 1:1 inclusion complex with CB[7].36,40 Thus, the spermine guest will be encapsulated by the CB[7] host through competitive binding, and it will eventually reach equilibrium after the replacement.
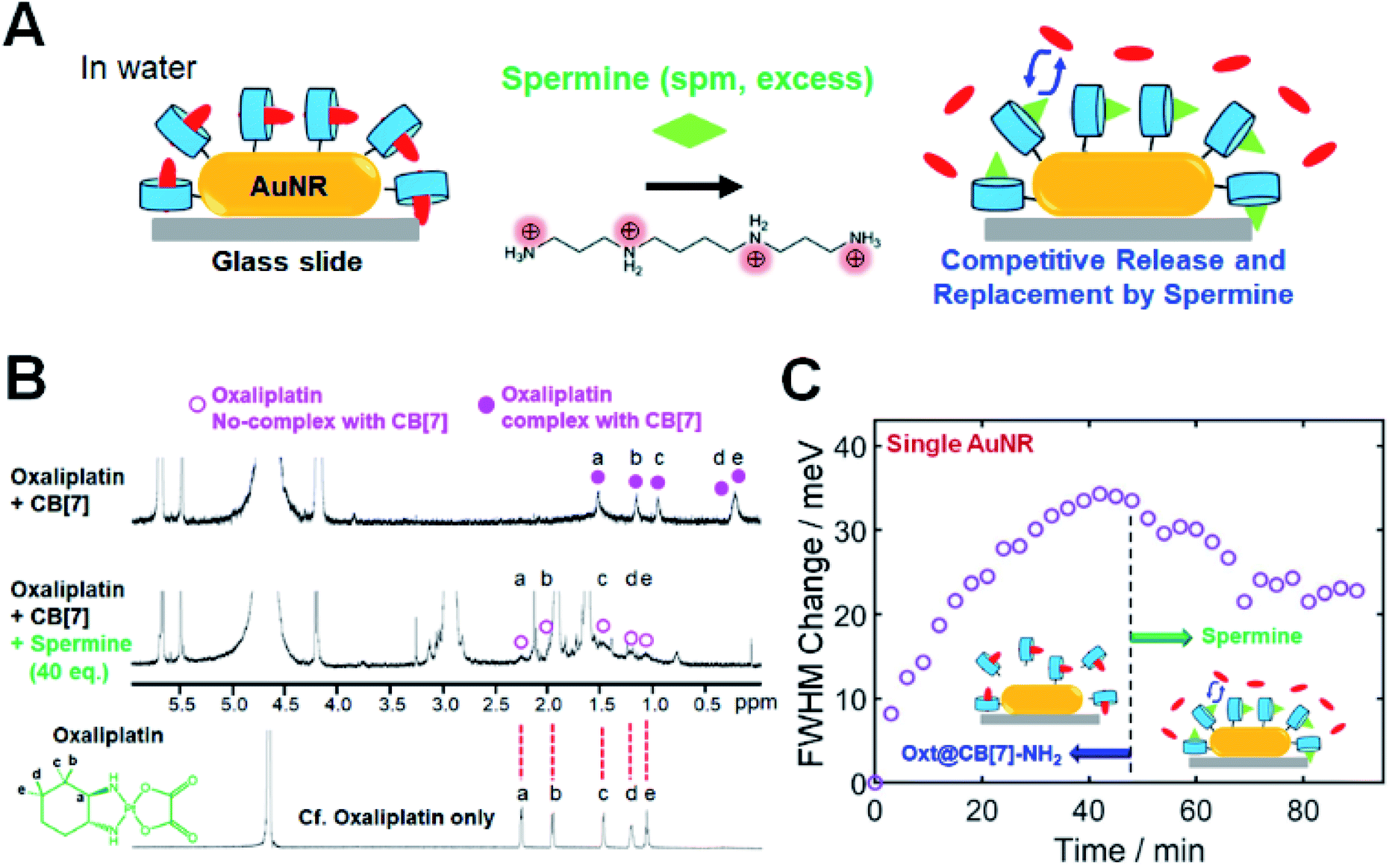 | ||
| Fig. 3 In situ tuning of CID through the competitive release and replacement by spermine. (A) Schematic showing the release of oxaliplatin from its host–guest complex with CB[7] and replacement by a competitive guest of spermine in sufficient amounts. (B) NMR spectra to reveal the release of oxaliplatin from the oxt@CB[7] complex and replacement by spermine at high concentration (40 eq.). (C) Time dependence of the oxt@CB[7]-NH2 adsorption observed via the FWHM change for a single AuNR with respect to the starting value. At the saturation point after 45 min, a competitive guest of spermine at high concentration (50 eq.) was introduced through a flow cell, and the FWHM change was decreased by spermine to ∼23 meV in the same AuNR as a function of time. | ||
First, 1H NMR spectroscopic measurements were conducted to experimentally confirm the formation of the oxt@CB[7] complex and the competitive replacement process by spermine. As shown in Fig. 3B, the protons of 1,2-diaminocyclohexane as the ligand of oxaliplatin showed significant upfield shifts after its complexation with CB[7], which indicated that the 1,2-diaminocyclohexane group was incorporated into CB[7]. Spermine was then quantitatively added to the oxt@CB[7]-NH2 complex at ratios of 10.0, 20.0, and 40.0 eq. (Fig. S17†). As the amount of spermine increased, the NMR signals (a–e) of the protons of 1,2-diaminocyclohexane on oxaliplatin gradually decreased and shifted downfield to the peak positions of oxaliplatin only, which indicated a dynamic exchange of oxaliplatin and spermine in the cavity of CB[7] and the release of oxaliplatin by spermine (Fig. 3B and S17†). The results indicate that the competitive replacement of oxaliplatin by spermine is a concentration-dependent process and that the competitive binding process is dependent on a sufficient amount of spermine.
To further demonstrate and clarify the capability of the competitive replacement of oxaliplatin from the oxt@CB[7] complex by a competitive guest, we investigated the host–guest chemistry using 1-trimethylammoniomethylferrocene (FcA, another competitor guest), which has been identified to have a much stronger binding affinity (Ka of >1012 M−1 in water)33 for CB[7] than spermine (Ka of 106 M−1). After adding FcA into oxt@CB[7] complexes, the NMR signals of protons of 1,2-diaminocyclohexane on oxaliplatin were observed to have shifted downfield to the peak positions of oxaliplatin only, which indicated the release of oxaliplatin and replacement by FcA in the cavity of CB[7] (Fig. S18†). Therefore, the NMR results show that the competitive guests (FcA and spermine) can replace oxaliplatin from the oxt@CB[7] complex on single AuNRs.
Then, we performed single-particle spectroscopic measurements to verify the in situ variation of CID through the competitive release and replacement by using a competitor guest of spermine in sufficient amounts in a flow cell. In an aqueous solution at physiological pH, spermine has a polycationic character, and all of its amino groups are positively charged, as shown in Fig. 3A. Fig. 3C shows the time dependence of the oxt@CB[7]-NH2 adsorption observed via the FWHM change for a single AuNR, with respect to the starting value. Fig. 3C clearly shows that the FWHM change of the single AuNR increased after introducing an oxt@CB[7]-NH2 solution (1 mM) through a flow cell, and saturation was observed after 45 min. Then, at the saturation point, we introduced a competitive guest (spermine) at high concentration (50 eq.) through a flow cell. As observed in Fig. 3C, the FWHM change decreased to ∼23 meV in the same AuNR over time, saturating again after the competitive release and replacement by spermine. The FWHM change value at saturation was further evaluated and confirmed by single-particle DF measurements on the time-dependent adsorption of the spm@CB[7]-NH2 complex, which was prepared using the 1:1 reaction of spermine and CB[7]-NH2 in water and observed via the FWHM change for single AuNRs (Fig. S19 and S20†). Therefore, our findings show that the oxt@CB[7]-NH2 complex could be changed either to the spm@CB[7]-NH2 complex or to the FcA@CB[7]-NH2 complex by manipulating the host–guest supramolecular interactions on the same AuNR. This strategy using the CB[7]-based host–guest chemistry enables the in situ change and tuning of CID (or FWHM broadening) in the same AuNR.
Further discussion is necessary regarding Fig. 2 and 3. Our NMR data to support the formation of an inclusion complex were acquired in free solution (Fig. S15, S17 and S18†). Thus, host–guest chemistry might be modified by the proximity of the metal (or Au). However, single-particle experimental data (Fig. 2D and 3C) support that guest molecules actively interact with CB[7]-NH2 on the AuNR surfaces and that the metal has a minor effect on CB[7]-based host–guest interactions.
Computational study of inclusion host–guest complexes, M@CB[7]
To gain a detailed insight into the experimental observations in the previous sections, a computational study using semi-empirical tight-binding and DFT calculations were performed (see Experimental methods for computation details). More specifically, the computational study was carried out to gain insight into the CID investigations using CB-based host–guest chemistry, to deepen our understanding of the increased CID in the oxt@CB[7] complex (Fig. 2D), and to better understand why CID decreased in the spm@CB[7]complex (Fig. 3C and S20†) compared to the oxt@CB[7] complex.In this study, we assumed that the geometric and electronic properties of M@CB[7]-NH2 (guest molecule, M = oxaliplatin and spermine) adsorbed on AuNR can be examined with an isolated M@CB[7] complex because the M@CB[7] complex is connected with AuNR through a rather long chain, i.e., –O(CH2)3S(CH2)2NH2– (see Scheme 1B). The molecular structures of M@CB[7] complexes were optimized with the self-consistent tight-binding (GFN2-xTB) method (Fig. S21 and S22†),50 and their electronic properties and encapsulation energies were examined using DFT calculations with the PBE0-D3 method.51 The influence of water medium (dielectric constant, ε = 78.4) was considered using the conductor-like screening model (COSMO) technique52 and polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM)53 during the geometry optimization and single point calculation, respectively. Fig. 4A shows the most stable geometries for optimized oxt@CB[7] and spm@CB[7] and reveals that hydrogen bonding plays a crucial role in the formation of both complexes, as indicated with black dotted lines. For the optimized oxt@CB[7] complex, the cyclohexyl ring of oxaliplatin is located deep inside the CB[7] cavity, and its amine groups form two hydrogen bonds (dHB < 2.5 Å) with portal oxygen atoms of CB[7], which is consistent with the previous experimental result.48 Compared to oxt@CB[7], the flexible geometry of spermine can provide a greater opportunity to maximize hydrogen bonding by twirling its sp3-based chain, and thus, 11 hydrogen bonds are formed between the four protonated amine groups of spermine and portal oxygen atoms of CB[7]. The electrostatic interaction between the positive Pt2+ ion of oxaliplatin and a negatively polarized portal oxygen atom of CB[7] contributes to the formation of oxt@CB[7] (red dotted line, Fig. 4A; electrostatic potential (ESP) map, Fig. 4B). However, the highly positive molecular charge (+4) of spermine can lead to higher electrostatic interactions with negatively polarized portal oxygen atoms (Fig. 4B). The ESP maps for isolated CB[7], oxaliplatin, and spermine are provided in Fig. S23.† The stronger host–guest interaction of spm@CB[7] than that of oxt@CB[7] leads to an exothermic encapsulation energy of 70.57 kcal mol−1 for spm@CB[7] (Fig. S22†), which is about 3.5 times higher than that of oxt@CB[7] (20.78 kcal mol−1) (Fig. S21†). Therefore, the experimental observation shown in Fig. 3A, i.e., the competitive replacement of guest molecules in oxt@CB[7] with excess spermine, can be easily understood with the difference in the host–guest interaction between the CB[7] cavitand and guest molecules.
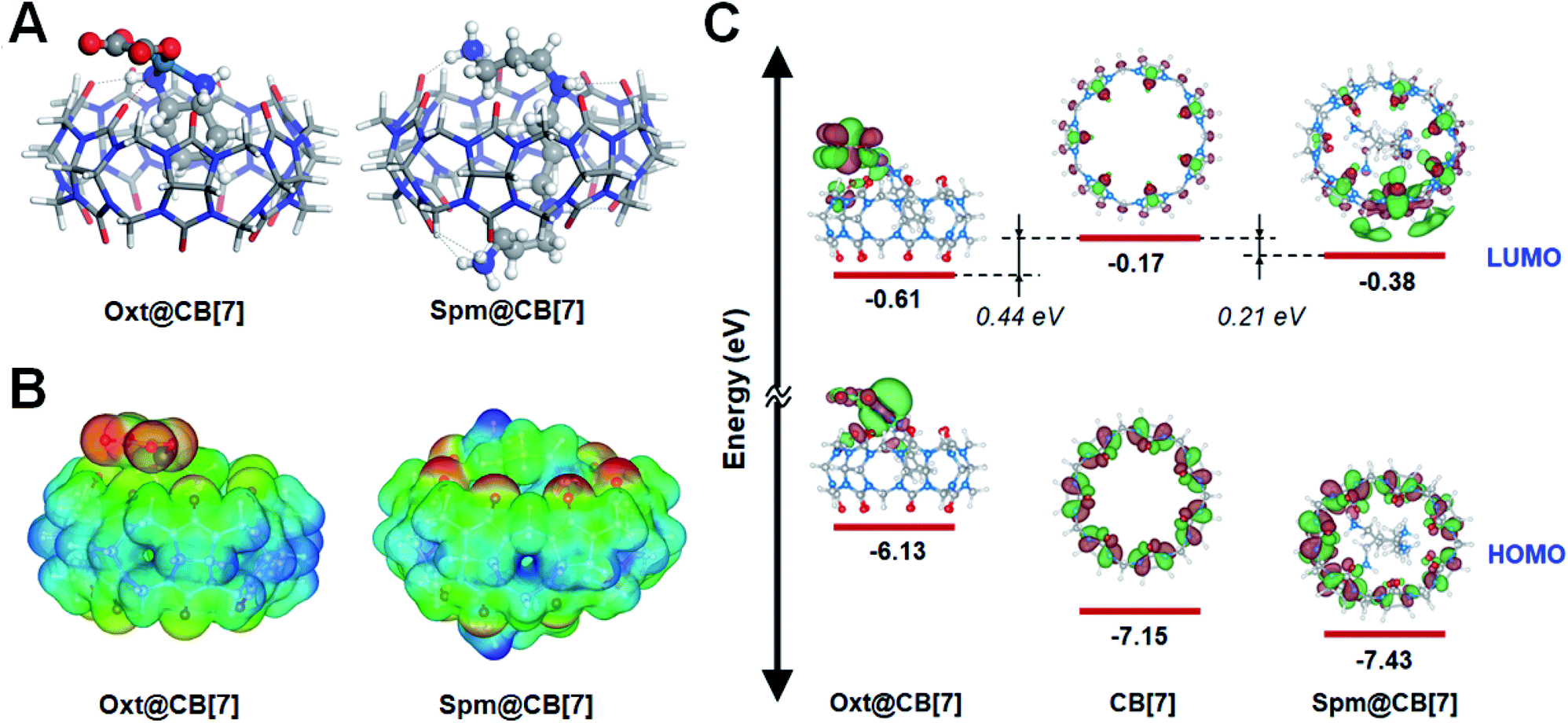 | ||
| Fig. 4 DFT calculations of CB[7] and the inclusion complexes (oxt@CB[7] and spm@CB[7]). (A) Optimized structures of oxt@CB[7] (left) and spm@CB[7] (right) complexes. Host (CB[7]) and guest (oxaliplatin and spermine) molecules are displayed with stick and ball-and-stick models, respectively. The black and red dotted lines denote N–H⋯O hydrogen bonding and Pt⋯O electrostatic interactions, respectively. (B) Electrostatic potential (ESP) maps of oxt@CB[7] (left) and spm@CB[7] (right) complexes. Color scale from red to blue corresponds to negative to positive. (C) Molecular orbital energy diagram for CB[7], the oxt@CB[7] complex, and spm@CB[7] complex. | ||
Fig. 4C shows the energy level diagram evaluated with the PBE-D3 method for the frontier molecular orbitals of CB[7] and both M@CB[7] complexes, which can further be used for elucidating the relative increase of CID depending on the guest molecule of M@CB[7]-NH2 compared with CID of CB[7] only (see Fig. 2D, 3C, and S20†). As assumed in our calculation, the energy level alignment of M@CB[7]-NH2 with respect to the Fermi level of the AuNR is expected to be marginally influenced by the guest molecule because CB[7] is not directly in contact with the AuNR. Therefore, considering the hot-electron transfer from the AuNR to the vacant CB[7] or its inclusion complexes in the CID mechanism (Scheme 1A), the energy level of the LUMO can be attributed to the relative strength of CID. The energy levels of the LUMO for CB[7], oxt@CB[7], and spm@CB[7] were evaluated to be −0.17, −0.61, and −0.38 eV, respectively, using the PBE-D3 method. The more stable LUMO energy can be understood as a closer distribution of the unoccupied electronic staes of the Fermi level of the AuNR, and thus, it can suggest the higher feasibility of the CID process. The LUMO energies of oxt@CB[7] and spm@CB[7] are more stable than that of CB[7] by 0.44 and 0.21 eV, respectively. As shown in Fig. 3C, the FWHM change as an indicator of CID gradually increases with the encapsulation of oxaliplatin and then decreases with the competitive replacement by spermine. The FWHM change with respect to CB[7] is still observed in the saturated stage of spm@CB[7], although its saturated value is lower than that of oxt@CB[7]. Therefore, the DFT calculations reasonably describe that the interfacial direct transfer of plasmon-induced energetic hot electrons generated in AuNRs to the LUMO of M@CB[7] is more efficient than that to CB[7] only, and furthermore, oxt@CB[7] induces more effective CID compared with spm@CB[7], which is consistent with the experimental results (Fig. 3C and S20†).
In situ reversible tuning of CID through manipulation of host–guest interactions in single AuNRs
In the previous CID investigations using 1-alkanethiol or thiophenol, which are strongly adsorbed on AuNR surfaces, in situ reversible tuning of FWHM broadening has not been considered and accomplished.2,4,15 Thus, in this study, we attempted to achieve in situ reversible tuning of CID by controlling CB[7]-based host–guest interactions in single AuNRs. In addition to the release and replacement by a competitive guest, as described in the previous section, another way of releasing encapsulated guest molecules is to use inorganic cations such as salts (NaCl).34,54,55 Cations have been determined to competitively displace cationic guests from the CB cavity by binding at the portals. As a result, the effective binding constants of the guests will be reduced in the presence of salts. Therefore, salts can trigger the release of guests from CB complexes; adding salt results in the release of the guest in the case of the 1:1 complex, as shown in Fig. 5A.34
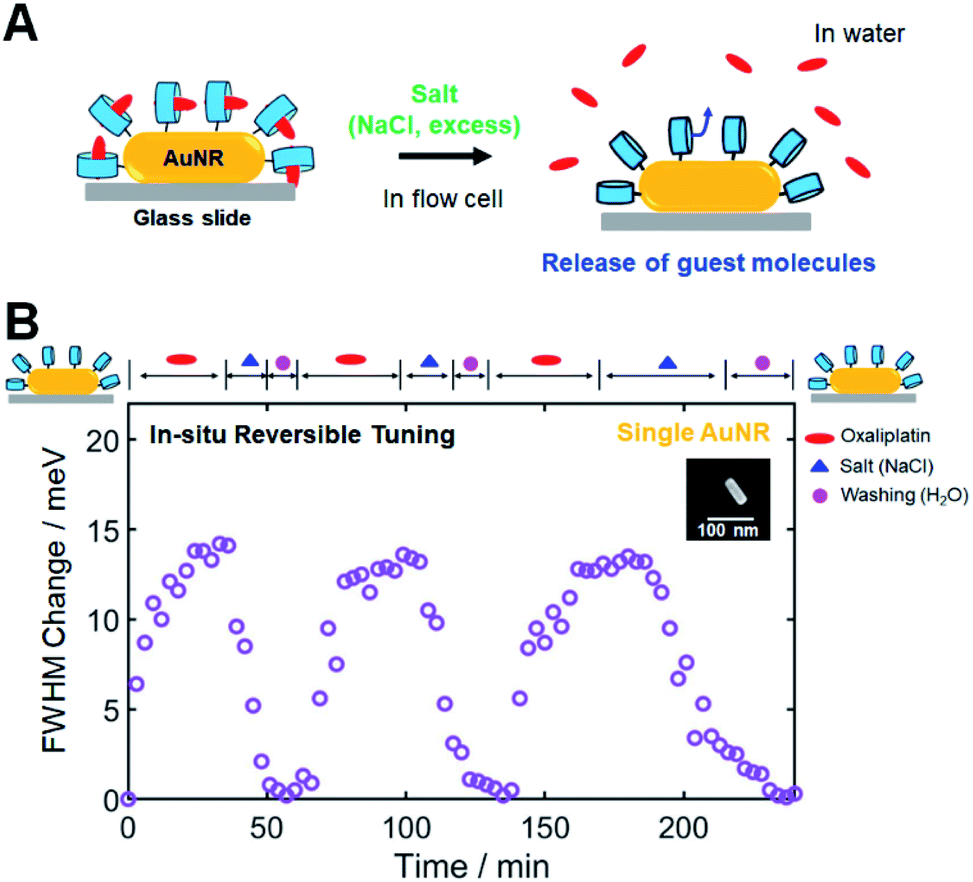 | ||
| Fig. 5 In situ reversible tuning of CID through manipulation of CB[7]-based host–guest interactions in single AuNRs. (A) Schematic to show the release of guest molecules by the addition of salt (NaCl) in a flow cell. Salt at high concentration can trigger the release of the encapsulated guest from the inclusion host–guest complex. (B) Reversible tuning of CID in a single AuNR through a series of injections of oxaliplatin, salt, and water in a flow cell. The time-dependent adsorption and release of the guest molecule (oxaliplatin) observed via the FWHM change for the single AuNR with respect to the starting value of the CB[7]-NH2@AuNR. | ||
First, we conducted NMR spectroscopic measurements to confirm that adding salt (NaCl) can release oxaliplatin, which is used as a guest in this study. By gradually increasing the concentration of NaCl, the NMR signals of the protons of 1,2-diaminocyclohexyl moiety on oxaliplatin shifted downfield toward the peak positions of oxaliplatin only, which indicated the release of oxaliplatin from the CB[7] cavity (Fig. S24†). Therefore, we confirmed that salt, when added in high concentrations, effectively released the guest used in this study from its CB inclusion complex, as illustrated in Fig. 5A.
Then, single-particle DF spectroscopic measurements were performed to check the reversible tuning of CID in a single AuNR through a series of injections of oxaliplatin, salt, and water in a flow cell. Fig. 5B shows the time-dependent repeated inclusion and release of the guest molecule (oxaliplatin) observed via the FWHM change for the single AuNR with respect to the starting value of the CB[7]-NH2@AuNR. When oxaliplatin was introduced, the FWHM change of the single AuNR shown in the SEM image started to increase, reaching saturation at ∼35 min (Fig. 5B). Then, encapsulated oxaliplatin was released from the CB complex after NaCl was added at high concentration, and the FWHM change value returned to the starting value of ∼0 after the consecutive steps of adding salt (excess) and flushing the cell with pure water in a flow cell. After the first cycle, we repeated the experiment in the same order of injections on an identical AuNR, and a similar result was observed for the oxaliplatin guest during the second and third cycles (Fig. 5B). Finally, we further confirmed that the AuNRs were stably fixed to the surface of the glass slide during single-particle measurements for 4 h in a flow cell (Fig. S25†). Therefore, we demonstrated that in situ reversible tuning of CID can be achieved through the successive steps of encapsulation and release of guest molecules in the same CB[7]-NH2@AuNR, which has not been considered in previous CID studies.
However, an important observation warrants further discussion. Fig. 5B clearly shows another advantage of using CB[7]-based host–guest chemistry, i.e., a single CB[7]-NH2@AuNR can be used as a recyclable platform after completing all necessary experiments and releasing the guest from the CB inclusion complex. To further demonstrate its recyclability and reproducibility, we performed single-particle DF spectroscopic measurements on a single CB[7]-NH2@AuNR through the processes of addition and release of oxaliplatin in a flow cell and repeated single-particle DF experiments for 7 consecutive days under the same experimental conditions. Fig. S26† shows the equilibrium ΔΓCID values obtained by fitting a Langmuir adsorption process in a single AuNR for 7 days. Fig. S26† shows that the equilibrium ΔΓCID values measured from the same CB[7]-NH2@AuNR were similar and reproducible over 7 days. Therefore, the result demonstrates that a single CB[7]-NH2@AuNR can be used as a reusable platform in CID investigations by employing various host–guest interactions on the same AuNR.
In addition, AuNRs tend to reshape by exposure to femtosecond laser pulses.56,57 Thus, it is also important to check if the shape transformation of single AuNRs occurs over long measurement times and under white light illumination (Fig. 3D, 5B and S26†). As shown in Fig. S27,† no noticeable spectral change was observed in the single particle scattering spectrum obtained after white light illumination for 4 h. The result supports that the shape transformation can be ruled out under our experimental conditions.
Conclusions
In summary, we performed SEM-correlated DF spectroscopic experiments to examine the CID process using CB[7]-based host–guest supramolecular interactions in single AuNRs. The increased LSPR linewidth broadening of single AuNRs was observed when oxaliplatin was encapsulated into CB[7]-NH2 attached to the AuNR surface. Using detailed DFT calculations, we were able to determine that the result can be attributed to the lowered LUMO energy value of the oxt@CB[7] complex compared to that of CB[7] only. In addition, in situ tuning of the chemical nature and electronic properties of the adsorbate was achieved through the competitive release of oxaliplatin from the CB complex and replacement by using a competitor guest of spermine in sufficient amounts. The spm@CB[7]complex showed a greater decrease in CID than the oxt@CB[7] complex, which can be explained by the increased LUMO energy value of the spm@CB[7]complex. Finally, we presented the release of the guest from the 1:1 host–guest complex by the addition of salt at high concentration and achieved in situ reversible tuning of CID in single AuNRs through successive steps of inclusion and release of the guest on the same AuNR in a flow cell. Furthermore, single CB[7]-NH2@AuNRs without any guest molecules were identified as a new recyclable platform for CID investigations by employing various host–guest interactions. Therefore, this study has paved a new route to investigate and tune reversible CID processes using CB[7]-based host–guest supramolecular chemistry in single AuNRs, and this new strategy using host–guest chemistry can be further applied to various guest molecules in reversible CID investigations for efficient hot-electron photochemistry and sensing applications.
Author contributions
Hui Bin Jeon, Kyeng Min Park, and Ji Won Ha conceived the project. Hui Bin Jeon and Kyeong Rim Ryu performed single particle spectroscopic experiments. Sehoon Park and Jaehoon Jung conducted the DFT calculations. Suman Kr Ghosh and Kyeng Min Park conducted the NMR experiments. Hui Bin Jeon, Kyeng Min Park, and Ji Won Ha analysed the data. Ji Won Ha wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by two National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIP) (No. 2018R1C1B3001154 and No. 2019R1A6A1A11053838).Notes and references
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- S. Y. Lee, P. V. Tsalu, G. W. Kim, M. J. Seo, J. W. Hong and J. W. Ha, Nano Lett., 2019, 19, 2568–2574 CrossRef CAS PubMed.
- M. Hu, J. Chen, M. Marquez, Y. Xia and G. V. Hartland, J. Phys. Chem. C, 2007, 111, 12558–12565 CrossRef CAS PubMed.
- B. Foerster, A. Joplin, K. Kaefer, S. Celiksoy, S. Link and C. Sönnichsen, ACS Nano, 2017, 11, 2886–2893 CrossRef CAS PubMed.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632–635 CrossRef CAS PubMed.
- Y. Tian, X. Wang, D. Zhang, X. Shi and S. Wang, J. Photochem. Photobiol., A, 2008, 199, 224–229 CrossRef CAS.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489 CrossRef CAS PubMed.
- Y. Tian, X. Shi, C. Lu, X. Wang and S. Wang, Electrochem. Commun., 2009, 11, 1603–1605 CrossRef CAS.
- G. W. Kim and J. W. Ha, Chem. Phys. Lett., 2018, 697, 38–42 CrossRef CAS.
- L. Rodríguez-Lorenzo, R. A. Álvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stéphan, M. Kociak, L. M. Liz-Marzán and F. J. García de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef PubMed.
- S. D'Agostino and F. Della Sala, J. Phys. Chem. C, 2011, 115, 11934–11940 CrossRef.
- S. W. Moon, P. V. Tsalu and J. W. Ha, Phys. Chem. Chem. Phys., 2018, 20, 22197–22202 RSC.
- S. W. Moon and J. W. Ha, Phys. Chem. Chem. Phys., 2019, 21, 7061–7066 RSC.
- P. Zijlstra, P. M. R. Paulo, K. Yu, Q. H. Xu and M. Orrit, Chemical Interface Damping in Single Gold Nanorods and Its Near Elimination by Tip-Specific Functionalization, 2012 Search PubMed.
- P. M. R. Paulo, P. Zijlstra, M. Orrit, E. Garcia-Fernandez, T. C. S. Pace, A. S. Viana and S. M. B. Costa, Langmuir, 2017, 33, 6503–6510 CrossRef CAS PubMed.
- R. C. Ferrier, H.-S. Lee, M. J. A. Hore, M. Caporizzo, D. M. Eckmann and R. J. Composto, Langmuir, 2014, 30, 1906–1914 CrossRef CAS PubMed.
- S. W. Moon and J. W. Ha, Analyst, 2019, 144, 2679–2683 RSC.
- E. de la Llave, R. Clarenc, D. J. Schiffrin and F. J. Williams, J. Phys. Chem. C, 2014, 118, 468–475 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- C. Boerigter, U. Aslam and S. Linic, ACS Nano, 2016, 10, 6108–6115 CrossRef CAS PubMed.
- M. J. Seo, G. W. Kim, P. V. Tsalu, S. W. Moon and J. W. Ha, Nanoscale Horiz., 2020, 5, 345–349 RSC.
- J. W. Ha, Chem. Phys. Lett., 2017, 676, 65–69 CrossRef CAS.
- P. Zijlstra, P. M. R. Paulo and M. Orrit, Nat. Nanotechnol., 2012, 7, 379 CrossRef CAS PubMed.
- T. Lünskens, A. von Weber, M. Jakob, T. Lelaidier, A. Kartouzian and U. Heiz, J. Phys. Chem. C, 2017, 121, 9331–9336 CrossRef.
- A. Hoggard, L.-Y. Wang, L. Ma, Y. Fang, G. You, J. Olson, Z. Liu, W.-S. Chang, P. M. Ajayan and S. Link, ACS Nano, 2013, 7, 11209–11217 CrossRef CAS PubMed.
- C. Sonnichsen, T. Franzl, T. Wilk, G. von Plessen, J. Feldmann, O. Wilson and P. Mulvaney, Phys. Rev. Lett., 2002, 88, 077402 CrossRef CAS PubMed.
- C. Novo, D. Gomez, J. Perez-Juste, Z. Zhang, H. Petrova, M. Reismann, P. Mulvaney and G. V. Hartland, Phys. Chem. Chem. Phys., 2006, 8, 3540–3546 RSC.
- M. Pelton, M. Liu, S. Park, N. F. Scherer and P. Guyot-Sionnest, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155419 CrossRef.
- K. R. Ryu, D. H. Nam, S. Lee and J. W. Ha, J. Phys. Chem. C, 2020, 124, 14818–14825 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- D. Das, K. I. Assaf and W. M. Nau, Front. Chem., 2019, 7, 619 CrossRef CAS PubMed.
- D. Shetty, J. K. Khedkar, K. M. Park and K. Kim, Chem. Soc. Rev., 2015, 44, 8747–8761 RSC.
- Y. Chen, Z. Huang, H. Zhao, J.-F. Xu, Z. Sun and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8602–8608 CrossRef CAS PubMed.
- X. Zhou, X. Su, P. Pathak, R. Vik, B. Vinciguerra, L. Isaacs and J. Jayawickramarajah, J. Am. Chem. Soc., 2017, 139, 13916–13921 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- H. Bai, H. Yuan, C. Nie, B. Wang, F. Lv, L. Liu and S. Wang, Angew. Chem., Int. Ed., 2015, 54, 13208–13213 CrossRef CAS PubMed.
- S.-R. Wang, J.-Q. Wang, G.-H. Xu, L. Wei, B.-S. Fu, L.-Y. Wu, Y.-Y. Song, X.-R. Yang, C. Li, S.-M. Liu and X. Zhou, Adv. Sci., 2018, 5, 1800231 CrossRef PubMed.
- J. An, S. Kim, A. Shrinidhi, J. Kim, H. Banna, G. Sung, K. M. Park and K. Kim, Nat. Biomed. Eng., 2020, 4, 1044–1052 CrossRef CAS PubMed.
- M. H. Jazayeri, H. Amani, A. A. Pourfatollah, H. Pazoki-Toroudi and B. Sedighimoghaddam, Sensing and Bio-Sensing Research, 2016, 9, 17–22 CrossRef.
- R. C. Hoft, M. J. Ford, A. M. McDonagh and M. B. Cortie, J. Phys. Chem. C, 2007, 111, 13886–13891 CrossRef CAS.
- G. W. Kim and J. W. Ha, Phys. Chem. Chem. Phys., 2020, 22, 11767–11770 RSC.
- L. Xiao, Y. Qiao, Y. He and E. S. Yeung, Anal. Chem., 2010, 82, 5268–5274 CrossRef CAS PubMed.
- E. Toprak, J. Enderlein, S. Syed, S. A. McKinney, R. G. Petschek, T. Ha, Y. E. Goldman and P. R. Selvin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6495–6499 CrossRef CAS PubMed.
- M. Böhmer and J. Enderlein, J. Opt. Soc. Am. B, 2003, 20, 554–559 CrossRef.
- Y. Jin Jeon, S.-Y. Kim, Y. Ho Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122–2125 RSC.
- B. Foerster, V. A. Spata, E. A. Carter, C. Sönnichsen and S. Link, Sci. Adv., 2019, 5, eaav0704 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- Z. Miskolczy, M. Megyesi, L. Biczók, A. Prabodh and F. Biedermann, Chem.–Eur. J., 2020, 26, 7433–7441 CrossRef CAS PubMed.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed.
- A. B. Taylor, A. M. Siddiquee and J. W. M. Chon, ACS Nano, 2014, 8, 12071–12079 CrossRef CAS PubMed.
- S. Link, C. Burda, M. B. Mohamed, B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. A, 1999, 103, 1165–1170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/d1sc01204e |
| This journal is © The Royal Society of Chemistry 2021 |