
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid and ultrasensitive electrochemical detection of circulating tumor DNA by hybridization on the network of gold-coated magnetic nanoparticles†
Dongfei
Chen
abc,
Yanfang
Wu
 abc,
Sharmin
Hoque
abc,
Richard D.
Tilley
abd and
J. Justin
Gooding
*abc
abc,
Sharmin
Hoque
abc,
Richard D.
Tilley
abd and
J. Justin
Gooding
*abc
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: justin.gooding@unsw.edu.au
bAustralian Centre for NanoMedicine, The University of New South Wales, Sydney, NSW 2052, Australia
cThe ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, The University of New South Wales, Sydney, NSW 2052, Australia
dElectron Microscope Unit, Mark Wainwright Analytical Centre, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 3rd March 2021
Abstract
An accurate and robust method for quantifying the levels of circulating tumor DNA (ctDNA) is vital if this potential biomarker is to be used for the early diagnosis of cancer. The analysis of ctDNA presents unique challenges because of its short half-life and ultralow abundance in early stage cancers. Here we develop an ultrasensitive electrochemical biosensor for rapid detection of ctDNA in whole blood. The sensing of ctDNA is based on hybridization on a network of probe DNA modified gold-coated magnetic nanoparticles (DNA-Au@MNPs). This DNA-Au@MNPs biosensor can selectively detect short- and long-strand DNA targets. It has a broad dynamic range (2 aM to 20 nM) for 22 nucleotide DNA target with an ultralow detection limit of 3.3 aM. For 101 nucleotide ctDNA target, a dynamic range from 200 aM to 20 nM was achieved with a detection limit of 5 fM. This DNA-Au@MNPs based sensor provides a promising method to achieve 20 min response time and minimally invasive cancer early diagnosis.
Introduction
Circulating cell-free tumor DNA (ctDNA) is small genomic fragments (typically 90–150 base pairs long1,2) released by tumor cells into the bloodstream.3–5 Importantly, ctDNA carries cancer-specific genetic and epigenetic aberrations. As ctDNA can be found in the blood stream, it is potentially an exceedingly important liquid biopsy based biomarker for the early detection of cancer that can be acquired minimally invasively.2,6,7 The analytical challenges are that ctDNA is found in extremely low abundance (fM or lower) and has a short half-life (16 min to 2.5 h) in complex biological fluids such as blood.2,8,9 As such, there is an unmet need for rapidly responding, ultrasensitive biosensor for the detection of ctDNA in blood.The twin challenges of fast responses and low detection limits are often contradictory. This is because to achieve sub-picomolar detection limits, the underlying challenge to be overcome is the slow mass transport of the analyte species to the sensing interface, which results in slow response times.10 We reported the concept of ‘dispersible electrodes’ to rise to these twin challenges.11–14 It utilizes gold-coated magnetic nanoparticles (Au@MNPs) that are distributed throughout the sample such that diffusional pathlengths are short, effectively seeking out the analyte, rather than the traditional sensing paradigm where the analyte must find the sensing interface.15,16 Application of a magnetic field allows the rapid reassembly of the sensor from the dispersed Au@MNPs in a similar manner to magnetic preconcentration. In magnetic preconcentration the magnetic nanoparticles only transport the analyte. However, with the dispersible electrode concept, the same Au@MNPs are also the electrochemical transducers of a sensor. The reduced diffusional pathlengths and magnetic field give rapid responses. Additionally, the fact that more of the analyte is captured than with a conventional sensor also gives higher sensitivities and lowered detection limits.11 This performance has been shown to be even more impressive with DNA modified Au@MNPs for the detection of microRNA, where the outstanding electrochemical measurement performance was suggested to be due to the network of the DNA-Au@MNPs, in electric fields used in the electrochemistry, to create an in-built amplification system.14
Herein, this study unlocks the potential of the dispersible electrodes concept to develop an electrochemical sensor for the detection of ctDNA. The sensor has the twin benefits of rapid response and low detection limits that are so badly needed. The Au@MNPs were synthesized and then modified with methylene blue (MB)-labeled probe DNA molecules that are complementary to the target ctDNA to provide the sensor specificity. These sensing nanoparticles are referred to as MB-DNA-Au@MNPs. The MB at the end of thiolated probe DNA strands on MB-DNA-Au@MNPs served as redox probes. The magnitude of the current from the MB is sensitive to the amount of target ctDNA that hybridizes with the probe strands. To enhance the sensor sensitivity, over the previous version of the electrically-reconfigurable Au@MNP sensors for miRNA,14 the surface of the macroscale gold electrode was modified with probe MB-DNA and 6-mercapto-1-hexanol (MCH). This means that the introduction of potassium ferricyanide (K3[Fe(CN)6]) into electrolyte solution allows a MB/ferricyanide amplification system to be established. The signal amplification is achieved through the electroreduction of ferricyanide by MB tags from both surfaces of the electrode and Au@MNPs. The feasibility of the sensing strategy was validated by firstly applying the developed sensor to detect a short-strand target DNA (22 nucleotides) as the model analyte. The dynamic range, detection limit and sensor specificity of the developed sensor were evaluated. Afterwards, this sensor was employed to detect 101 nucleotide ctDNA sequence indicative of non-small-cell lung cancer (NSCLC) in buffer solutions and human blood samples with a 20 min response time.
Results and discussion
The principle of the proposed sensing strategy is illustrated in Fig. 1. As seen in Fig. 1a, the surfaces of the Au@MNPs were modified with a MB modified DNA probe sequence, complementary to the target ctDNA. The Au@MNPs, shown by the representative TEM image in Fig. S1,† were synthesized using a previously developed method.15 The characterization information of Au@MNPs according to the MIRIBEL principle17 are summarized in Table S3.† 10 μL of Au@MNP solution (1.56 × 1010 particles per mL) was added to each sample, which resulted in a total of 1.56 × 108 particles in each sample. The DNA probe had a thiol modification at the 5′ end and MB redox label at the 3′ end. After exposing the MB-DNA-Au@MNPs to complementary ctDNA target for 30 min, the hybridized MB-DNA-Au@MNPs were separated from the supernatant solution by applying a centrifugation step (5000 rpm for 5 min) and then removal of the supernatant by pipette with a magnet placed at the bottom of the sample tube. The hybridized MB-DNA-Au@MNPs were rinsed twice with phosphate buffered saline (PBS) solution (pH 7.4), discarding the supernatant, and then re-dispersed into the 0.5 mM potassium ferricyanide (K3[Fe(CN)6]) in PBS in a custom-made glass electrochemical cell. The hybridized MB-DNA-Au@MNPs were collected onto the surface of gold electrode using a magnet for electrochemical measurements.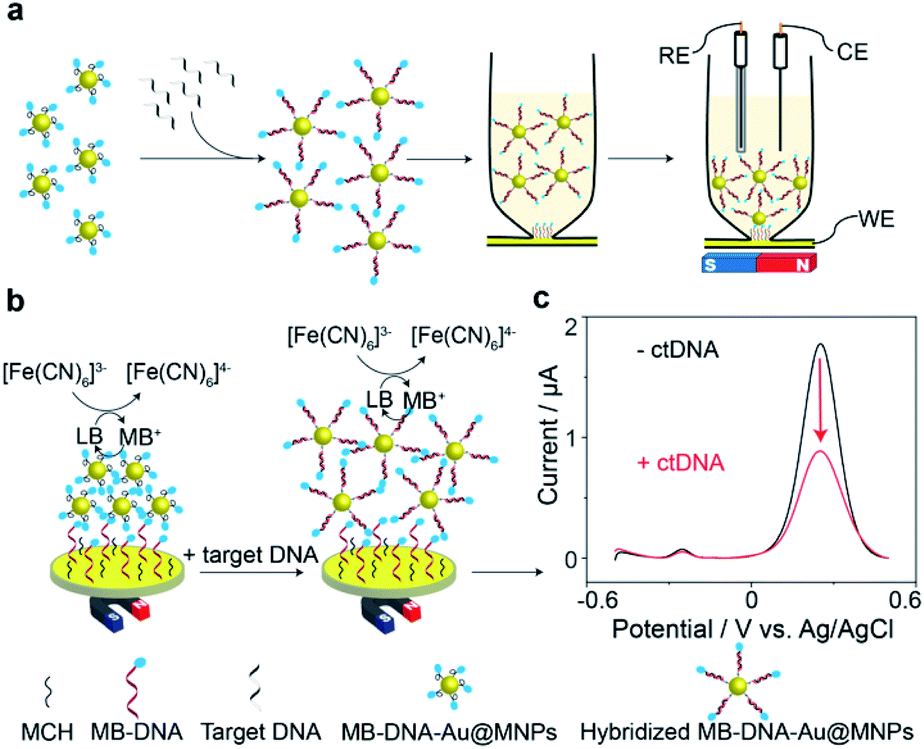 | ||
| Fig. 1 Schematic illustration of DNA-mediated reduction of potassium ferricyanide (K3[Fe(CN)6]) by methylene blue (MB). (a) Workflow for the measurement of target ctDNA. (b) Electrochemical sensing mechanism of the DNA sensor. (c) Background-subtracted square wave voltammograms (SWVs) in the absence and presence of target ctDNA (101 nucleotides). The frequency and pulse amplitude of square wave voltammograms were 2 Hz and 25 mV, respectively. | ||
The electrochemical sensing mechanism of the DNA sensor is presented in Fig. 1b. Under a magnet, the MB-DNA-Au@MNPs were brought to the gold surface wherein a mixed monolayer of single-strand DNA and MCH was immobilized with an upright conformation of the MB-DNA strands.18,19 A redox amplification cycle,20,21 where MB is reduced to leucomethylene blue (LB) which is then oxidized back to MB by [Fe(CN)6]3− in the detection solution, induces the signal amplification.20 Square wave voltammograms (SWVs) representing the electrochemical signals, before and after hybridization with the target ctDNA are shown in Fig. 1c. An average (50.7 ± 3.7)% decrease in the magnitude of electrochemical current for detecting 20 nM target ctDNA was observed. According to our previous work,14 after hybridizing with target DNA strands, the formed duplex on the Au@MNP caused an increased distance between the MB-DNA-Au@MNPs, thus impeding the electron tunnelling through the network of the MB-DNA-Au@MNPs. It was confirmed that even when there was a very limited number of hybridized DNA strands, and hence duplex structures formed on the Au@MNPs, this process is enough to cause changes in the electronic communication in the network of MB-DNA-Au@MNPs and thus change the electrochemical response correspondingly. Furthermore, the pulsing of the applied electric field was also hypothesized to influence the re-directing of the magnetically assembled network of the Au@MNPs to be closer to the electrode surface, which acts to further suppress the electrochemical current and enhance the sensitivity of current change accounted for a small number of target molecules. It is these two processes operating together that enable the ultrasensitive detection of ctDNA by the network of MB-DNA-Au@MNPs. Therefore, the decrease in peak current can serve as a measure of the presence of target ctDNA.
Surface modification on the gold electrode surface is expected to have an impact on the electrochemical redox cycling and thus the sensing performance. Herein, five different interfacial designs were investigated to obtain the maximum signal change for the ultrasensitive detection of 22 nucleotide DNA (Fig. 2). In panel (a), the gold foil is not modified, MB only exists on the surface of Au@MNPs. In panel (b and d), MB only exists on either the surface of Au@MNPs or the surface of gold foil; in panel (c), MB is presented on both surfaces of gold foil and Au@MNPs; in panel (e), no MB exists in the whole system. The change in current shows that the self-assembled monolayer of thiolated DNA and MCH on the underlying bulk gold electrode as well as the Au@MNPs increases the sensitivity as there is a larger change in current (panel (c)). Therefore, the system with MB-DNA modified on both the gold foil and Au@MNPs was chosen for further experiments. Note the presence of [Fe(CN)6]3− is also shown to be necessary (see Fig. S2 in the ESI†). From the data presented in Fig. S2(e, f, h and i),† the amplitude of changed current for detecting 20 nM target DNA in the absence and presence of K3[Fe(CN)6] was 0.016 μA and 0.96 μA, respectively. This gives a sixtyfold increase in signal intensity for the detection of 20 nM DNA.
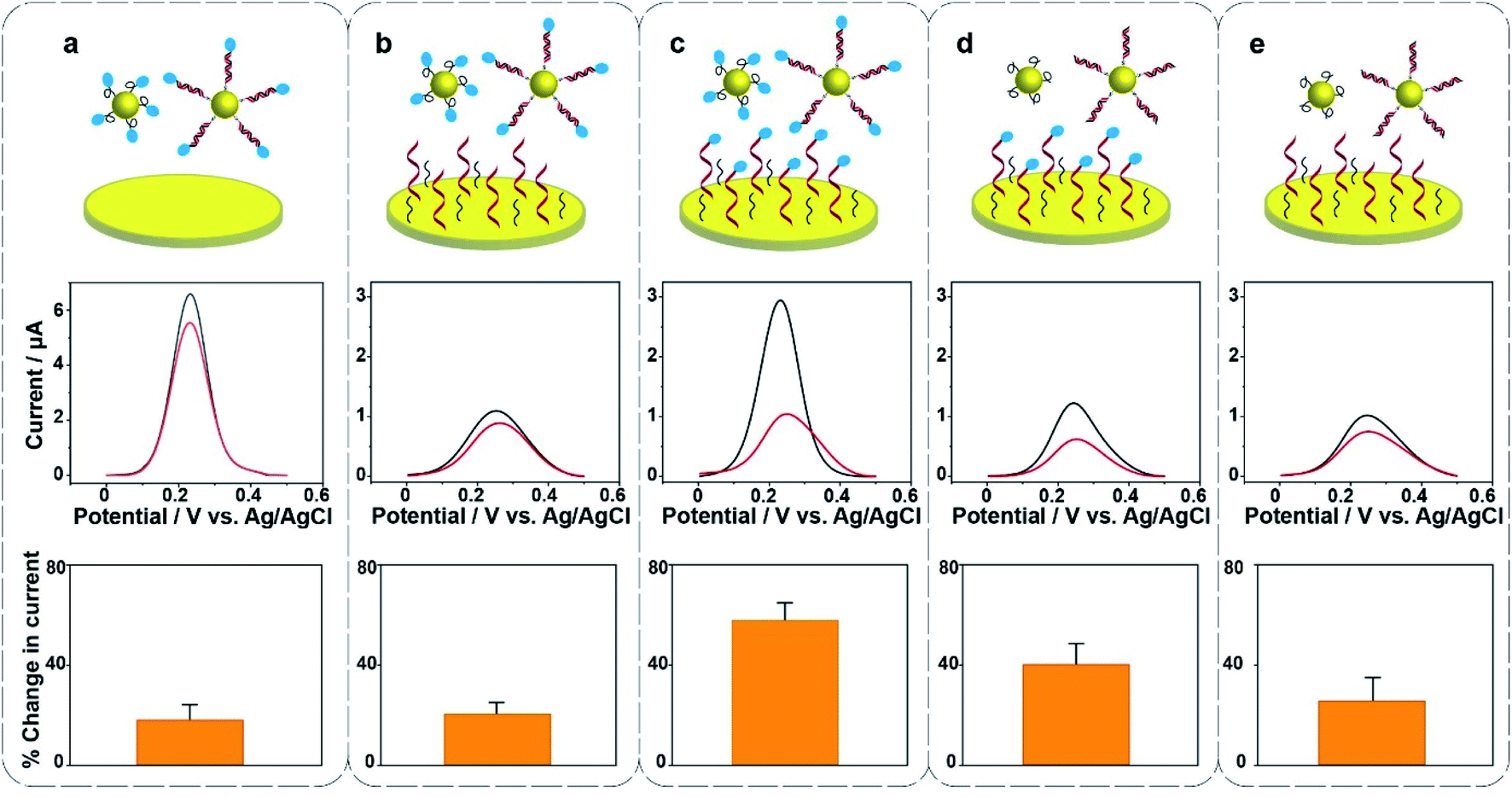 | ||
| Fig. 2 Hybridization-induced change in background-subtracted SWVs and percentage change in current after exposing the sensor to 20 nM complementary target DNA (22 nucleotides) in five different systems as indicated by the cartoons. Electrolyte solution was 0.5 mM K3[Fe(CN)6] in phosphate buffered saline (pH 7.4). The frequency and pulse amplitude of SWVs were 2 Hz and 25 mV, respectively. Error bars represent standard deviations from at least 3 independent measurements. | ||
To examine the feasibility of the sensing platform for nucleic acid detection, a 22 nucleotide DNA strand was selected as the target. The dependence of the change in peak current in the SWVs on target DNA concentration is shown in Fig. 3. The change in current is linear with the logarithm of the concentration of target DNA. The linear regression equation was Y = 4.32X + 88.97 with R2 = 0.993 and the standard error of the regression was 1.72%. A broad dynamic range with ten orders of concentration crossing 20 nM to 2 aM was achieved with an extremely low detection limit of 3.3 aM. Usually, the concentration of ctDNA in the bloodstream from cancer patients lies in fM or lower.2,9 Our diagnostic method with the 3.3 aM detection limit indicates a reliable sensitivity and great potential of this proposed sensor for the ultrasensitive detection of cancer-relevant nucleic acid targets in early stages. This broad linear range is consistent with other reported studies.22,23
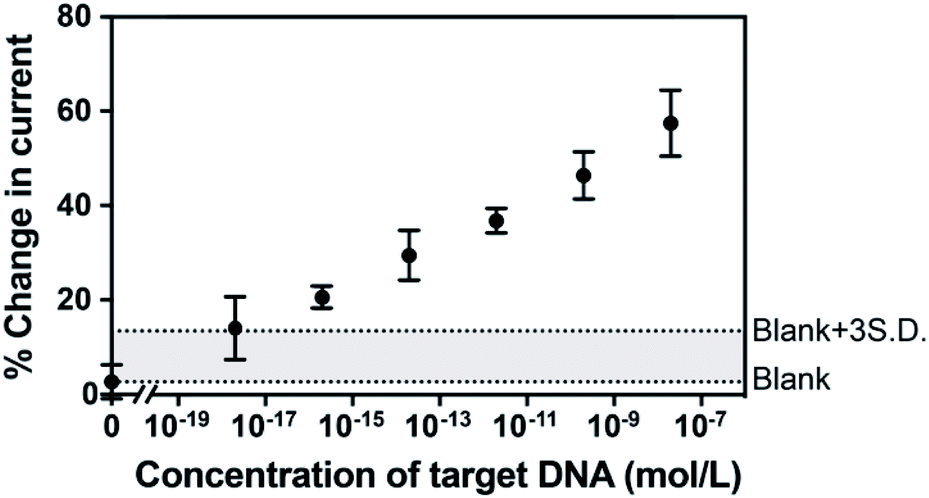 | ||
| Fig. 3 The dependence of hybridization-induced current change from SWVs upon complementary target 22 nucleotide DNA concentration (0 aM, 2 aM, 200 aM, 20 fM, 2 pM, 200 pM and 20 nM). Two dash lines are provided to indicate the levels of the average blank and the three times standard deviation above the averaged blank. Error bars represent standard deviations from 5 independent measurements. | ||
The sensor was next challenged by detecting a long-strand ctDNA (101 nucleotides) indicative of human non-small-cell lung cancer (NSCLC) which stands for 80–85% of lung cancer.24 The hybridization efficiency is dependent on the position of the target nucleic acid where the probe DNA binds to. To investigate the position-dependent hybridization efficiency, we compared the performances of two kinds of probe DNA sequences targeted to 3′ end and to the middle of ctDNA target, respectively (Fig. 4a). A much lower hybridization efficiency for the probe DNA targeting to the middle of target DNA was observed compared with that binding to the 3′ end of target DNA. These results demonstrate the importance of choosing the appropriate nucleic acid to improve the sensitivity and specificity of sensing platform. This is particularly important for long strand ctDNA detection. In further experiments below, the probe DNA that targets to 3′ end of ctDNA was used.
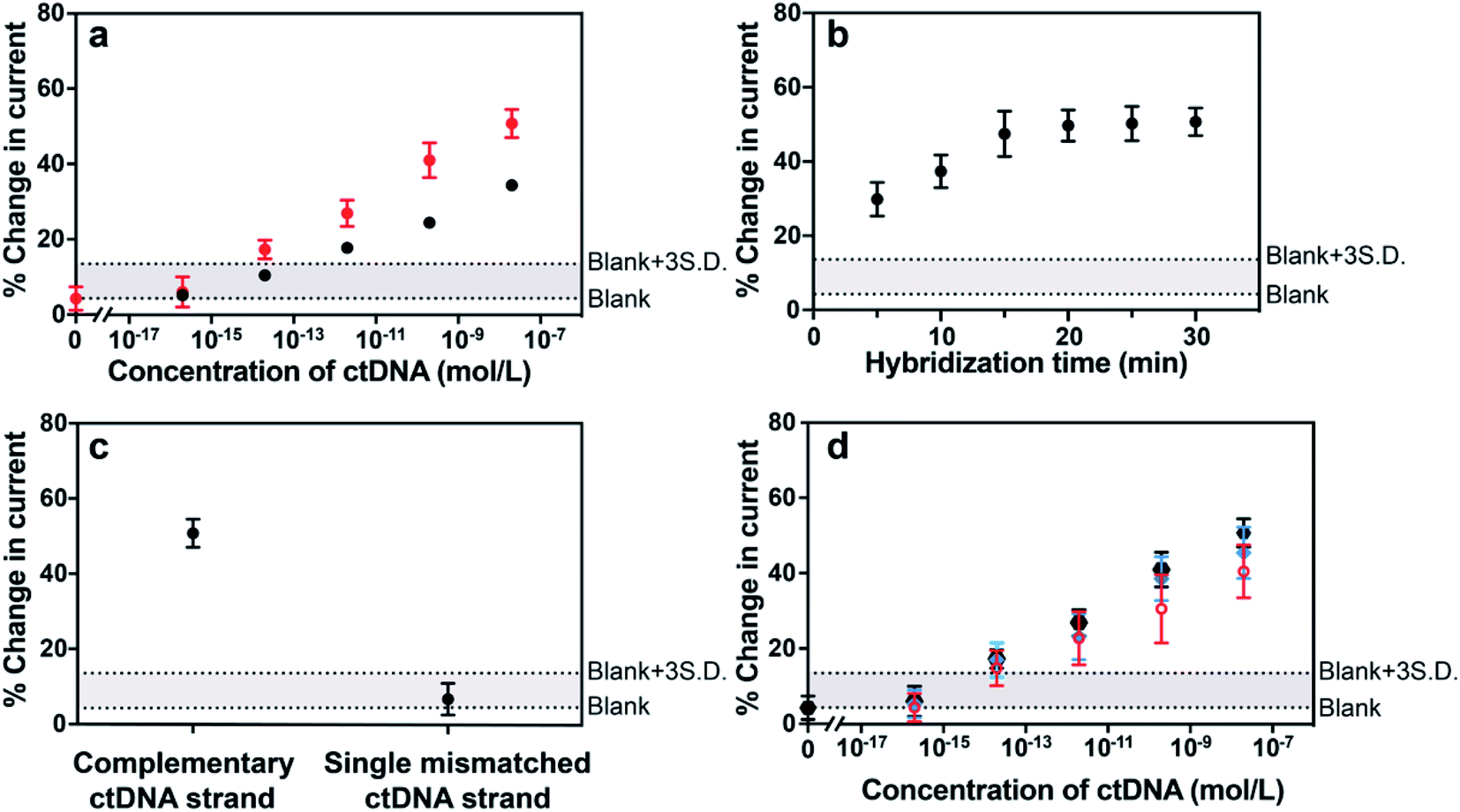 | ||
| Fig. 4 (a) Hybridization-induced change in the SWVs after exposing the sensor to different concentrations of complementary ctDNA target (101 nucleotides) wherein the probe DNA hybridized to the 3′ end (red data points) and the middle (black data points) of the ctDNA. (b) Effect of hybridization time after exposing the sensor to 20 nM complementary ctDNA target (101 nucleotides) on the SWV current change. (c) Hybridization-induced change in the SWVs after exposing the sensor to complementary and single mismatched circulating tumor DNA target sequences. (d) Hybridization-induced change in SWVs after exposing the sensor to different concentrations of complementary ctDNA target (101 nucleotides) spiked in phosphate buffered saline (black data points), human plasma (blue data points) and 50% human blood (red data points). Two dash lines are provided to indicate the levels of the average blank and the three times standard deviation above the averaged blank. Error bars represent standard deviations from at least 5 independent experiments. | ||
There are three major challenges in achieving ultrasensitive detection of ctDNA, that is, short half-life, low abundance in the early stages of cancer and the complex biological environment of whole blood when focusing on liquid biopsies. The half-life of ctDNA in the blood circulation is typically less than 2.5 h,2,9 therefore, assay and hybridization time is a critical factor for ctDNA detection. Digital PCR, as the gold standard method for the quantification of ctDNA, however, presents a relatively good sensitivity which is mainly limited by the maximum number of droplets available for analysis. Moreover, this method requires time (up to several hours) from the DNA extraction from blood samples to the quantification of DNA.25,26 Therefore, it is of great significance to demonstrate this electrochemical detection platform having the capability of rapidly analyzing ctDNA, especially from blood samples directly. With the need for rapid responses in mind, the impact of hybridization time on the signal change of the sensor was investigated. Fig. 4b showed that the hybridization-induced current change increased gradually with the hybridization time from 0 to 20 min and thereafter remained steady. This 20 min response time makes the MB-DNA-Au@MNP sensor the most rapidly responding ctDNA biosensor we are aware of. Prior to our study, the fastest ctDNA biosensor we found at a response time of 65 min for a 69 base-pair long sequence.27 Other studies use shorter DNA sequences as models of ctDNA and have response times of the order of 1.5 to 5.5 h.28–30
The specificity of the biosensor was explored using the long ctDNA (101 nucleotides) strands by investigating single point mutation of the NSCLC-related sequence. The average changes of (50.7 ± 3.7)% and (6.6 ± 4.2)% decrease in the current were observed after hybridization with 20 nM complementary ctDNA target and single mismatched 101 nucleotide ctDNA sequence (Fig. 4c), indicating that this sensor can differentiate the tumor DNA sequence from the wild-type (healthy) sequence. This biosensor was also shown to be able to discriminate short-strand complementary and single-mismatched DNA target (22 nucleotides) (see Fig. S3 in the ESI†). Averaged values of (1.36 ± 0.03) μA and (0.83 ± 0.03) μA were obtained from 10 consecutive measurements of background-subtracted SWVs for the sensor before and after exposure to 200 pM 101 nucleotide ctDNA target, indicating stable signals for analytical analysis. Also, the robustness of the measurement methodology was reflected by the small variation among measurements in different concentrations, indicated by the narrow error bars in the results of Fig. 4a.
Additionally, the detection limit and dynamic range of the sensor were evaluated when detecting different concentrations of 101 nucleotide ctDNA in phosphate buffered saline, undiluted human plasma and 50% whole human blood (Fig. 4d). Plasma and blood samples used in this experiment were obtained from a healthy human donor and were spiked with the 101 nucleotide ctDNA of different concentrations. We performed the detection experiment with 50% human blood due to the high viscosity of 100% whole blood impacting the efficiency of the magnetic re-collection of the nanoparticles.14 Compared with the detection performance of the 22 nucleotide DNA target, the sensing of 101 nucleotide ctDNA target presented a good sensitivity and a dynamic range from 200 aM to 20 nM with a detection limit of 5 fM in phosphate buffered saline, indicating that the length of the target did not significantly interfere the sensing performance. Importantly, this detection range is expected to align well with the levels of the ctDNA in patients for early diagnosis. Furthermore, the sensor performance in 50% human blood showed only minor decrease relative to phosphate buffered saline and human plasma, exhibiting the robustness of the sensor in complex biological media and the exciting potential in clinically relevant analysis.
Conclusions
In summary, this study introduces a new electrochemical sensing assay for the direct detection of ctDNA from whole blood in combination with the DNA-Au@MNPs with high sensitivity and excellent selectivity. This approach improves the detection limit and response time by applying dispersible electrodes to preconcentrate and collect analyte of interest to the sensing interface, thus addressing the limited mass transfer rate and achieving direct analysis of blood samples in ultralow amounts. Moreover, the sensing platform can be applied to not only short strand DNA sensing but also long strand ctDNA sensing with a broad dynamic range and ultralow detection limits. Additionally, the significantly reduced analysis time, which can be as short as about 20 min, makes the approach more attractive compared to PCR-based methods. Taken together, the electrochemical sensing strategy reported here demonstrates the potential to be used for minimally invasive analysis of ctDNA, which could be used as an alternative to cancer tissue biopsy for monitoring the dynamics of cancer.Conflicts of interest
J. J. G., D. C., and Y. W. have filed a provisional patent application pertaining to the results presented in this paper. The authors declare no other competing interests.Acknowledgements
This research was supported by the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology (CE14100036), the ARC Australian Laureate Fellowship (FL150100060) and the NHMRC Investigator Award (APP1196648).Notes and references
- F. Mouliere, D. Chandrananda, A. M. Piskorz, E. K. Moore, J. Morris, L. B. Ahlborn, R. Mair, T. Goranova, F. Marass, K. Heider, J. C. M. Wan, A. Supernat, I. Hudecova, I. Gounaris, S. Ros, M. Jimenez-Linan, J. Garcia-Corbacho, K. Patel, O. Ostrup, S. Murphy, M. D. Eldridge, D. Gale, G. D. Stewart, J. Burge, W. N. Cooper, M. S. van der Heijden, C. E. Massie, C. Watts, P. Corrie, S. Pacey, K. M. Brindle, R. D. Baird, M. Mau-Sorensen, C. A. Parkinson, C. G. Smith, J. D. Brenton and N. Rosenfeld, Sci. Transl. Med., 2018, 10, eaat4921 CrossRef PubMed.
- J. C. M. Wan, C. Massie, J. Garcia-Corbacho, F. Mouliere, J. D. Brenton, C. Caldas, S. Pacey, R. Baird and N. Rosenfeld, Nat. Rev. Cancer, 2017, 17, 223–238 CrossRef CAS PubMed.
- A. M. Aravanis, M. Lee and R. D. Klausner, Cell, 2017, 168, 571–574 CrossRef CAS PubMed.
- J. Das and S. O. Kelley, Angew. Chem. Int. Ed., 2020, 59, 2554–2564 ( Angew. Chem. , 2020 , 132 , 2574–2784 ) CrossRef CAS PubMed.
- A. E. Rodda, B. J. Parker, A. Spencer and S. R. Corrie, ACS Sens., 2018, 3, 540–560 CrossRef CAS PubMed.
- M. Murtaza, S. J. Dawson, D. W. Tsui, D. Gale, T. Forshew, A. M. Piskorz, C. Parkinson, S. F. Chin, Z. Kingsbury, A. S. Wong, F. Marass, S. Humphray, J. Hadfield, D. Bentley, T. M. Chin, J. D. Brenton, C. Caldas and N. Rosenfeld, Nature, 2013, 497, 108–112 CrossRef CAS PubMed.
- J. Das, I. Ivanov, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2016, 138, 11009–11016 CrossRef CAS PubMed.
- A. K. Mattox, C. Bettegowda, S. Zhou, N. Papadopoulos, K. W. Kinzler and B. Vogelstein, Sci. Transl. Med., 2019, 11, eaay1984 CrossRef PubMed.
- F. Diehl, K. Schmidt, M. A. Choti, K. Romans, S. Goodman, M. Li, K. Thornton, N. Agrawal, L. Sokoll, S. A. Szabo, K. W. Kinzler, B. Vogelstein and L. A. Diaz Jr, Nat. Med., 2008, 14, 985–990 CrossRef CAS.
- Y. Wu, R. D. Tilley and J. J. Gooding, J. Am. Chem. Soc., 2019, 141, 1162–1170 CrossRef CAS PubMed.
- I. Y. Goon, L. M. Lai, M. Lim, R. Amal and J. J. Gooding, Chem. Commun., 2010, 46, 8821–8823 RSC.
- K. Chuah, L. M. Lai, I. Y. Goon, S. G. Parker, R. Amal and J. Justin Gooding, Chem. Commun., 2012, 48, 3503–3505 RSC.
- L. M. Lai, I. Y. Goon, K. Chuah, M. Lim, F. Braet, R. Amal and J. J. Gooding, Angew. Chem. Int. Ed., 2012, 51, 6456–6459 ( Angew. Chem. , 2012 , 124 , 6562–6565 ) CrossRef CAS.
- R. Tavallaie, J. McCarroll, M. Le Grand, N. Ariotti, W. Schuhmann, E. Bakker, R. D. Tilley, D. B. Hibbert, M. Kavallaris and J. J. Gooding, Nat. Nanotechnol., 2018, 13, 1066–1071 CrossRef CAS PubMed.
- I. Y. Goon, L. M. H. Lai, M. Lim, P. Munroe, J. J. Gooding and R. Amal, Chem. Mater., 2009, 21, 673–681 CrossRef CAS.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 31, e1904385 CrossRef PubMed.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777–785 CrossRef CAS PubMed.
- B. Liu and J. Liu, Anal. Methods, 2017, 9, 2633–2643 RSC.
- T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119, 8916–8920 CrossRef CAS.
- S. O. Kelley, E. M. Boon, J. K. Barton, N. M. Jackson and M. G. Hill, Nucleic Acids Res., 1999, 27, 4830–4837 CrossRef CAS PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- Y. An, R. Li, F. Zhang and P. He, Anal. Chem., 2020, 92, 5404–5410 CrossRef CAS PubMed.
- S. Su, H. Sun, W. Cao, J. Chao, H. Peng, X. Zuo, L. Yuwen, C. Fan and L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 6826–6833 CrossRef CAS PubMed.
- A. M. Newman, S. V. Bratman, J. To, J. F. Wynne, N. C. Eclov, L. A. Modlin, C. L. Liu, J. W. Neal, H. A. Wakelee, R. E. Merritt, J. B. Shrager, B. W. Loo Jr, A. A. Alizadeh and M. Diehn, Nat. Med., 2014, 20, 548–554 CrossRef CAS PubMed.
- M. Baker, Nat. Methods, 2012, 9, 541–544 CrossRef CAS.
- B. O'Leary, S. Hrebien, J. P. Morden, M. Beaney, C. Fribbens, X. Huang, Y. Liu, C. H. Bartlett, M. Koehler, M. Cristofanilli, I. Garcia-Murillas, J. M. Bliss and N. C. Turner, Nat. Commun., 2018, 9, 896 CrossRef PubMed.
- C. Cai, Z. Guo, Y. Cao, W. Zhang and Y. Chen, Nanotheranostics, 2018, 2, 12–20 CrossRef PubMed.
- H. F. Wang, R. N. Ma, F. Sun, L. P. Jia, W. Zhang, L. Shang, Q. W. Xue, W. L. Jia and H. S. Wang, Biosens. Bioelectron., 2018, 122, 224–230 CrossRef CAS PubMed.
- D. Li, Y. Xu, L. Fan, B. Shen, X. Ding, R. Yuan, X. Li and W. Chen, Biosens. Bioelectron., 2020, 148, 111826 CrossRef CAS PubMed.
- Y. Huang, M. Tao, S. Luo, Y. Zhang, B. Situ, X. Ye, P. Chen, X. Jiang, Q. Wang and L. Zheng, Anal. Chim. Acta, 2020, 1107, 40–47 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01044a |
| This journal is © The Royal Society of Chemistry 2021 |