
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-CGRP disrupts amylin fibrillization and regulates insulin secretion: implications on diabetes and migraine†
Amber L. H.
Gray‡
 a,
Aleksandra
Antevska‡
a,
Benjamin A.
Link§
a,
Bryan
Bogin
bc,
Susan J.
Burke
d,
Samuel D.
Dupuy
f,
J. Jason
Collier
e,
Zachary A.
Levine
bc,
Michael D.
Karlstad
f and
Thanh D.
Do
*a
a,
Aleksandra
Antevska‡
a,
Benjamin A.
Link§
a,
Bryan
Bogin
bc,
Susan J.
Burke
d,
Samuel D.
Dupuy
f,
J. Jason
Collier
e,
Zachary A.
Levine
bc,
Michael D.
Karlstad
f and
Thanh D.
Do
*a
aDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA. E-mail: tdo5@utk.edu
bDepartment of Pathology, Yale School of Medicine, New Haven, CT 06520, USA
cDepartment of Molecular Biophysics & Biochemistry, Yale University, New Haven, CT 0652, USA
dLaboratory of Immunogenetics, Pennington Biomedical Research Center, Baton Rouge, LA 70808, USA
eLaboratory of Islet Biology and Inflammation, Pennington Biomedical Research Center, Baton Rouge, LA 70808, USA
fDepartment of Surgery, Graduate School of Medicine, University of Tennessee Health Science Center, Knoxville, TN 37920, USA
First published on 24th March 2021
Abstract
Despite being relatively benign and not an indicative signature of toxicity, fibril formation and fibrillar structures continue to be key factors in assessing the structure–function relationship in protein aggregation diseases. The inability to capture molecular cross-talk among key players at the tissue level before fibril formation greatly accounts for the missing link toward the development of an efficacious therapeutic intervention for Type II diabetes mellitus (T2DM). We show that human α-calcitonin gene-related peptide (α-CGRP) remodeled amylin fibrillization. Furthermore, while CGRP and/or amylin monomers reduce the secretion of both mouse Ins1 and Ins2 proteins, CGRP oligomers have a reverse effect on Ins1. Genetically reduced Ins2, the orthologous version of human insulin, has been shown to enhance insulin sensitivity and extend the life-span in old female mice. Beyond the mechanistic insights, our data suggest that CGRP regulates insulin secretion and lowers the risk of T2DM. Our result rationalizes how migraine might be protective against T2DM. We envision the new paradigm of CGRP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amylin interactions as a pivotal aspect for T2DM diagnostics and therapeutics. Maintaining a low level of amylin while increasing the level of CGRP could become a viable approach toward T2DM prevention and treatment.
:amylin interactions as a pivotal aspect for T2DM diagnostics and therapeutics. Maintaining a low level of amylin while increasing the level of CGRP could become a viable approach toward T2DM prevention and treatment.
Introduction
The onset and progression of many neurodegenerative diseases and chronic metabolic disorders are correlated with the accumulation of insoluble proteinaceous deposits.1–4 The oligomeric antecedents have been considered as the cause of disease pathogenesis due to their cytotoxicity.5–9 The possible toxic species are not limited to oligomers of individual proteins but cross-seeding of several distinct species.10–13 Thus, it is important to account for the intervention of molecules present in the same environment and how they possibly affect the aggregation. When two peptides co-assemble, the new system may acquire unique properties that neither homologous counterparts could have, including cytotoxicity and/or aggressive fibrillization,10,14,15 which can contribute to or exacerbate disease pathology.For example, the pancreatic islets secrete several important prohormones to regulate blood glucose levels. Amylin, also known as islet amyloid polypeptide, is a 37-residue regulatory peptide belonging to the calcitonin peptide family and is co-secreted with insulin from pancreatic β-cells.16–18 Amylin plays a role in glycemic regulation through delaying gastric emptying, promoting satiety, and suppressing insulin secretion. Amylin aggregation has long been considered a hallmark of type-2 diabetes mellitus (T2DM).19,20 Human amylin is an insulin suppressor of which aggregates damage islet-cells,21,22 whereas rodent amylin, which is non-amyloidogenic, is not a modulator of insulin biosynthesis or secretion.23 There are several hypotheses on what can trigger amylin aggregation (e.g., pH,24,25 Zn2+ concentration,26–29 impaired insulin secretion,26,30,31etc.), but there have been limited studies on the roles of calcitonin gene-related peptides (CGRPs) in the process.
Within the calcitonin gene family, CGRPs are widely expressed in the brain, spinal cord, and pancreas.32 Two major isoforms of CGRPs are α-CGRP and β-CGRP, which have been identified and are encoded from two distinct genes.33,34 α-CGRP is found primarily in the central nervous system and peripheral nervous system tissues while β-CGRP is predominately located in the enteric nervous system.35 Despite these differences, α- and β-CGRPs are both released from the sensory nerves36 and in a subpopulation of δ-cells of islets.37 CGRPs play an important role in both neural and vascular actions.38,39
On the other hand, anti-CGRP treatments were recently approved to prevent and treat migrane,40 a disease that may reduce the risk of T2DM.41 In migraine, sensory nerve fibers convey the “pain” signal to the brain and evoke the release of vasoactive peptides including CGRPs.42,43 Therefore, the biological role of CGRPs certainly deserves thorough investigation in a broad context.
CGRPs and amylin share high sequence similarity (Scheme 1). Similar to amylin, CGRPs also inhibit insulin secretion in both rodent and pigs,44–47 possibly through interactions with β-cell receptors such as RDC1 and CRLR;96 however, in a few studies, they were shown to stimulate insulin secretion.48–50 Both peptides induce several proinflammatory actions. For example, both can dilate blood vessels51 and compete for binding sites in the brain, skeletal muscle, and liver.52 Surprisingly, their possible roles and synergistic effects in T2DM have not been thoroughly explored.
 | ||
| Scheme 1 Primary sequences of CGRPs (red), amylin, (blue), and insulin (green). For mouse insulins, the residual difference between Ins1 and Ins2 is indicated in bold. In this work, we study human α-CGRP and human amylin. | ||
Studies of protein aggregation and characterization of oligomers existing in a dynamic equilibrium require sensitive analytical techniques capable of simultaneously isolating individual species. Ion mobility spectrometry-mass spectrometry (IMS-MS) has been demonstrated as a reliable technique for this task.53–55 In IMS-MS, oligomers are identified via orthogonal mass to charge (m/z) and drift time dimensions; the latter can yield valuable structural information. Amylin has been extensively studied with IMS-MS,30,56–60 and the main focus lies on structures of the monomers and dimers.30,57,58,60 Here, in addition to traditional IMS-MS analysis of amylin and CGRP homo- and hetero-oligomers in solution, we perform liquid chromatography (LC)-IMS-MS of single mouse islets incubated with each peptide or their mixture to evaluate their synergistic effect on insulin secretion. This work is further complemented by atomistic replica exchange molecular dynamics (REMD) simulations, transmission electron microscopy (TEM), and through evaluating multiple pathways of toxicity using cultured mouse β-cells. In this contribution, we provide a comprehensive view on how CGRP disrupts amylin fibrillization, and at the same time, how CGRP regulates secretion of mouse Ins1 and Ins2 proteins, with implications on the protective effect of migraine against T2DM.
Materials and methods
Ion mobility spectrometry-mass spectrometry
The chemicals were purchased and used without further purification. Human amylin and human α-CGRP peptides were purchased from AnaSpec (California, USA) at 98% purity. The peptide powders were pretreated with hexafluoroisopropanol (TCI America) and the solvents were left to evaporate in a desiccator overnight. Prior to IMS-MS analysis, stocks of each peptide were prepared by reconstituting the lyophilized peptide in LC-grade water (Honeywell) and diluting to 10 μM in a buffer of 20 mM ammonium acetate (VWR) pH = 6.8. The amylin:CGRP mixture was prepared by mixing the two peptide stocks in a 1:1 ratio and diluting the solution with 20 mM ammonium acetate to the final concentration of each peptide of 5 μM. For the peptide samples containing ZnCl2, we used 50:50 methanol:H2O instead of ammonium acetate. IMS-MS experiments were performed on an Agilent 6560 IMS-QTOF (Agilent, Santa Clara). Briefly, the peptides were ionized by using an electrospray ionization source and their ions were then pulsed into a helium filled drift cell (P ≈ 3.94 Torr). After exiting the drift cell, the ions were guided through the QTOF mass spectrometer by a hexapole ion guide. Data were collected in positive polarity with ΔV = 890, 790, 690, 590, and 490 V. Additional instrument parameters are listed in Table S1.†
The resulting arrival time distributions (ATDs) and mass spectra were extracted with Agilent IM-MS Browser software (v. 10.0) and graphed with Origin Pro. The collisional cross section (CCS, σexp) of the ions was calculated from the ion's reduced mobility (K0) according to the Mason–Schamp equation (eqn (1)):97
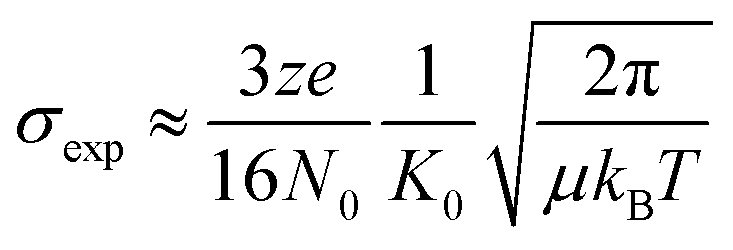 | (1) |
Replica exchange molecular dynamics
Atomistic molecular dynamics simulations were run using a GROMACS 2020.1 integrator61 and an AMBER99sb-ILDN protein force field62 solvated with TIP3P water,63 in order to match our prior work with amylin.64The disulfide bond between cysteines was held constant using rigid constraints between CYX residues, which groups deprotonated CYS pairs into a single joint amino acid. Additionally, histidines were neutralized and only contained a hydrogen attached to the epsilon nitrogen atom. Newton's equations of motion were integrated over a 2 fs time step using a leapfrog algorithm. Short-range van der Waals and Coulomb potentials were truncated at 1.2 nm, while longer-ranged electrostatics were tabulated with particle mesh Ewald summation, which reduces computation with fast Fourier transforms. Cartesian periodic boundary conditions were also implemented in each direction to minimize the effects from unit-cell boundaries. Initial monomers of amylin and CGRP were generated using AVOGADRO 1.1.165 in a random-coil conformation, since monomers are intrinsically disordered. Seeding geometries for each replicate were selected at random from 20 ns equilibration simulations, which populated a wide range of structures, and thereby increased the diversity between replicates. Box dimensions ranged from 6–7 nm in length, while dimers were constructed by randomly pairing permutations of monomeric proteins and confirming that they remained dimerized for at least an additional 20 ns.
Temperature replica-exchange MD (T-REMD)66 was carried out under an NVT ensemble using a Nose–Hoover thermostat67 at temperatures between 290 and 370 K for 200 ns per replica (approx. 8.2 μs of the total simulation time). This resulted in about 41 replicates per system, and the Monte Carlo exchange rate between replicates was empirically tuned to 25% (with exchanges attempted every 3 ps). While each replicate was run for 200 ns, analysis was only carried out from 100–200 ns, in order to minimize the impact of specific initial conditions. Convergence of REMD simulations was confirmed when the number of new protein conformations plateaued as a function of simulation time, indicating that only prior states were being sampled. Analysis of the room temperature replicate (300 K) from REMD was used to determine the structural and energy landscapes of protein monomers and dimers. Collision cross sections were calculated for each frame using the MOBCAL software package,68 and protein clustering was carried out by grouping Cα atoms together within a root-mean-square deviation of 2 nm. Analysis of the room temperature replicate (300 K) was used to determine the secondary structural propensities and energy landscapes of CGRP monomers, in addition to homo- and heterodimers. Additional computational details can be found in our prior work.64
Transmission electron microscopy
The peptides for TEM analysis were prepared at the same concentrations as for IMS-MS analysis and stained with 1% phosphotungstic acid stain on a piece of parafilm. Additional information can be found in the ESI.† TEM images were acquired on a JEOL 1400-Flash transmission electron microscope (JEOL, Peabody) at 80 kV and recorded with a Gatan OneView camera.Mouse islet culturing and harvesting
Six 10 week-old male C57BL/6J mice (stock #000664) were ordered from the Jackson Laboratory (Bar Harbor, Maine) and allowed to acclimate to the animal facility conditions on a 12 h light cycle at 22 ± 1 °C. The mice were provided standard chow and drinking water ad libitum until they reached 12 weeks of age. At 12 weeks of age, the mice were euthanized via CO2 asphyxiation followed by a cervical dislocation. All procedures were approved by the University of Tennessee Institutional Animal Care and Use Committee. Additional details regarding pancreatic perfusion and islet isolation can be found in the ESI.†69 Following isolation, the islets were incubated at 37 °C and 5% CO2 for 24 hours in RPMI + D-glucose. After the 24 hour incubation, the islets were individually pipetted into Eppendorf tubes containing the blank solvent, solutions of individual peptides, or a mixture of the peptides.Here we used a total of six mice for two sets of experiments. In the first set of experiments (3 mice), islets from the same mouse, except for those used as controls, were incubated with fresh amylin, fresh CGRP, or fresh mixture of amylin and CGRP. We used at least 3 islets from different mice per peptide condition. Because our LC-IMS-MS workflow was optimized for sub-single islet measurements, we can test multiple conditions on the same mouse, and obtain multiple (at least three) technical replicates from a single islet. In the second set of experiments (3 mice), we followed the same procedure as above. The only difference is that the islets were incubated with 4 day old amylin, CGRP, or the mixture.
Liquid chromatography IMS-MS of single mouse islets
Stocks of amylin and CGRP were prepared as previously described and diluted with a 5% MeOH/0.1% formic acid solution to a final concentration of 10 μM. The concentration of each peptide in the amylin:CGRP mixture was 10 μM. Aliquots of 50 μL of each peptide solution (amylin, CGRP, and amylin:CGRP) were placed into Eppendorf tubes, wherein single islets were manually placed. Two sets of peptide solutions were used: a fresh solution, where the peptide solutions were prepared approximately one day before acquiring the islets and contains primarily peptide monomers, and then an “aged” solution, where peptide solutions were left at room temperature four days before islet acquisition and consists primarily of peptide oligomers. The “aged” solution was characterized with IMS-MS to determine the relative ratios of oligomers to monomers. This was used to evaluate the effect of oligomers on the same islet system. For LC-IMS-MS analysis, an Agilent 1260 Infinity coupled to the Agilent 6560 IMS-MS was employed. In each run, 4.0 μL of the sample was injected into a 2.1 × 50 mm Zorbas Extended C18 reverse phase column (Agilent, Santa Clara). The mobile phase was composed of an aqueous solution of 0.1% formic acid in LC-grade water (solvent A) and acetonitrile (solvent B). The mobile phase gradient is as follows: 98% solvent A for 3 minutes, 80% solvent A (3–5 minutes), 65% solvent A (5–18 minutes), 50% solvent A (18–23 minutes), 25% solvent A (23–26 minutes), 10% solvent A (26–33 minutes), 10% solvent A was held for 2 minutes then followed by a rise to 98% (35–40 minutes) and kept for 5 minutes (40–45 minutes) for column equilibration. The column compartment was set to 40 °C. Three technical replicates were performed for each sample, with two consecutive blank injections between each sample to clean the column.
Cell viability assays
Stock solutions of amylin and CGRP in LC-grade water were added to cultures of 832/13 rat insulinoma cells to obtain the desired concentrations of 20, 200 nM, and 5 μM and incubated overnight at 37 °C. Adenylate kinase release into the culture media was assessed using the Adenylate Kinase Assay Kit (Abcam #: ab228557) which measures the activity of the adenylate kinase released by damaged cells. Interleukin-1β (IL-1β) and camptothecin (CT) were used as controls.Results and discussion
IMS-MS reveals a CGRP α-helical intermediate that is stable upon interacting with Zn2+
Fig. 1A shows the representative ESI-mass spectrum of amylin. The mass spectral peaks are annotated with their nominal oligomer-to-charge ratio (n/z). The top mass spectrum shows several peaks corresponding to small-order oligomers that are high in abundance. The bottom mass spectrum highlights the less intense mass spectral peaks that correspond to higher-order oligomers. In the designated mass range, amylin can form up to a heptamer. However, upon analysis of the isotopic spacing and ion mobility data, oligomers larger than what is initially seen within each mass spectral peak can be observed. Isobaric oligomers may be hidden under the same nominal m/z. For example, the inset in Fig. 1A shows the isotopic spacing of n/z = 1/3, indicating the presence of a monomer with a +3 charge state and a dimer with a +6 charge state (i.e. n/z = 2/6 = 1/3).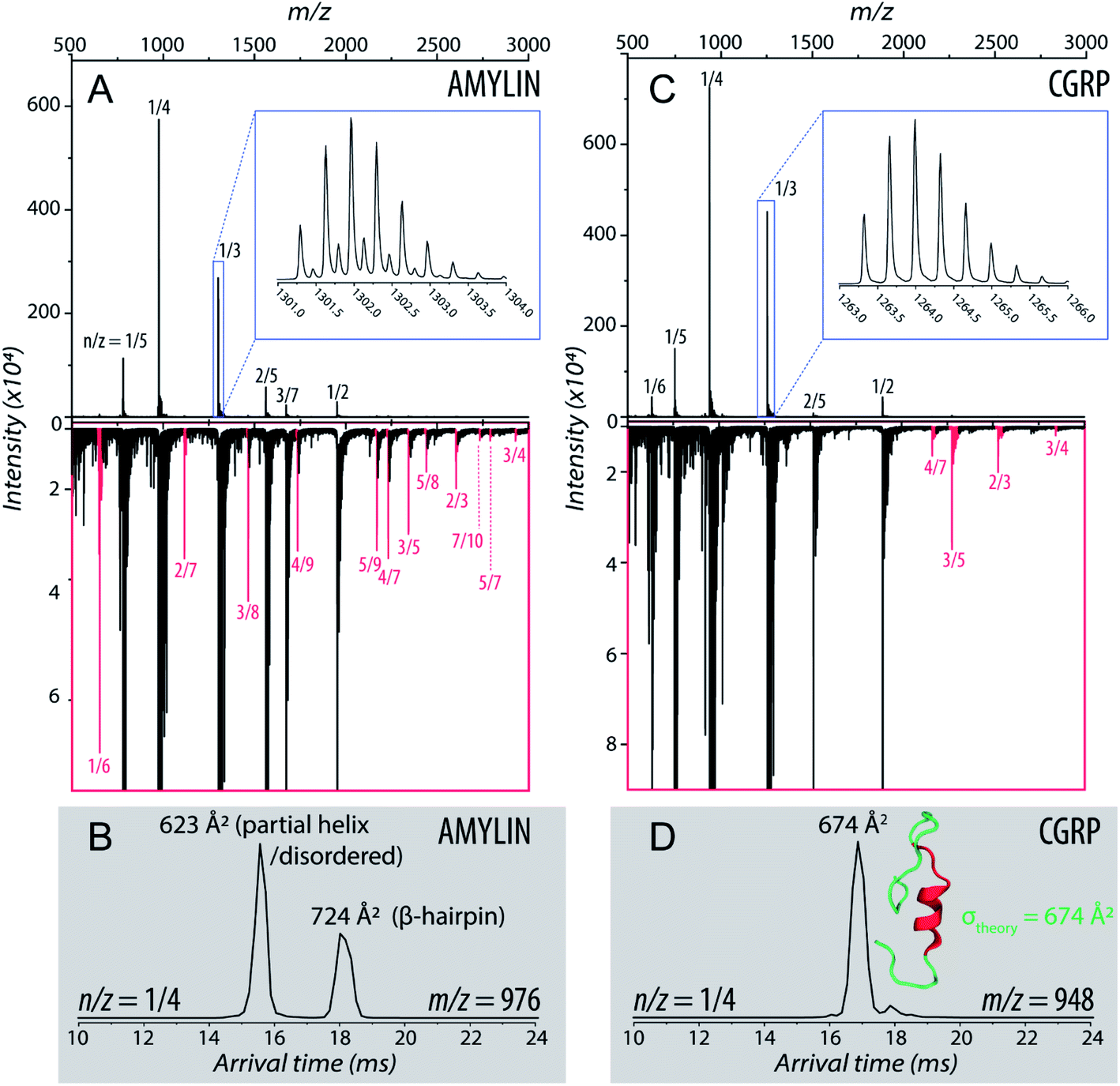 | ||
| Fig. 1 (A) Mass spectrum of amylin (10 μM) in 20 mM ammonium acetate. Each peak is labeled by its nominal n/z ratio. The bottom spectrum exhibits oligomeric species with a relatively low intensity that is not easily identifiable in the top spectrum. The inset shows the isotopic spacing of n/z = 1/3 which exhibits the presence of a dimer with z = +6. (B) The ATD of amylin at m/z = 976. (C) Mass spectrum of CGRP (10 μM) in 20 mM ammonium acetate. The insrt shows the isotopic spacing of n/z = 1/3 which indicates that this mass spectral peak correlates to a triply charged dimer. (D) The ATD of CGRP at m/z = 948 and a REMD conformation of CGRP monomer with the segment 8–18 colored in red. This conformation is consistent with previous NMR and CD studies of CGRP in structure-inducing solvent.70,72,73 | ||
Amylin, specifically the monomer (z = +4), has been characterized and studied extensively with IMS-MS in the past.30,57,58,60 It has been shown by Bowers and Shea57,58,60 that amylin populates two distinct families of stable conformations of amyloidogenic β-hairpin and a coil-helix. We note that the terms “helix” and “β-hairpin” were taken from the same studies. Fig. 1B shows the arrival time distribution (ATD) of the amylin monomer (z = +4) illustrating these two families of conformations. The collisional cross sections (CCSs) of these species were found to be 623 Å2 for the coil-helix and 724 Å2 for the β-hairpin. The difference in the CCS between these two structures is in good agreement with the data by Bowers and Shea.57,58,60
Unlike amylin, α-CGRP (which we will refer to as “CGRP” hereafter) has not been characterized with IMS-MS. The mass spectrum of CGRP is shown in Fig. 1C, indicating that this peptide formed several small oligomers with the largest one being a tetramer. Upon initial observation of the mass spectra, it is evident that CGRP formed smaller oligomers than amylin. Our data indicate that amylin readily assembles into oligomeric species and is much more prone to aggregation than CGRP.
Fig. 1D shows the ATD of the CGRP monomer (z = +4), which contains only one prominent feature with an arrival time at 16.89 ms. When compared to the amylin data, the CGRP structure falls in between the two major populations of amylin (coil-helix at 15.58 ms and β-hairpin at 18.05 ms). If CGRP were to adopt the same or similar conformations as amylin, then CGRP should have a slightly shorter arrival time (smaller CCS) than the respective amylin conformers due to having less mass. Therefore, this feature could correlate to a coil-helical CGRP conformer (CCSexp = 674 Å2). This is not pure speculation; previous studies of CGRP in structure-inducing solvents (e.g., trifluoroethanol (TFE)/water) demonstrated that residues 8–18 of the peptides exhibit a strong α-helical propensity.70–73 This segment encompasses the chameleon sequence “LAGLL” found in 8 different proteins.74 However, it is entirely possible for several conformations with different secondary structures to have the same CCS, as discussed below together with the REMD simulations. Nonetheless, Fig. 1D suggests that the structural preference of CGRP prevents the formation of the β-hairpin motif, thus inhibiting extensive CGRP aggregation.
Zinc complexation promotes β-hairpin of amylin but favors the native structure of CGRP
To further investigate the aggregation of CGRP and amylin under biologically relevant conditions, we added 0.5 mM ZnCl2 to the samples. We used the amylin data, which were previously reported,60 as the control. Zn2+ is involved in the packaging, processing, and the secretion of insulin from β-cells. Zn2+ is found in mM concentration in pancreatic β-cells, reaching 10–20 mM in dense-core granules,75,76 and has been found to affect amylin fibrillization upon complexation. Previous studies showed that Zn2+ inhibited amylin fibril formation at low, physiologically relevant extracellular concentrations27,29 but promoted aggregation at high (intracellular) concentrations.60
Fig. 2A shows the mass spectrum of amylin in the presence of Zn2+ (50:1 ZnCl2:amylin) which illustrates many Zn–amylin complexes. A majority of these species, such as the complexes of amylin containing Zn and Zn(H2O)5, were observed by Bowers and coworkers.60 The same types of complexes were observed for CGRP (Fig. 2B).
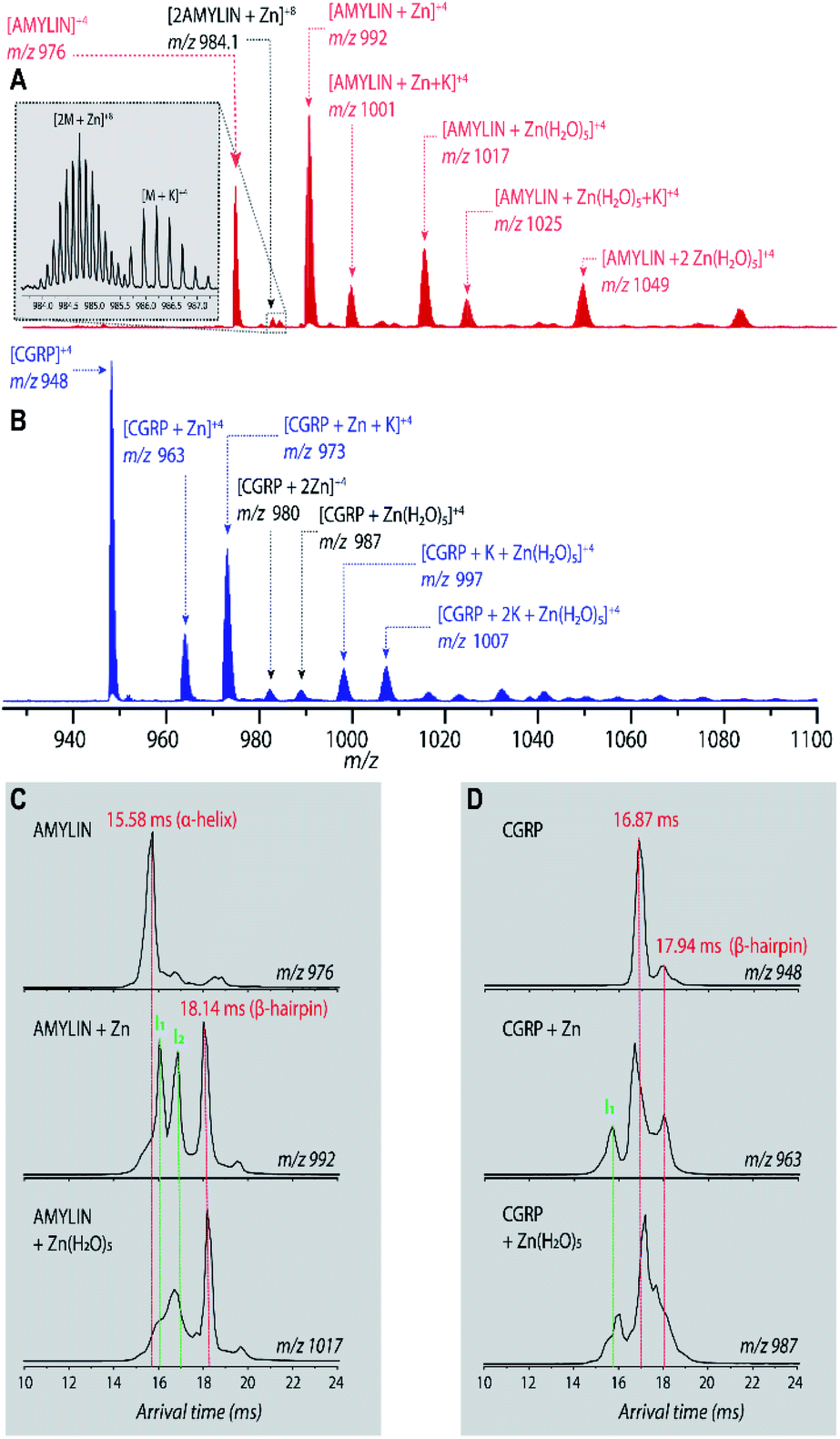 | ||
| Fig. 2 (A and B) Representative ESI-mass spectra (in 50:50 MeOH:H2O) of (A) 10 μM amylin and (B) 10 μM CGRP, both in the presence of 0.5 mM Zn2+. (C) Representative ATDs of amylin, amylin:Zn, and amylin:Zn(H2O)5. (D) Representative ATDs of CGRP, CGRP:Zn, and CGRP:Zn(H2O)5. | ||
Bowers and coworkers suggested that the amplification of amylin aggregation upon coordination with Zn2+ could be attributed to stabilization of the β-hairpin motif.60 Similar findings are illustrated in Fig. 2C where we show the ATDs of the bare amylin monomer (z = +4) and two Zn:amylin complexes. The ATD of the bare amylin monomer has one major feature which corresponds to the coil-helix conformation. However, upon complexation of amylin with Zn2+, the conformational distribution shifts to favor the β-hairpin, with this effect becoming more pronounced when complexed with the Zn(H2O)5 adduct.
We detected two intermediate species upon Zn–amylin complexation (annotated as I1 and I2 in Fig. 2C). These intermediates were favored by Zn2+ complexation, competing with the amyloidogenic β-hairpin. Such competition could lead to an inhibition of fibril formation.27,28 These intermediates were not observed in the previous study60 due to low mobility resolution of the instruments used. Here, their presence provide a rational explanation for the moderate inhibitory effect of Zn2+ on amylin aggregation. However, fibril formation was still observed in months-old samples of amylin with ZnCl2 as shown in Fig. 3D.
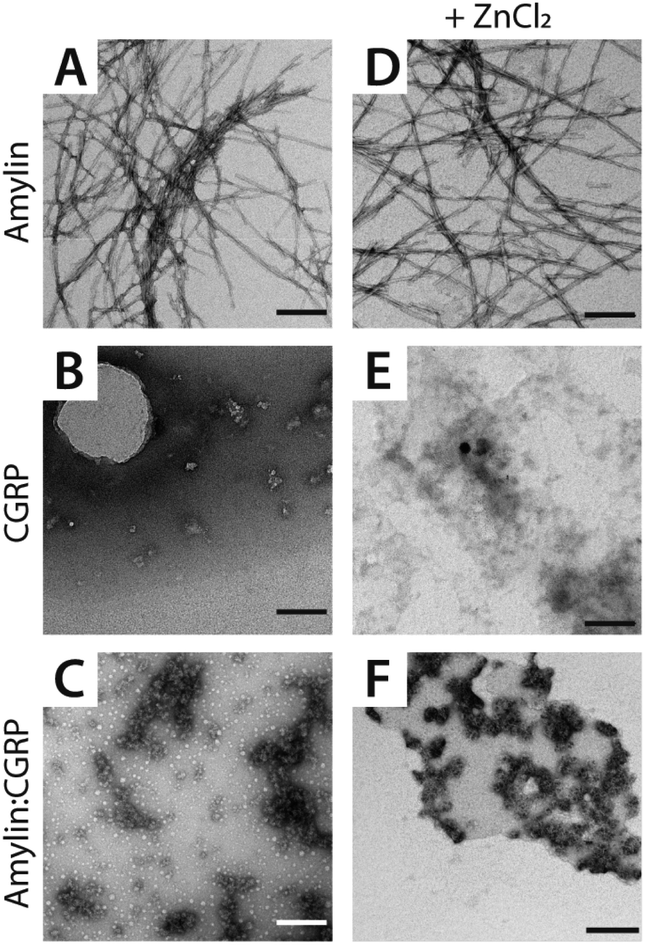 | ||
| Fig. 3 Representative TEM images of (A and D) amylin, (B and E) CGRP, and (C and F) the mixture. The scale bar is 150 nm. | ||
Since CGRP shares similar biological properties with amylin, we evaluated its structures upon Zn complexation. Fig. 2D shows the ATDs of the bare monomer (z = +4), CGRP + Zn, and CGRP + Zn(H2O)5. For bare CGRP (Fig. 2D), there is only a single prominent feature, identical to that in Fig. 1D. However, once CGRP complexes with Zn, we note the addition of at least two new features. As mentioned in the previous section, the feature at ∼18 ms is assigned to the amyloidogenic β-hairpin motif. Observing the ATDs of the Zn–CGRP complexes, Zn slightly promotes the formation of this β-hairpin, albeit not as much as what was observed for amylin. In addition, a new compact species (ta = ∼16 ms) in the same ATDs (Fig. 2D; feature I1) exhibits a similar arrival time to that of the amylin's coil-helix. Given its short arrival time, this intermediate is likely a coil (disordered) structure. Notably, although Zn shifts the conformations of CGRP to a disordered and β-hairpin, the original structure of the CGRP is still the most dominant feature. Thus, the conformational space of CGRP is not dramatically changed upon Zn2+ complexation. The IMS-MS data help explain why CGRP does not aggregate as aggressively as amylin. Additionally, the TEM images (Fig. 3B and E) show that CGRP formed neither small observable aggregates nor fibrils under any of the conditions.
Amylin, CGRP, and their mixture do not show significant toxicity to cultured β-cells
Since both amylin and CGRP are present within pancreatic islets, it would not be surprising if these peptides interacted in vivo to form amylin–CGRP hetero-oligomers. In the first approach, we identify and characterize the types of hetero-oligomers in solution using IMS-MS. Fig. S1† shows the partial mass spectra of an amylin–CGRP heterodimer and heterotrimer. In each panel, the top mass spectrum is of amylin alone, while the bottom mass spectrum is of the amylin:CGRP mixture. Based on the nominal mass, the heterodimer consisted of amylin–CGRP in a 1:1 stoichiometric ratio and the heterotrimer was composed of 2:1 amylin–CGRP. Other species identified within the mass spectrum of the amylin–CGRP mixture is found in Fig. S2.† Based on m/z values, the hetero-oligomers contain more amylin than CGRP, suggesting that CGRP binds to amylin oligomers to remodel amylin aggregation. Additionally, the TEM images acquired from the amylin:CGRP mixture show the presence of small globular aggregates (Fig. 3C and F). Only indistinct and faint fibrillar species can be observed among the predominantly globular aggregates in the TEM images of the mixture without ZnCl2 samples (see Fig. S3†).
Furthermore, the mass spectrum of the amylin:CGRP mixture at t = 1 week does not contain the prominent amylin mass spectral peaks (Fig. S4†). This hints that amylin has fallen out of the solution to form insoluble species which connect to the observed globular aggregates. In other words, the globular aggregates are predominantly made of amylin. When Zn2+ is present, some distinct fibrils of various sizes can be observed along with the globular aggregates in the TEM images of the mixture (Fig. S3†). This agrees with our previous observation of the changed conformational spaces of the peptides upon Zn2+ coordination, which subsequently lessened the cross-talk between them.
To determine the cytotoxicity of the amylin–CGRP aggregates, we studied the release of adenylate kinase, an intracellular enzyme marker of cell death, from cultured β-cells when exposed to amylin–CGRP aggregates and amylin fibrils. We assessed the toxicity of amylin and the amylin:CGRP mixture using two negative controls: the no-treatment condition and interleukin-1β (IL-1β) treatment. IL-1β was used to indicate that insulin secretion can be reduced without significant toxicity.77 For positive control, we used camptothecin (CT), a topoisomerase I toxin that damages eukaryotic cells, including β-cells.78
Previous investigations of amylin toxicity used micromolar concentrations (e.g., 0.5–8 μM in Shigihara et al.79 and ∼2–8 μM in Ritzel and Butler80) to assess toxicity. Here we performed three sets of experiments at 20 nM, 200 nM, and 5 μM (see Fig. 4 and S5†). In all cases, amylin, CGRP and their mixture did not result in significant toxicity. Nonetheless, amylin appeared to be more toxic than CGRP and amylin + CGRP. Regarding toxicity of amylin, our data are in line with those of Westwell-Roper et al.81 who suggested that in a living organism, if amylin accumulates, it acts on the islet resident macrophage to create toxicity but not directly on the β-cells.
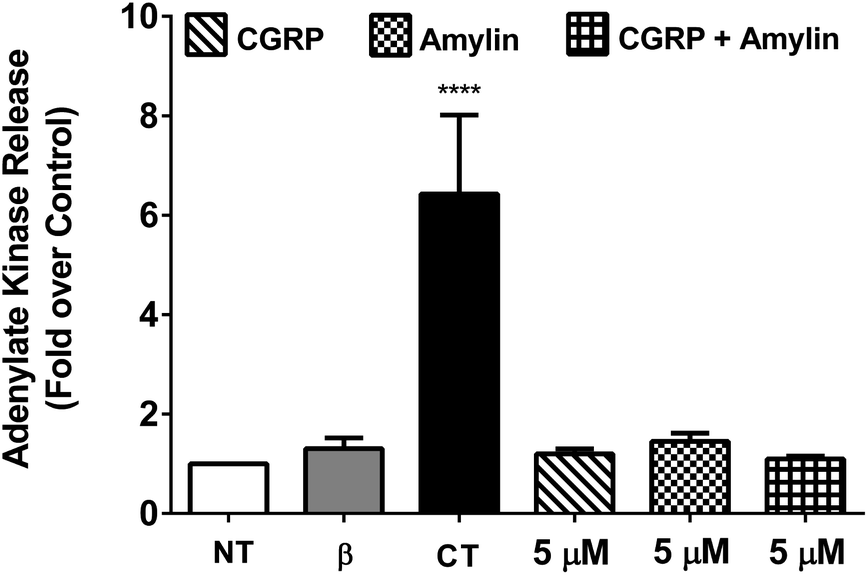 | ||
| Fig. 4 Cell viability assay using cultured β-cells exposed to CGRP, amylin, and amylin:CGRP. 832/13 rat β-cells were either left untreated (NT) or exposed to 1 ng mL−1 IL-1β and 2 mM camptothecin (CT) compared with 5 μM of amylin, CGRP, and amylin plus CGRP for 18 h. ****, p < 0.0001 versus NT using one-way ANOVA. n = 4–6 per group. When compared to CT, the effects of amylin, CGRP, or amylin:CGRP on cellular viability are minor, although amylin has a stronger effect than the other two conditions. | ||
In silico CGRP–amylin simulations support IMS-MS derived conformers
To gain structural CGRP and amylin interactions, we carried out REMD simulations on CGRP monomers and heterodimers in solution. For amylin, since the early implicit-solvent REMD simulations by Bowers and Shea,57,58 newer force fields and explicit solvent models have been utilized to better describe intrinsically the disordered nature of amylin monomers and the β-hairpin dimer, including a recent study by Levine and Shea64 on the interactions of amylin with mitochondrial derived peptides. Here the simulation protocol of CGRP simulations is identical to that of the reported amylin.While theoretical and experimental CCS comparison has been a common approach in peptide structural studies using IMS-MS,54,55,82 for intrinsically disordered peptides, matching experimental CCSs to model structures is challenging. In the gas phase, disordered structures can be denatured, while α-helical structures can be over-stabilized.83 Notably, the structure shown in the inset of Fig. 1D was obtained from our REMD simulation, which shows a helical content consistent with previous NMR/circular dichroism studies in PFE/water (a condition that stabilizes the α-helix).70,72,73 However, the population of this structure is very low given that the majority of CGRP monomers (from simulation) are disordered. Another way to understand the data is as follows. When we cluster protein states from REMD based just on residues 8–18 (corresponding to CGRP regions most likely to exhibit any secondary structures), we find that 60% of CGRP conformations adopt either a disordered + β-stranded structure, a completely disordered structure, or a disordered-helical structure (Fig. S6†). When we back-calculate CCS values for each of these eigenstates, we find an excellent agreement with the IMS-MS data. Finally, the range of theoretical CCSs for both CGRP/amylin monomers and dimers is in agreement with the experimental data (Fig. S7–S9†).
Here we used the REMD data to rationalize the inhibitory effect of CGRP on amylin aggregation. To better quantify these structural heterogeneities, we provide molecular contact maps (Fig. 5) that indicate the average proximity of amino acids in each structure. CGRP and amylin monomers exhibit no obvious contact patterns since they tend to remain strongly disordered. However, a characteristic “X” pattern on these contact maps indicates the presence of a hairpin, which is notable in the amylin homodimer. While largely disordered, one of the CGRP proteins also appears to adopt a partial hairpin in the homodimer. Interestingly, in the CGRP–amylin heterodimer, the positions of the hairpin move to the exterior of each protein. In CGRP, the hairpin moves more towards the N-terminus while in amylin, the hairpin moves towards the C-terminus. As for intermolecular interactions, amylin tends to bind CGRP towards its C-terminus almost directly on the hairpin location. This bolsters the hypothesis that disordered CGRP tends to bind β-stranded amylin, which likely inhibits the ability to form regular fibrillar structures. Specifically, Asn31 in amylin regularly forms hydrogen bonds with residues 16–18 on CGRP. This interaction provides both molecules with stable β-sheets that are prevalent in almost all states, while the rest of the molecule is highly disordered. Of note, Asn31 mutations have been shown to reduce the β-sheet content in simulations of amylin protofilaments, suggesting that such interactions may be important in forming oligomeric complexes with CGRP.84 It is also possible that CGRP–amylin heterodimers compete against amylin homodimers, thereby inhibiting fibril formation at high concentrations. Taken together, these models help contextualize the changes in CCS values from experiments, and provide mechanistic insights on how CGRP might bind to amylin to reduce fibrillization, thereby contributing to the formation of disordered oligomers.
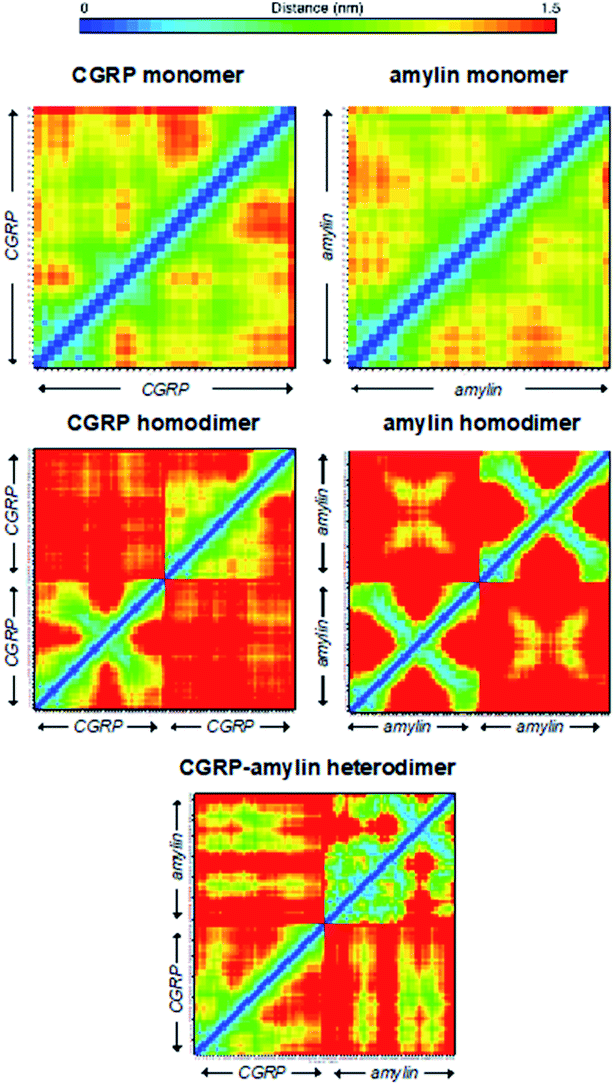 | ||
| Fig. 5 Molecular contact maps from REMD simulations reveal a range of disordered and hairpin conformations. | ||
The amylin–CGRP mixture impairs insulin secretion but CGRP oligomers increase the Ins1 level
As previously mentioned, it has been well-established that both amylin and CGRP independently inhibit insulin secretion. As we determined that hetero-oligomers of amylin and CGRPs exist in solution, the next step is to investigate their effect on insulin secretion using healthy mouse islets as the model. We chose single mouse islets as the biological system because multiple islets from the same animals can be collected and assayed under different conditions (amylin, CGRP, and the mixture). By analyzing single islets, we reduced the number of required pancreases (animals). To address islet heterogeneity, three islets per animal per condition were assayed, and a total of six animals were used. Furthermore, with LC-IMS-MS, multiple LC injections from a single islet can be performed to yield at least three technical replicates. Each analysis requires approximately 150 cells and can separate mouse Ins1 and Ins2 proteins as discussed below. In comparison to traditional ELISA, our method is less expensive and requires a fewer number of pancreases. Fig. S10† illustrates a schematic of the workflow.Upon LC-IMS-MS analysis of the single islets, we detected both mouse Ins1 and Ins2 (Fig. 6A). Ins1 was detected at an observed mass of 5799.668 Da (0.276 ppm difference from the theoretical mass of 5799.666 Da),85 and at four different charge states (Fig. 6B) which were identified unambiguously with isotopic spacings. The [M+6H]6+ species at m/z 967.619 is the most intense, and the other charge states were at m/z 829.963 (z = +7), 1060.943 (z = +5), and 1451.182 (z = +4). Similarly, Ins2 was detected at an observed mass of 5793.5974 Da (2.261 ppm difference from the theoretical mass, 5793.6105 Da)86 (Fig. S11†). In terms of CCSs, there is no reported experimental CCS of mouse insulins. However, there are reported values for human insulin by Bush and co-workers. The reported CCS of z = +4 human insulin monomer is 772 Å2,82 which agrees well with our experimental CCSs of Ins1 at the same charge state (CCSHe,calibrated = 753 Å2 and 783 Å2, CCSN2 = 902 Å2 and 936 Å2) (Table S2†).
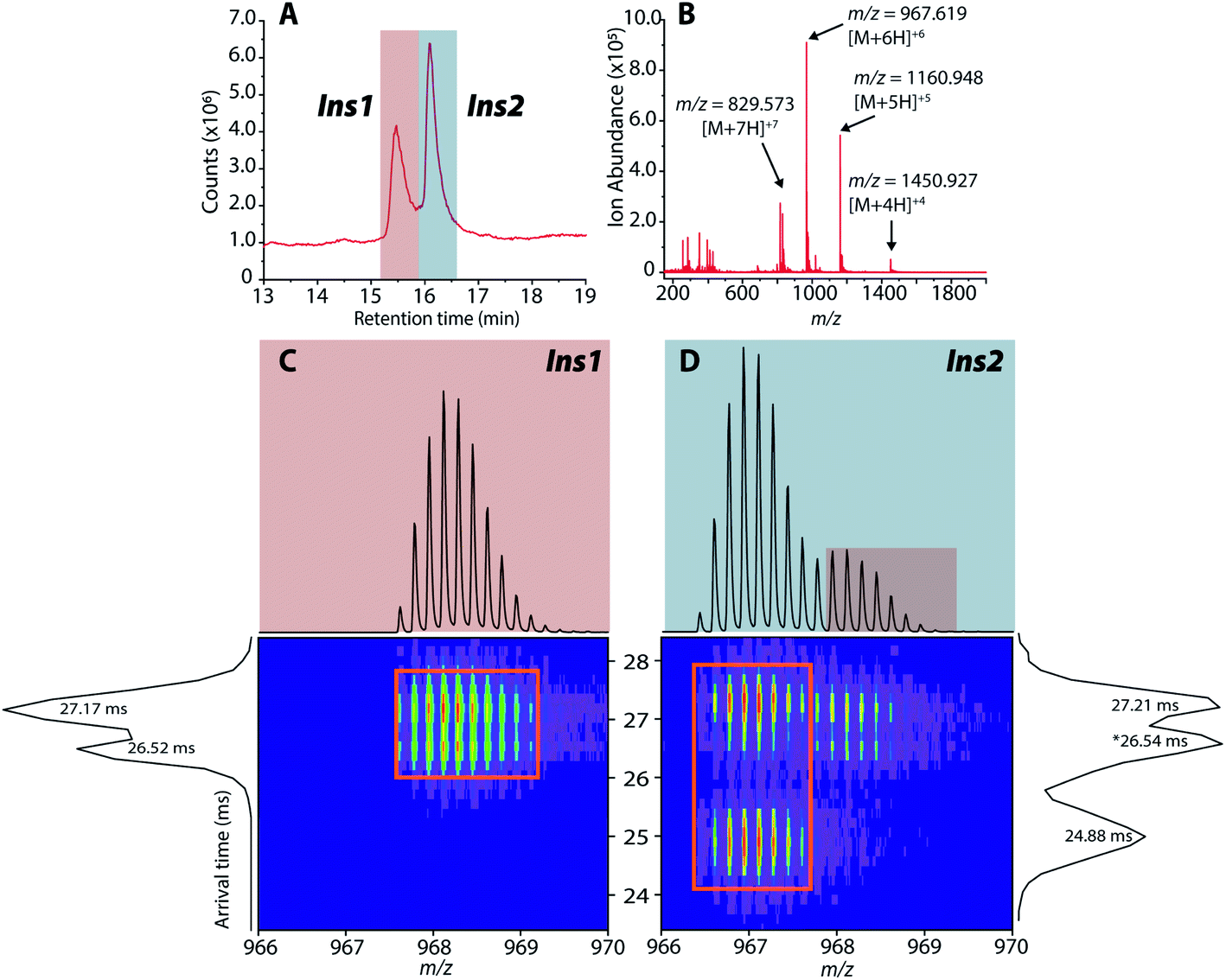 | ||
| Fig. 6 (A) Extracted LC chromatogram showing Ins1 and Ins2. (B) Extracted partial LC-IMS-MS mass spectrum showing only Ins1. (C and D) 2D plots of arrival time vs. m/z of Ins1 and Ins2 at z = +6. | ||
The rodent (mouse and rat) insulin two-gene system is well reported. In mouse islets, expressions of Ins1 and Ins2 are similar (57:43% total insulin mRNA).87 Ins1 differs from Ins2 by two amino acids in chain B (P9S and K29M) while other differences can be found at the proinsulin and gene levels.88 Ins1 was shown to be more sensitive to glucose stimulation than Ins2,87 and thus it had a more advantageous effect under scant food conditions. However, as the environment changes, Ins1 increased the risk of T1DM (insulin resistance), evidenced by nonobese diabetic (NOD) mice carrying only Ins1 gene developed T1DM as early as 10 weeks old.88 On the other hand, mouse Ins2 is the homologue of human insulin, which exerts a protective effect against T1DM in NOD mice.89 Therefore, in this work, the toxicity of amylin and the amylin:CGRP mixture, and their implications on T2DM should be evaluated based on the level of Ins2 rather than Ins1.
The 2D plots in Fig. 6C and D highlight the structural difference between Ins1 and Ins2. Additional 2D plots are shown in Fig. S12.† The arrival time at ∼24–25 ms in panel D suggests that Ins2 either adopts a more compact conformation or is more prone to form oligomers. The biological assembly of Ins2 is a hexamer stabilized by Zn2+–histidine coordination.95 Structural comparison of Ins1 and Ins2 is beyond the scope of this study, and will be a topic of future investigation.
Incubation of the islets with pure amylin and the amylin:CGRP mixture shows the lowest levels of insulin secretion, regardless of the peptide solution conditions (i.e., “aged” vs. “fresh”; Fig. 7). A one-way ANOVA analysis was performed to determine the statistical significance of our data followed by a Tukey test between each condition and the control. A qualitative comparison of amylin and CGRP oligomers in aged and fresh culture media is shown in Fig. S13.† The aged media contain significantly higher amounts of oligomers than the fresh media.
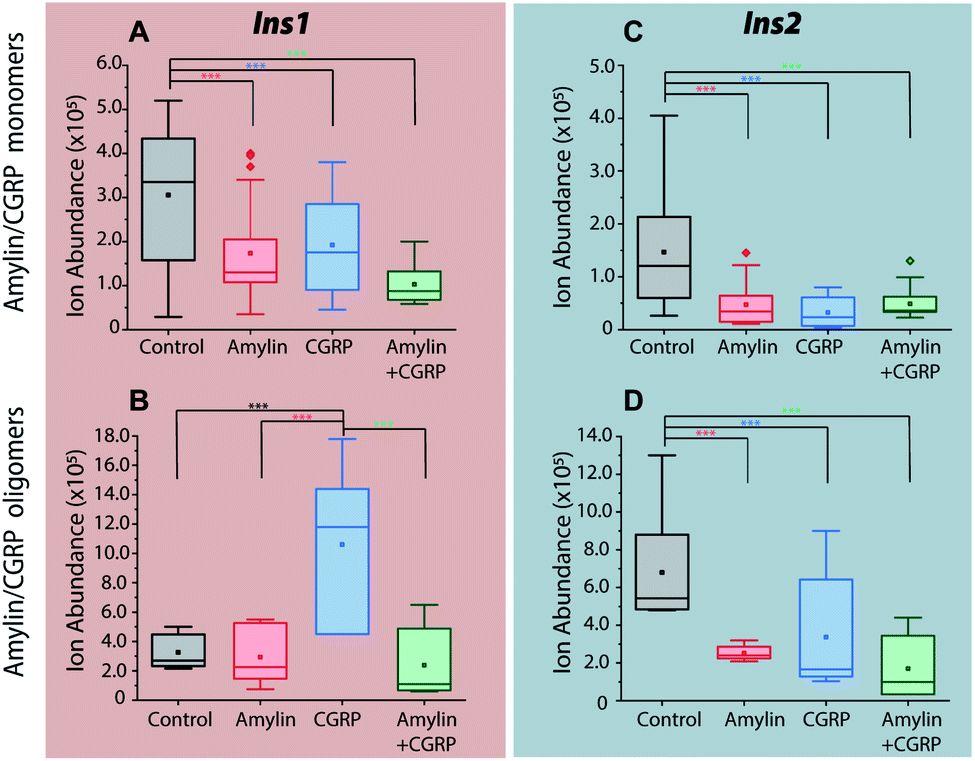 | ||
| Fig. 7 Comparisons of insulin signals obtained from single islets exposed to (A and C) media contain mostly amylin/CGRP monomers and (B and D) media contain mostly amylin/CGRP oligomers. *** indicates p < 0.05. | ||
Interestingly, once the islets were placed under the conditions containing aged CGRP (more CGRP dimers), the Ins1 level increased, which contradicts the inhibitory effect of fresh CGRP. To our knowledge, this is the first time the dual effect of CGRP on insulin secretion was observed in isolated mouse islets, and this effect is specific to Ins1 and not Ins2. Similar effects were observed in animals with one insulin-gene system like dogs and pigs.48,50 Notably, Hermansen et al.50 observed a similar trend to ours in which CGRP inhibits (dog) insulin secretion at low doses in a short time frame but increases it at high doses over a longer period. Unfortunately, earlier studies did not examine the possibility of how CGRP oligomers might behave differently from the CGRP monomer. The exact mechanism that allows for the dual effect of CGRP on Ins1 is not known.
In terms of Ins2, its inhibitory effect toward insulin secretion is consistent between the two media conditions. However, the effect is more profound in fresh CGRP. The data also confirm that CGRP is as effective as amylin in suppressing insulin secretion, but CGRP does not aggregate into fibrils.
CGRP in migraine and diabetes
There have been several studies that have concluded that there is an inverse relationship between diabetes and migraine prevalence. Bonnet and coworkers41 showed that women with active migraine had a lower risk of developing T2DM as opposed to women with no migraine history. Lund and co-workers90 demonstrated that there is a markedly reduced prevalence of migraine among older patients with diabetes. These studies prompted us to investigate neuropeptides that are upregulated in migraine to determine their roles in islet chemistry and diabetes progression, and CGRP is one of those peptides. The elevated concentration of CGRP in migraine patients may exert a protective effect via suppressing insulin release. Templeman et al.91 showed that decreased insulin led to lower fasting glucose and improved insulin sensitivity in aged mice. Thus, CGRP may play a critical role in regulating insulin release that can help lower the risk of diabetes.Regarding toxicity of amylin, there are two possibilities for this peptide to generate negative effects on islet β-cells. The first is by direct toxicity, which our data do not support. The second is by activation of islet resident macrophages, which in turn produce inflammatory molecules in response to amylin uptake. Based on these two possibilities, our data are in line with those of Westwell-Roper et al.81 who provided compelling evidence for amylin accumulation acting on the islet resident macrophage to promote inflammation-associated toxicity but not directly on the β-cells.
Our data show that CGRP could not regulate Ins1 effectively, and interestingly NOD mice with only the Ins1 gene develop T1DM as early as 10 weeks old.88 Replacing Ins1 with human insulin (the orthologue of mouse Ins2) protects the mice from diabetes.92 To our knowledge, this is the first time that the dual effect of CGRP on insulin secretion has been investigated using isolated (mouse) islets. Moreover, because the biochemistry of mouse Ins1 and Ins2 is not the same, diabetes research using mouse models may need to distinguish these two isoforms.
We speculate that the use of some anti-CGRP medications for migraine treatment may lower the level of CGRP and increase the risk of developing T2DM. Specifically, some monoclonal CGRP antibodies which bind to CGRP93,94 could hinder CGRP function. This is based on the premise that mouse Ins2 and human insulin behave similarly. Lowering the production of amylin while increasing the level of CGRP could be a viable approach toward T2DM prevention and treatment. Finally, we did not perform a clinical study to directly correlate migraine with diabetes, and this topic is beyond the scope of this paper. There have not been any clinical trials of this type. Nonetheless, we plan to further investigate the mechanism of CGRP on insulin secretion as well as other neuropeptides associated with migraine.
Summary and conclusion
In summary, we studied the interactions that occur between amylin and CGRP in the context of T2DM. First, we show that the conformational preference of CGRP cannot be overridden by Zn2+ complexation. Such properties may prevent the extensive aggregation of CGRP. We also identified intermediate amylin:Zn states that could prevent aggregation at low Zn2+ concentrations. Second, we demonstrate that CGRP can hinder amylin fibrillization, leading to the formation of globular aggregates made predominantly of amylin. We show that amylin and the amylin:CGRP mixture do not show significant toxicity to pancreatic β-cells. However, amylin toxicity could be due to inflammation associated with islet resident macrophage as recently suggested by Westwell-Roper et al.81 Since amylin is co-secreted with insulin, reducing the secretion of insulin is a strategy to lower amylin accumulation. Third, CGRP alone effectively reduces the secretion of Ins2, the orthologue of human insulin, but is less effective against Ins1. In fact, CGRP oligomers are able to stimulate the secretion of Ins1 which is linked to early diabetic development. Fourth, we demonstrate LC-IMS-MS of single islets as a viable and robust method to evaluate insulin secretion in the presence of exogenous peptides, which can become a platform to screen anti-aggregation drugs for T2DM. Finally, our study highlights the importance of surveying not only disease-implicated species, but also their probable co-assemblies that can arise in vivo, which provides a new look on disease etiology and novel insight into the diagnostics and treatments of diabetes.
Author contributions
TDD conceived the study. SDD and MDK performed mouse dissection, isolated mouse islets, and contributed to sample preparation. ALHG and BAL led efforts associated with IMS-MS and TEM experiments. AA collected and analyzed MS data on insulin secretion from isolated single islets. BB and ZAL performed REMD simulations and analyzed the data. SJB and JJC performed toxicity assays. TDD, ALHG, AA, and ZAL wrote the first draft of the manuscript. All authors contributed to revising that document.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TDD gratefully acknowledges the laboratory start-up research support from the University of Tennessee and Department of Chemistry, and the Global Academic Support Program from Agilent. Additionally, we would like to acknowledge John Dunlap and the University of Tennessee Advanced Microscopy and Imaging Center for acquiring the TEM images. ZAL was funded by the National Institutes of Health (grants K01AG062752 and R01AG068285) and thanks the Yale Center for Research Computing for guidance and the use of the re-search computing infrastructure. ZAL also acknowledges that this study was conducted, in part, at the Yale Claude D. Pepper Older Americans Independence Center (P30AG21342).Notes and references
- A. Aguzzi and T. O'Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Gruning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fandrich, R. F. Frank, B. Reif, S. Gunther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2011, 8, 93–101 CrossRef PubMed.
- C. G. Glabe, J. Biol. Chem., 2008, 283, 29639–29643 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, Nature, 2006, 440, 352–357 CrossRef CAS PubMed.
- L. M. Yan, A. Velkova and A. Kapurniotu, Curr. Pharm. Des., 2014, 20, 1182–1191 CrossRef CAS PubMed.
- A. I. Ilitchev, M. J. Giammona, C. Olivas, S. L. Claud, K. L. Lazar Cantrell, C. Wu, S. K. Buratto and M. T. Bowers, J. Am. Chem. Soc., 2018, 140, 9685–9695 CrossRef CAS PubMed.
- M. Baldassarre, C. M. Baronio, L. A. Morozova-Roche and A. Barth, Chem. Sci., 2017, 8, 8247–8254 RSC.
- T. D. Do, N. J. Economou, A. Chamas, S. K. Buratto, J.-E. Shea and M. T. Bowers, J. Phys. Chem. B, 2014, 118, 11220–11230 CrossRef CAS PubMed.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. V. D. Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. D. Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- A. Vandersteen, M. F. Masman, G. De Baets, W. Jonckheere, K. van der Werf, S. J. Marrink, J. Rozenski, I. Benilova, B. De Strooper, V. Subramaniam, J. Schymkowitz, F. Rousseau and K. Broersen, J. Biol. Chem., 2012, 287, 36732–36743 CrossRef CAS PubMed.
- D. L. Hay, S. Chen, T. A. Lutz, D. G. Parkes and J. D. Roth, Pharmacol. Rev., 2015, 67, 564–600 CrossRef CAS PubMed.
- O. Schmitz, B. Brock and J. Rungby, Diabetes, 2004, 53(suppl 3), S233–S238 CrossRef CAS PubMed.
- T. Sanke, G. I. Bell, C. Sample, A. H. Rubenstein and D. F. Steiner, J. Biol. Chem., 1988, 263, 17243–17246 CrossRef CAS.
- J. F. Paulsson, A. Andersson, P. Westermark and G. T. Westermark, Diabetologia, 2006, 49, 1237–1246 CrossRef CAS PubMed.
- Y. Kiriyama and H. Nochi, Cells, 2018, 7, 95 CrossRef PubMed.
- R. A. Ritzel, J. J. Meier, C. Y. Lin, J. D. Veldhuis and P. C. Butler, Diabetes, 2007, 56, 65–71 CrossRef CAS PubMed.
- D. Raleigh, X. Zhang, B. Hastoy and A. Clark, J. Mol. Endocrinol., 2017, 59, R121–R140 CAS.
- S. Nagamatsu, M. Nishi and D. F. Steiner, Diabetes Res. Clin. Pract., 1992, 15, 49–55 CrossRef CAS PubMed.
- A. Pithadia, J. R. Brender, C. A. Fierke and A. Ramamoorthy, J. Diabetes Res., 2016, 2016, 2046327 Search PubMed.
- S. B. Charge, E. J. de Koning and A. Clark, Biochemistry, 1995, 34, 14588–14593 CrossRef CAS PubMed.
- P. Nedumpully-Govindan and F. Ding, Sci. Rep., 2015, 5, 8240 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, R. P. Nanga, N. Popovych, R. de la Salud Bea, S. Vivekanandan, E. N. Marsh and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 8973–8983 CrossRef CAS PubMed.
- S. Salamekh, J. R. Brender, S. J. Hyung, R. P. Nanga, S. Vivekanandan, B. T. Ruotolo and A. Ramamoorthy, J. Mol. Biol., 2011, 410, 294–306 CrossRef CAS PubMed.
- X. Ge, A. Kakinen, E. N. Gurzov, W. Yang, L. Pang, E. H. Pilkington, P. Govindan-Nedumpully, P. Chen, F. Separovic, T. P. Davis, P. C. Ke and F. Ding, Chem. Commun., 2017, 53, 9394–9397 RSC.
- A. C. Susa, C. Wu, S. L. Bernstein, N. F. Dupuis, H. Wang, D. P. Raleigh, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2014, 136, 12912–12919 CrossRef CAS PubMed.
- J. R. Brender, E. L. Lee, K. Hartman, P. T. Wong, A. Ramamoorthy, D. G. Steel and A. Gafni, Biophys. J., 2011, 100, 685–692 CrossRef CAS PubMed.
- F. A. Russell, R. King, S. J. Smillie, X. Kodji and S. D. Brain, Physiol. Rev., 2014, 94, 1099–1142 CrossRef CAS.
- S. G. Amara, V. Jonas, M. G. Rosenfeld, E. S. Ong and R. M. Evans, Nature, 1982, 298, 240–244 CrossRef CAS PubMed.
- M. G. Rosenfeld, J. J. Mermod, S. G. Amara, L. W. Swanson, P. E. Sawchenko, J. Rivier, W. W. Vale and R. M. Evans, Nature, 1983, 304, 129–135 CrossRef CAS PubMed.
- P. K. Mulderry, M. A. Ghatei, R. A. Spokes, P. M. Jones, A. M. Pierson, Q. A. Hamid, S. Kanse, S. G. Amara, J. M. Burrin and S. Legon, et al. , Neuroscience, 1988, 25, 195–205 CrossRef CAS PubMed.
- B. Zhong, S. Ma and D. H. Wang, In Vivo, 2019, 33, 1431–1437 CrossRef CAS PubMed.
- B. Ahren and F. Sundler, Cell Tissue Res., 1992, 269, 315–322 CrossRef CAS PubMed.
- Z. Kee, X. Kodji and S. D. Brain, Front. Physiol., 2018, 9, 1249 CrossRef PubMed.
- S. D. Brain, T. J. Williams, J. R. Tippins, H. R. Morris and I. MacIntyre, Nature, 1985, 313, 54–56 CrossRef CAS PubMed.
- U. Arulmani, A. Maassenvandenbrink, C. M. Villalon and P. R. Saxena, Eur. J. Pharmacol., 2004, 500, 315–330 CrossRef CAS PubMed.
- G. Fagherazzi, D. El Fatouhi, A. Fournier, G. Gusto, F. R. Mancini, B. Balkau, M. C. Boutron-Ruault, T. Kurth and F. Bonnet, JAMA Neurol., 2019, 76, 257–263 CrossRef PubMed.
- P. L. Durham, Headache, 2006, 46, S3–S8 CrossRef PubMed.
- S. Iyengar, K. W. Johnson, M. H. Ossipov and S. K. Aurora, Headache, 2019, 59, 659–681 CrossRef PubMed.
- B. Ahren and M. Pettersson, Int. J. Pancreatol., 1990, 6, 1–15 CAS.
- M. Pettersson and B. Ahren, Regul. Pept., 1988, 23, 37–50 CrossRef CAS PubMed.
- H. Tanaka, R. Kashiwagi and T. Koizumi, Exp. Clin. Endocrinol. Diabetes, 2013, 121, 280–285 CrossRef CAS PubMed.
- B. Leighton and G. J. Cooper, Nature, 1988, 335, 632–635 CrossRef CAS PubMed.
- T. N. Rasmussen, M. Bersani, P. Schmidt, L. Thim, H. Kofod, P. N. Jorgensen, S. S. Poulsen and J. J. Holst, Pancreas, 1998, 16, 195–204 CrossRef CAS PubMed.
- T. Morishita, A. Yamaguchi, T. Yamatani, A. Nakamura, N. Arima, Y. Yamashita, H. Nakata, T. Fujita and T. Chiba, Diabetes Res. Clin. Pract., 1992, 15, 63–69 CrossRef CAS.
- K. Hermansen and B. Ahren, Regul. Pept., 1990, 27, 149–157 CrossRef CAS.
- S. D. Brain, S. Wimalawansa, I. MacIntyre and T. J. Williams, Am. J. Pathol., 1990, 136, 487–490 CAS.
- M. T. Galeazza, T. D. O'Brien, K. H. Johnson and V. S. Seybold, Peptides, 1991, 12, 585–591 CrossRef CAS.
- L. A. Woods, S. E. Radford and A. E. Ashcroft, Biochim. Biophys. Acta, 2013, 1834, 1257–1268 CrossRef CAS PubMed.
- G. Ben-Nissan and M. Sharon, Curr. Opin. Chem. Biol., 2018, 42, 25–33 CrossRef CAS PubMed.
- F. Lanucara, S. W. Holman, C. J. Gray and C. E. Eyers, Nat. Chem., 2014, 6, 281–294 CrossRef CAS PubMed.
- L. M. Young, R. A. Mahood, J. C. Saunders, L.-H. Tu, D. P. Raleigh, S. E. Radford and A. E. Ashcroft, Analyst, 2015, 140, 6990–6999 RSC.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2011, 133, 7240–7243 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 18283–18292 CrossRef CAS PubMed.
- L. M. Young, P. Cao, D. P. Raleigh, A. E. Ashcroft and S. E. Radford, J. Am. Chem. Soc., 2013, 136, 660–670 CrossRef PubMed.
- A. I. Ilitchev, M. J. Giammona, J. N. Schwarze, S. K. Buratto and M. T. Bowers*, J. Phys. Chem. B, 2018, 122, 9852–9859 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- Z. A. Levine, K. Teranishi, A. K. Okada, R. Langen and J.-E. Shea, J. Am. Chem. Soc., 2019, 141, 14168–14179 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1996, 314, 141–151 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef PubMed.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
- S. J. Burke, M. D. Karlstad, K. M. Regal, T. E. Sparer, D. Lu, C. M. Elks, R. W. Grant, J. M. Stephens, D. H. Burk and J. J. Collier, Biochim. Biophys. Acta, Gene Regul. Mech., 2015, 1849, 637–652 CrossRef CAS PubMed.
- M. C. Manning, Biophys. Res. Commun., 1989, 160, 388–392 CrossRef CAS PubMed.
- H. A. Watkins, D. L. Rathbone, J. Barwell, D. L. Hay and D. R. Poyner, Br. J. Pharmacol., 2013, 170, 1308–1322 CrossRef CAS PubMed.
- B. Lynch and E. T. Kaiser, Biochemistry, 1988, 27, 7600–7607 CrossRef CAS PubMed.
- A. L. Breeze, T. S. Harvey, R. Bazzo and I. D. Campbell, Biochemistry, 1990, 30, 575–582 CrossRef PubMed.
- W. Li, L. N. Kinch, P. A. Karplus and N. V. Grishin, Protein Sci., 2015, 24, 1075–1086 CrossRef CAS.
- B. Formby, F. Schmid-Formby and G. M. Grodsky, Diabetes, 1984, 33, 229–234 CrossRef CAS PubMed.
- M. C. Foster, R. D. Leapman, M. X. Li and I. Atwater, Biophys. J., 1993, 64, 525–532 CrossRef CAS PubMed.
- S. J. Burke, K. Stadler, D. Lu, E. Gleason, A. Han, D. R. Donohoe, R. C. Rogers, G. E. Hermann, M. D. Karlstad and J. J. Collier, Am. J. Physiol.: Endocrinol. Metab., 2015, 309, E715–E726 CrossRef CAS PubMed.
- J. J. Collier, S. J. Burke, M. E. Eisenhauer, D. H. Lu, R. C. Sapp, C. J. Frydman and S. R. Campagna, PLoS One, 2011, 6, e22485 CrossRef CAS PubMed.
- N. Shigihara, A. Fukunaka, A. Hara, K. Komiya, A. Honda, T. Uchida, H. Abe, Y. Toyofuku, M. Tamaki, T. Ogihara, T. Miyatsuka, H. J. Hiddinga, S. Sakagashira, M. Koike, Y. Uchiyama, T. Yoshimori, N. L. Eberhardt, Y. Fujitani and H. Watada, J. Clin. Invest., 2014, 124, 3634–3644 CrossRef CAS PubMed.
- R. A. Ritzel and P. C. Butler, Diabetes, 2003, 52, 1701–1708 CrossRef CAS PubMed.
- C. Y. Westwell-Roper, J. A. Ehses and C. B. Verchere, Diabetes, 2014, 63, 1698–1711 CrossRef CAS PubMed.
- R. Salbo, M. F. Bush, H. Naver, I. Campuzano, C. V. Robinson, I. Pettersson, T. J. Jorgensen and K. F. Haselmann, Rapid Commun. Mass Spectrom., 2012, 26, 1181–1193 CrossRef CAS.
- A. Tkatchenko, M. Rossi, V. Blum, J. Ireta and M. Scheffler, Phys. Rev. Lett., 2011, 106, 118102 CrossRef PubMed.
- F. Tofoleanu, Y. Yuan, F. C. t. Pickard, B. Tywoniuk, B. R. Brooks and N. V. Buchete, J. Phys. Chem. B, 2018, 122, 5657–5665 CrossRef CAS PubMed.
- E. T. Jansson, T. J. Comi, S. S. Rubakhin and J. V. Sweedler, ACS Chem. Biol., 2016, 11, 2588–2595 CrossRef CAS PubMed.
- M. Ramstrom, C. Hagman, Y. O. Tsybin, K. E. Markides, P. Hakansson, A. Salehi, I. Lundquist, R. Hakanson and J. Bergquist, Eur. J. Biochem., 2003, 270, 3146–3152 CrossRef PubMed.
- H. Roderigo-Milne, A. C. Hauge-Evans, S. J. Persaud and P. M. Jones, Biochem. Biophys. Res. Commun., 2002, 296, 589–595 CrossRef CAS.
- N. Babaya, M. Nakayama, H. Moriyama, R. Gianani, T. Still, D. Miao, L. Yu, J. C. Hutton and G. S. Eisenbarth, Diabetologia, 2006, 49, 1222–1228 CrossRef CAS PubMed.
- C. M. Elso, N. A. Scott, L. Mariana, E. I. Masterman, A. P. R. Sutherland, H. E. Thomas and S. I. Mannering, PLoS One, 2019, 14, e0225021 CrossRef CAS PubMed.
- L. I. Berge, T. Riise, O. B. Fasmer, O. Hundal, K. J. Oedegaard, K. Midthjell and A. Lund, Epidemiology, 2013, 24, 129–134 CrossRef PubMed.
- N. M. Templeman, S. Flibotte, J. H. L. Chik, S. Sinha, G. E. Lim, L. J. Foster, C. Nislow and J. D. Johnson, Cell Rep., 2017, 20, 451–463 CrossRef CAS PubMed.
- C. M. Elso, N. A. Scott, L. Mariana, E. I. Masterman, A. P. R. Sutherland, H. E. Thomas and S. I. Mannering, PLoS One, 2019, 14, e0225021 CrossRef CAS PubMed.
- H. Deng, G. G. Li, H. Nie, Y. Y. Feng, G. Y. Guo, W. L. Guo and Z. P. Tang, BMC Neurol., 2020, 20, 57 CrossRef CAS PubMed.
- R. Manoukian, H. Sun, S. Miller, D. Shi, B. Chan and C. Xu, J. Headache Pain, 2019, 20, 44 CrossRef PubMed.
- T. Blundell, G. Dodson, D. Hodgkin and D. Mercola, Adv. Protein Chem., 1972, 26, 279–402 CrossRef CAS.
- A. Martínez, S. Kapas, M. J. Miller, Y. Ward and F. Cuttitta, Endocrinology, 2000, 141(1), 406–411 CrossRef PubMed.
- E. A. Mason and H. W. Schamp, Ann. Phys., 1958, 4(3), 233–270 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental protocols, TEM images, mass spectra and ATDs. See DOI: 10.1039/d1sc01167g |
| ‡ Equal contribution. |
| § Current address: Department of Chemistry, University of Washington, Seattle, WA 98195. |
| This journal is © The Royal Society of Chemistry 2021 |