
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic enhancement of the emergency treatment effect of organophosphate poisoning by a supramolecular strategy†
Junyi
Chen‡
ab,
Yadan
Zhang‡
a,
Yao
Chai
a,
Zhao
Meng
a,
Yahan
Zhang
a,
Longming
Chen
a,
Dongqin
Quan
 *a,
Yongan
Wang
*a,
Qingbin
Meng
*a and
Chunju
Li
*bc
*a,
Yongan
Wang
*a,
Qingbin
Meng
*a and
Chunju
Li
*bc
aState Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, P. R. China. E-mail: nankaimqb@sina.com; qdqwzb@163.com; yonganw@126.com
bCollege of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, P. R. China
cKey Laboratory of Inorganic-Organic Hybrid Functional Material Chemistry, Ministry of Education, Tianjin Key Laboratory of Structure and Performance for Functional Molecules, College of Chemistry, Tianjin Normal University, Tianjin 300387, P. R. China. E-mail: cjli@shu.edu.cn
First published on 1st March 2021
Abstract
Poisoning by organophosphorus agents (OPs) is a serious public health issue across the world. These compounds irreversibly inhibit acetylcholinesterase (AChE), resulting in the accumulation of acetylcholine (ACh) and overstimulation of ACh receptors. A supramolecular detoxification system (SDS) has been designed with a view to deliver pyridine-2-aldoxime methochloride (PAM) with a synergistic inhibition effect on the ACh-induced hyperstimulation through host–guest encapsulation. NMR and fluorescence titration served to confirm the complexation between carboxylatopillar[6]arene (CP6A) and PAM as well as ACh with robust affinities. Patch-clamp studies proved that CP6A could exert an inhibition effect on the ACh-induced hyperstimulation of ACh receptors. Support for the feasibility of this strategy came from fluorescence imaging results. In vivo studies revealed that complexation by CP6A serves to increase the AChE reactivation efficiency of PAM. The formation of the PAM/CP6A complex contributed to enhance in a statistically significant way the ability of PAM not only to relieve symptoms of seizures but also to improve the survival ratio in paraoxon-poisoned model rats. These favorable findings are attributed to synergistic effects that PAM reactivates AChE to hydrolyze ACh and excess ACh is encapsulated in the cavity of CP6A to relieve cholinergic crisis symptoms.
Introduction
Organophosphorus agents (OPs) are extremely poisonous chemicals.1,2 Acute OPs poisoning represents a persistent world health threat to general populations under pesticide exposure or terrorist attacks.3–7 Countless deaths have been caused by a system-wide cholinergic crisis within hours of OPs ingestion before receiving medical care.8,9 OPs can bind to and phosphorylate various enzymes around the body but the clinical relevance of most of these interactions is ambiguous. Instead, irreversibly inhibiting synaptic acetylcholinesterase (AChE) via the formation of aged phosphyl–AChE conjugates appears to be pivotal for toxicity.10,11 AChE, a serine hydrolase, regulates the breakdown of the neurotransmitter acetylcholine (ACh) at neuronal synapses and neuromuscular junctions.12,13 The aging of AChE results in an accumulation of ACh, inducing hyperstimulation of the cholinergic system and then causing paralysis, seizures, respiratory failure or even immediate death.14–17 In clinic practice, reactivators have been used to reactivate AChE of victims suffering from OPs poisoning.18,19 Pyridinium aldoxime derivatives are widely used reactivators, but oxime alone is not efficient against OPs due to the lack of a direct effect on ACh-induced hyperstimulation.20,21 Thus, anticholinergic and anticonvulsant drugs are used in combination for the emergency treatment protocol.22–25This research is aimed at developing a new supramolecular strategy to cooperatively enhance the emergency treatment effect of OPs poisoning by host–guest encapsulation. In principle, by introducing a water-soluble macrocyclic container, clinical reactivators can be encapsulated based on reversible noncovalent interactions. Metabolically disordered ACh induced by OPs can competitively bind with the macrocycle to inhibit hyperstimulation of the cholinergic system; meanwhile, ACh-triggered release of reactivators occurs to recover the hydrolysis function of AChE.
As a proof of concept, pyridine-2-aldoxime methochloride (PAM), the reference oxime generally counteracting paraoxon (POX) poisoning served as the model reactivator.26 A popular macrocycle, carboxylated pillar[6]arene (CP6A), was chosen as the container due to its high water solubility, good biocompatibility, and size/charge matching with PAM and ACh.27–30 In this supramolecular detoxification system (SDS), intranasal administration allows direct delivery of the complex of PAM and CP6A from nasal mucosa to the central nervous system (CNS).31,32 In the CNS, PAM was competitively replaced from CP6A, accompanied by the engulfing of ACh molecules in the host cavities (Fig. 1). Particularly, this scheme was found to synergistically alleviate poisoning symptoms and increase survival rates in model animals. This work thus serves to highlight the inherent promise of the SDS as a candidate scheme for emergency treatment of OPs poisoning.
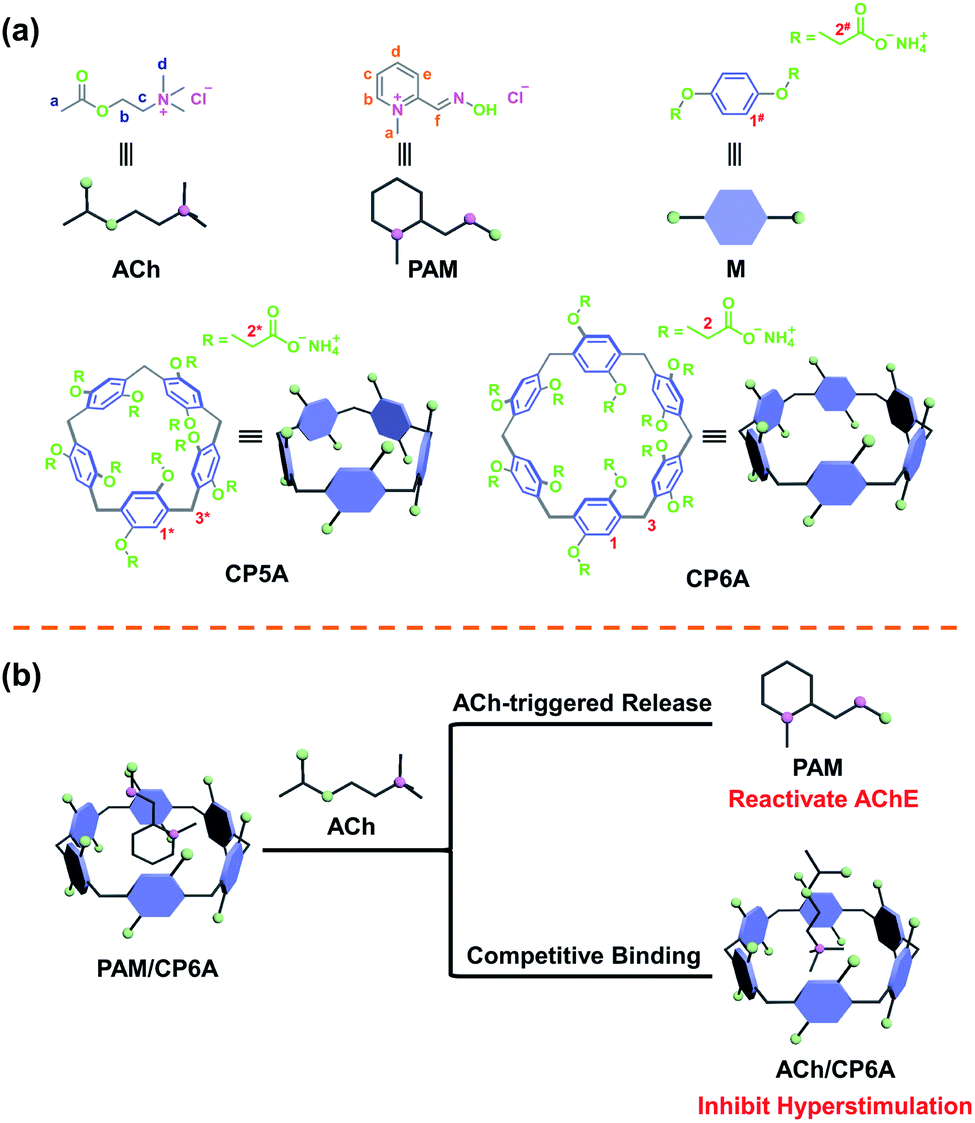 | ||
| Fig. 1 Schematic presentation of compounds and the supramolecular detoxification system (SDS) used in this study. (a) Chemical structure of ACh, PAM, macrocycle monomer (M), carboxylated pillar[5]arene (CP5A) and CP6A. (b) SDS using PAM and CP6A to form the host–guest complex and its synergistic therapeutic mechanism. | ||
Results and discussion
Host–guest complexation studies
The design predicate underlying our proposed SDS is that CP6A could bind PAM and release it in the presence of ACh. Therefore, the host–guest complexation behaviors between the host (CP6A) and guests (ACh and PAM) were first verified by 1H NMR experiments. Fig. 2a–c shows the 1H NMR spectra of PAM in deuterated phosphate buffer (pD = 7.4) in the absence and presence of one equivalent of CP6A. Upon the addition of CP6A, all the protons of guest PAM exhibited remarkable upfield shifts and broadening effects compared with free PAM. Meanwhile, the peaks for CP6A shifted downfield (Δδ = 0.09 and 0.13 ppm for H1 and H2) due to the complexation-induced deshielding effect. In addition, the host shows similar binding behaviors toward ACh (Fig. S11†). The proton signals of ACh underwent a substantial upfield shift (Δδ = −0.12, −1.34, −1.57 and −1.30 ppm for Ha, Hb, Hc and Hd) and experienced considerable broadening when mixed with CP6A, presumably as a consequence of the inclusion-induced shielding effect.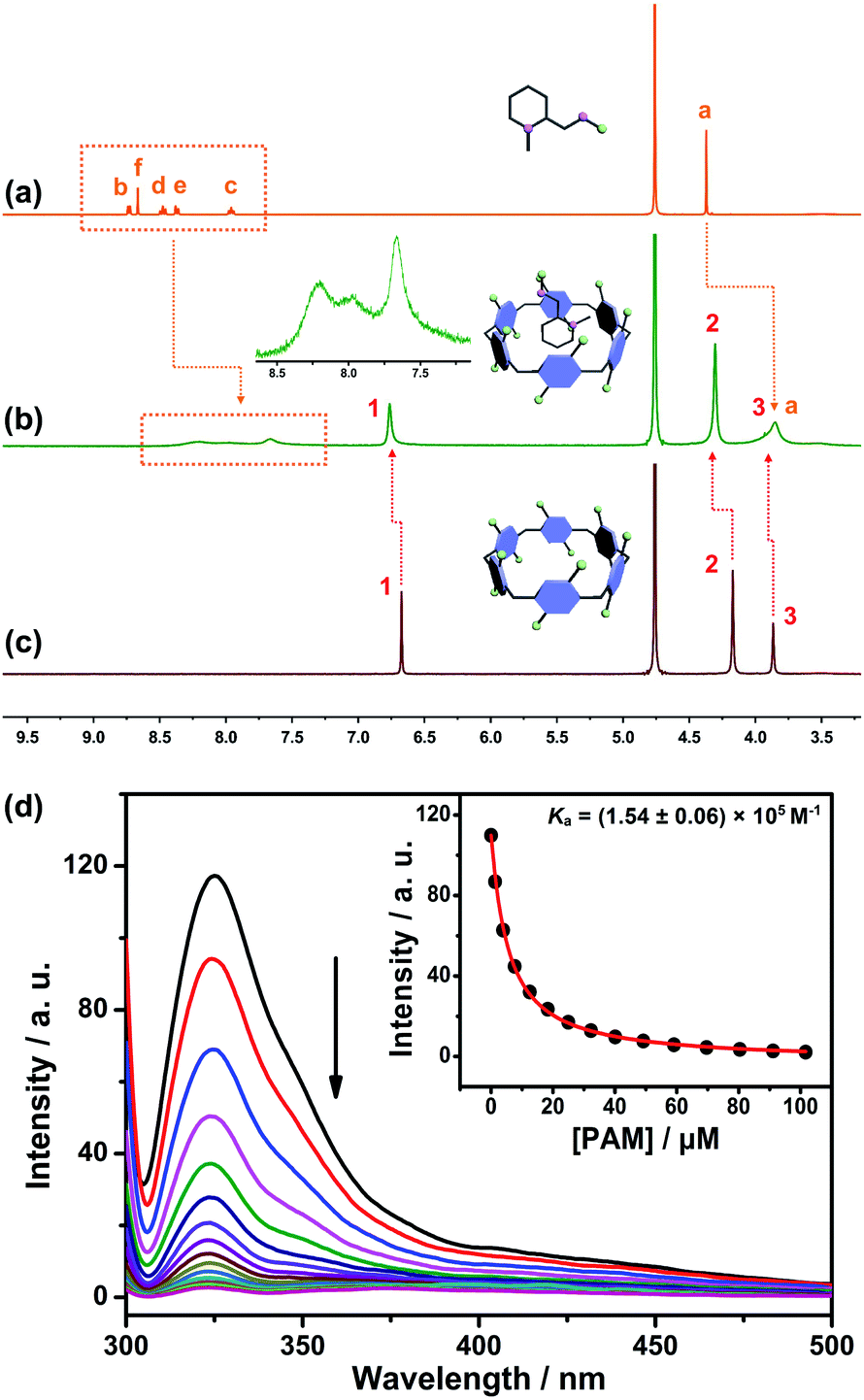 | ||
Fig. 2
1H NMR spectrum (400 MHz, D2O) of (a) PAM (5 mM), (b) PAM (5 mM) + CP6A (5 mM) and (c) CP6A (5 mM). (Inset) Partially enlarged 1H NMR spectrum of the host–guest complex. (d) Fluorescence titration of CP6A (1.00 μM) with PAM in 10 mM PBS buffer at pH 7.4, λex = 290 nm. (Inset) The associated titration curve at λem = 330 and fit according to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry. Data were from n = 3 independent experiments and are presented as mean ± SD. :1 binding stoichiometry. Data were from n = 3 independent experiments and are presented as mean ± SD. | ||
Quantitative data for the host–guest complexation were obtained from fluorescence titration experiments. The results of the continuous variation method (Job's plot) proved consistent with a 1:1 binding stoichiometry between the hosts and guests. Upon the addition of PAM, the fluorescence intensity of CP6A gradually decreased due to photo-induced electron transfer.33 The association constant (Ka) was determined to be (1.54 ± 0.06) × 105 M−1 by a standard curve fitting protocols (Fig. 2d). Although this value was slightly higher than that of ACh/CP6A [(7.03 ± 0.10) × 104 M−1, Fig. S18†], accumulative ACh induced by OP poisoning was expected to replace the PAM from the host–guest complex.
For comparison, the interactions of ACh and PAM with carboxylated pillar[5]arene (CP5A) were studied. Upon the addition of CP5A, the peaks for Hb, Hc, Hd and He of PAM displayed upfield shifts and broadening effects, while the signal of protons Ha and Hf exhibited small shifts, suggesting that PAM was partially engulfed by the cavity of CP5A (Fig. S9†). As for ACh, slight NMR spectral changes were observed in the presence of the host (Fig. S10†). The Ka value of PAM/CP5A was determined to be (3.59 ± 0.11) × 104 M−1 (Fig. S13†), while that for ACh was only 28.0 ± 5.8 M−1 (Fig. S15†). The differences of Ka values are reasonable considering the internal cavity diameters of pillar[5]arene (about 4.7 Å) and pillar[6]arene (about 6.7 Å).27 The cavity size of CP6A was capacious enough to encapsulate both PAM and ACh to form stable host–guest complexes, while CP5A cannot efficiently bind ACh because its cavity is too small in comparison with the guest's spherical trimethylammonium cation. Controlled 1H NMR experiments of mixtures of the macrocycle monomer (M, six equivalent) and two guests (PAM and ACh) were also conducted (Fig. S7 and S8†), showing that PAM and ACh could not interact with M.
Inhibition of ACh-induced nAChR responses by CP6A
The design of this synergistic detoxification strategy depends on the following two expectations: (i) quick release of PAM can be achieved owning to ACh-triggered competitive binding and (ii) CP6A could inhibit ACh-induced hyperstimulation of the cholinergic system via host–guest inclusion. To determine whether CP6A could relieve stimulation of the ACh receptors via encapsulating excess ACh, HEK293 cells expressing the nicotinic ACh receptor (nAChR) α7 subunit were chosen to perform patch-clamp experiments. The cells were clamped at a holding potential of −70 mV, and inward currents invoked by 30 μM of ACh were compared with those observed on co-application of the ACh with the test compounds. Control experiments showed that M, CP5A or CP6A alone generated no stimulus effect towards nAChR (Fig. S21†). The peak current amplitude activated by 30 μM ACh was −7893.5 pA and co-application of M or CP5A could not antagonize the ACh-activated current without changing the shape and amplitude of the signal (Fig. 3a). It is reasonable that M does not interact with ACh and the Ka value of ACh/CP5A is quite low. While adding CP6A to ACh-containing test solution, a reduction of amplitude of ∼30% occurred, suggesting that CP6A can exert an inhibition effect on the ACh-induced hyperstimulation of nAChR owning to their strong host–guest interactions.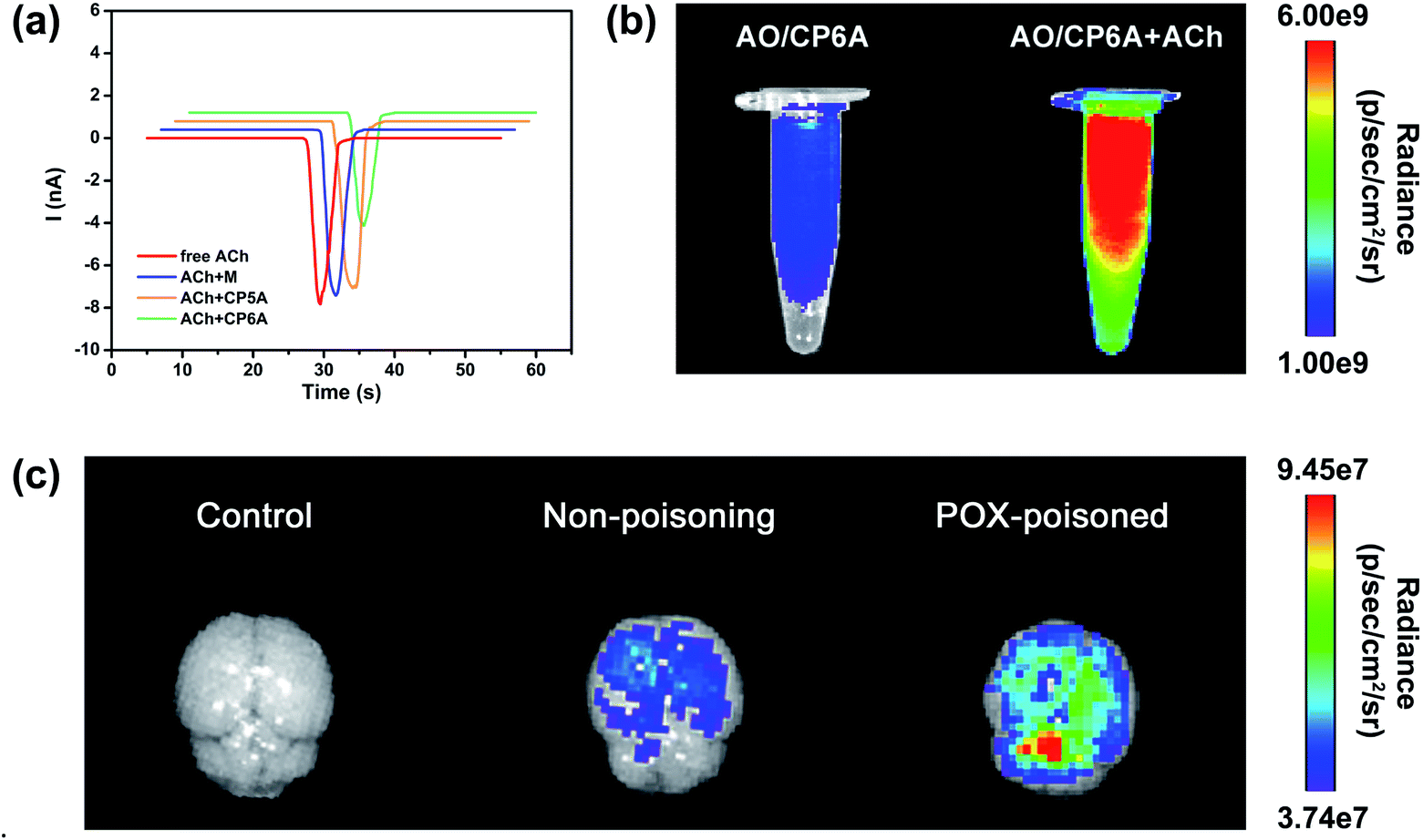 | ||
| Fig. 3 Confirmation of the feasibility of the SDS. (a) Currents in response to ACh (30 μM), ACh + M (30 μM + 180 μM), ACh + CP5A (30 μM + 30 μM) and ACh + CP6A (30 μM + 30 μM) respectively. Currents were recorded with whole-cell patch-clamp electrophysiology from a HEK-293 cell expressing nAChRs (α7) and voltage-clamped to −70 mV. (b) Fluorescence images of AO/CP6A (10 μM/50 μM) and AO/CP6A (10 μM/50 μM) +ACh (1 mM) in aqueous PBS at pH 7.4. (c) Ex vivo imaging of the brain harvested from SD rats at 0.5 h after intracerebroventricular injection of AO/CP6A (10 μM/50 μM). | ||
Feasibility of competitive release
The ACh-triggered release was then examined via an indicator displacement assay. Acridine orange (AO) was screened as the optimal reporter dye since the Ka for AO/CP6A (Fig. S20†) was comparable to that of PAM/CP6A at the same order of magnitude. In this regard, we examined the fluorescence intensity of AO/CP6A in aqueous phosphate buffered saline (PBS) at pH 7.4. Complexation-induced fluorescence quenching of chromophore could be observed, which validated the robust complexation stability of AO/CP6A. While excess ACh was added to AO/CP6A for simulation of OPs poisoning, pronounced regeneration of intrinsic emission of AO was observed (Fig. 3b). Fluorescence spectroscopy was also performed to further investigate the release behavior (Fig. S22†). A weak fluorescence signal was seen for AO/CP6A in PBS over the spectral range corresponding to the AO-based emission. Upon adding ACh, the fluorescence intensity of AO was significantly enhanced, which was taken as evidence that AO/CP6A underwent ACh-triggered competitive binding with the concomitant release of AO.In vivo, a rat model for OPs toxicity using POX was set up to test whether ACh-triggered release of the reporter dye could occur in brain tissues. Accordingly, after 1 h of poisoning with 0.8 × LD50 of POX (600 μg kg−1, i.p.), AO/CP6A was directly injected into the brain, whereas the same amount of the complex was administrated in non-poisoned rat as a comparison. As displayed in Fig. 3c, no detectable fluorescence signal could be observed in the control group and a weak AO fluorescence was seen at 0.5 h postinjection in the non-poisoning group. In sharp contrast, within the brain, a significant enhancement of AO fluorescence clearly occurred in the POX-poisoned group (Fig. 3c and S23†). Accumulative ACh ascribed to POX poisoning tended toward the greater number of cargo release via competitive replacement. Taken together, these findings provided support for this competitive complexation process that released cargo in an ACh-dependent manner.
Safety profile of CP6A
Prior to pharmacodynamics analysis, the cytotoxicity of CP6A in mouse brain microvascular endothelial cells (bEnd.3) was assessed using a Cell Counting Kit-8 (CCK-8) assay. CP6A displayed minimal cytotoxicity against bEnd.3 cells even at relatively high concentrations (Fig. S24†), which agreed well with reported work.34,35 Further support for the low toxicity inferred for CP6A came from histological analyses of brain slices, including the cerebral cortex and hippocampal region. Compared with the control group, no detectable lesions or histopathological abnormalities could be noted (Fig. S25†). The above cytotoxicity and histopathology results led us to consider that CP6A could be applied safely to construct the SDS.Content of PAM in cerebrospinal fluid
Based on the above results, the in vivo emergency treatment efficiency of the SDS was expected to be enhanced as the result of ACh-triggered release of PAM and competitive binding between CP6A and Ach (Fig. 4a). To test this hypothesis, the content of PAM in cerebrospinal fluid was first tested using high-performance liquid chromatography (HPLC). SD rats were intranasally administrated with PAM and a mixture of PAM/M, PAM/CP5A and PAM/CP6A, respectively, with an equivalent PAM dose of 10 mg kg−1. At predetermined intervals of 10 min and 30 min after administration, cerebrospinal fluid was collected. First, an appropriate calibration curve was derived to calculate the PAM concentration (Fig. S26†). Analysis of the results revealed that no appreciable changes of PAM content were seen for the M + PAM group, while co-application with CP5A and CP6A led to increased PAM concentration within 30 min (Fig. 4b). For example, at a time interval of 10 min, the PAM concentration in the cerebrospinal fluid of animals co-dosed with CP5A (404.6 ± 128.0 ng mL−1) proved to be 96.6% higher than in its absence (205.8 ± 35.7 ng mL−1). Especially, it was notable that at this dose level, administration of PAM/CP6A was much more efficacious in terms of the PAM content than PAM alone. The drug concentration of the PAM/CP6A group (521.0 ± 89.8 ng mL−1) was about 2.5-fold larger than that of the corresponding PAM-free group.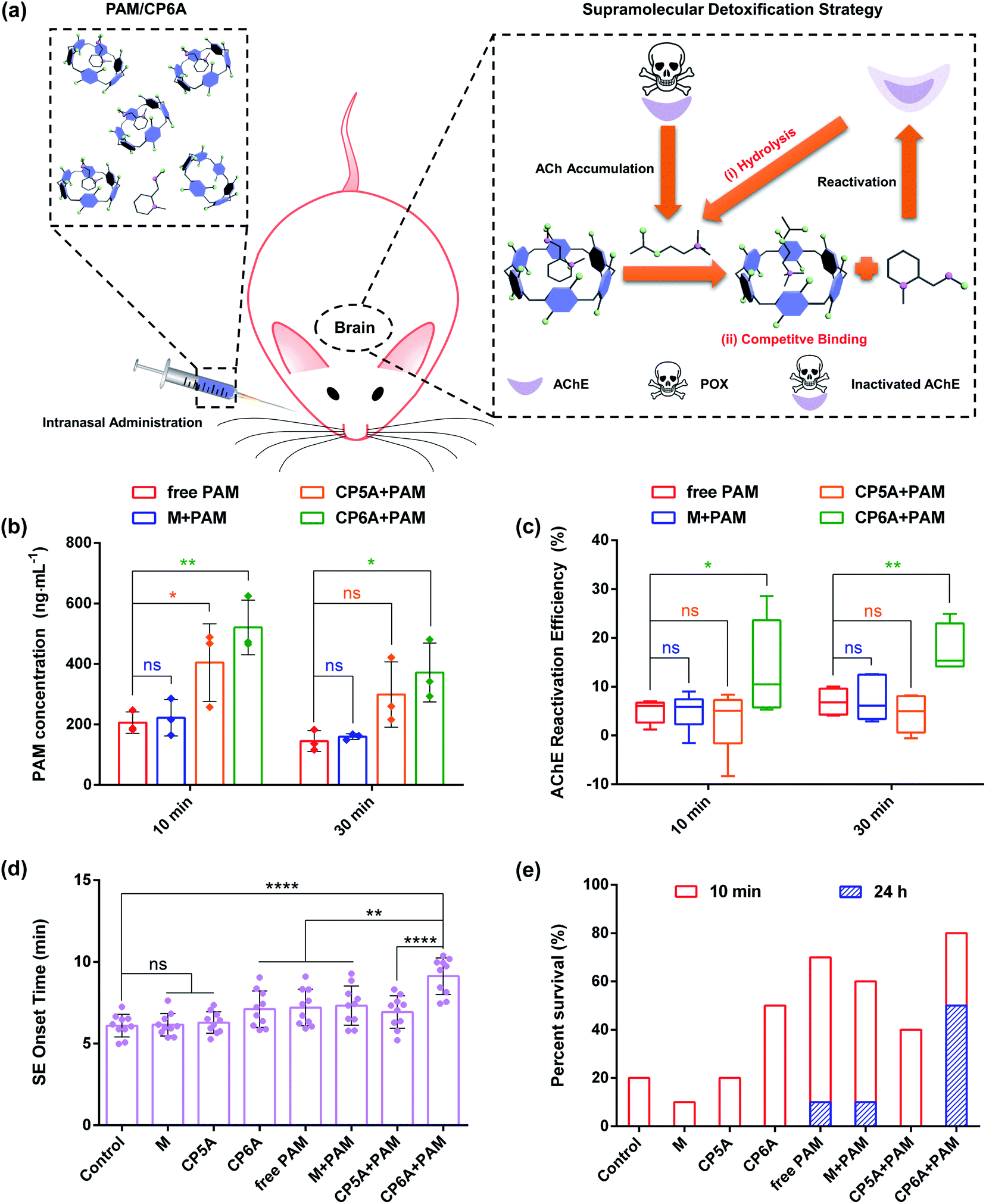 | ||
| Fig. 4 In vivo detoxification experiments. (a) Schematic illustration of the supramolecular detoxification strategy. Note: (i) ACh-triggered release of PAM to reactivate AChE and (ii) engulfing of ACh by CP6A to inhibit hyperstimulation toward AChE receptors. (b) Concentration of PAM in rat cerebrospinal fluid after intranasal administration of free PAM, M + PAM, CP5A + PAM and CP6A + PAM (mean ± SD, n = 3). The dose of PAM is 10 mg kg−1. (c) Determination of brain AChE reactivation efficiency in vivo. Mean AChE activity of brain homogenates was measured in a control group of rats (taken as 100%), after poisoning with 0.8 × LD50 of POX (600 μg kg−1, i.p.) and after intranasal administration of M + PAM, CP5A + PAM and CP6A + PAM (mean ± SD, n = 6). (d) Efficiency of several formulations against 2 × LD50 of POX-induced SE. The graph shows the SE onset time of rats after intranasal administration of PBS, M, CP5A, CP6A, free PAM, M + PAM, CP5A + PAM and CP6A + PAM, respectively (mean ± SD, n = 10). (e) Survival rate of rats poisoned with 2 × LD50 of POX (1500 μg kg−1, i.p.) and after treatment with PBS, M, CP5A, CP6A, free PAM, M + PAM, CP5A + PAM and CP6A + PAM (n = 10 rats per group). Note: after POX poisoning for 24 h, no rat has survived in the control group, M group, CP5A group, CP6A group and CP5A + PAM group. Significant differences were assessed using one-way ANOVA tests with multiple comparisons. ns, not significant. *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
Reactivation of AChE in the brain
As noted in the introduction, PAM, a powerful reactivator, served as a reference, generally reactivating POX-induced phosphoryl AChE. Thus, the abilities of several formulations to reactivate AChE in the brain were measured using a standard method of assessing the efficacy of antidotes. Analysis of AChE viability by HPLC revealed that almost no changes in the AChE content were observed for rats treated with host molecules alone (Fig. S28†). In addition, after 1 h of poisoning with 0.8 × LD50 of POX (600 μg kg−1, i.p.), the corresponding doses of M, CP5A and CP6A were intranasally administrated. This sublethal dose of POX inhibited 83.1 ± 3.1% AChE activity in the brain for a total of 70 min and a similar level of AChE inhibition was observed after 90 min. Treatment with M, CP5A and CP6A led to no effect on reactivating AChE at both time points (Fig. S29†). Then AChE reactivation efficiency was assessed for the various formulations (Fig. 4c). The reactivation ratio of brain AChE was only 5% in the case of PAM alone over the course of 10 min. Compared to the PAM group, co-application with M showed no remarkable influence on the AChE reactivation efficiency, consistent with no change in the PAM content. The CP5A + PAM group resulted in a 3% reactivation efficiency. This result was quite reasonable because the Ka value of PAM/CP5A was three orders of magnitude higher than that of ACh/CP5A and PAM could not release effectively from its host–guest complex via competitive replacement with ACh. It was notable that a large and statistically significant increase in AChE reactivation efficiency was seen for the PAM/CP6A group. A similar tendency was also found at 30 min after treatment.In vivo pharmacodynamics analysis
POX affects cholinergic transmission by inhibiting AChE. This inhibition causes accumulation of ACh in the synaptic cleft, resulting in a cholinergic crisis and ultimately intense status epilepticus (SE) which produce brain damage and death. Therefore, the effect on emergency treatment of OPs poisoning was assessed using this SDS. In the present study, animals that received 2 × LD50 of POX (1500 μg kg−1, i.p.) were monitored behaviourally within 24 h. Without antidotal treatment of the severe cholinergic crisis, SE and death ensued rapidly in all animals for PBS groups (negative control), demonstrating the lethal effect of this dose of exposure. Intranasal administration with M and CP5A was invalid in the incidence of SE and mortality. Compared to the PBS control group, CP6A resulted in delaying the onset of SE and increasing survival rate within 10 min (Fig. 4d and e and Table S1†), leading us to suggest that CP6A could inhibit the overstimulation of ACh receptors in vivo via forming a robust host–guest complex between ACh and CP6A. Furthermore, treatment with M + PAM was found to retain the pharmacological action of PAM and the therapeutic effect of CP5A + PAM proved to be lower than that of the PAM group. In contrast, a significant increase of the survival rate accompanied by postponing SE time was observed for the rats treated with CP6A + PAM (Fig. 4d and e and Table S1†). We also counted the seizure score of rats at 7 min after administration. The seizure responses were scored on a five point Racine scale, which could be found in the ESI† for details.36 In the group of rats treated with PBS, all the rats achieved a seizure score of 3 or above, while various degrees of symptom relief was seen for the other groups (Fig. S30†). It was noted that most of the CP6A + PAM treated rats reached less than Racine level 3 seizure stage. These results were taken as further evidence that CP6A + PAM was effective at emergency treatment of acute POX poisoning.In addition to evaluate the therapeutic effect with different treatments, hematoxylin and eosin (H&E) assay was performed to assess the emergency treatment effect of various groups (Fig. S31†). Morphological assessment of brain sections of POX group revealed the formation of edemas and shrinkage of neurons in the cerebral cortex and hippocampal region, features that were characteristic of severe brain damage. As compared to the POX group, images from PAM, M + PAM and CP5A + PAM also exhibited similar edemas and apoptosis. In contrast, treatment with CP6A + PAM showed much lower brain damage as inferred from the corresponding histological analyses. On this basis, we propose that the SDS embodied in PAM/CP6A can be used to increase reactivation efficacy of AChE comparing with PAM alone and relieve the hyperstimulation of the cholinergic system via host–guest encapsulation. To the extent this favorable augury translates into practice, it is expected to provide a significant and salutary benefit for the emergency treatment of OPs poisoning.
Conclusions
In summary, we have put forward an available SDS that involves the direct encapsulation of PAM, a reference oxime generally counteracting POX, by an appropriately sized receptor, CP6A. Accumulative ACh caused by POX poisoning competitively bound with CP6A to replace PAM. The partial formation of an inclusion complex decreased the current amplitude activated by ACh at the cellular level. Ex vivo imaging of the brain provided support for the expectation that ACh-triggered release of cargo occurred in the POX-poisoned rat model. In vivo, a statistically significant improvement of AChE reactivation efficiency was found upon co-application with CP6A. Intranasal administration of PAM/CP6A could efficiently relieve POX-induced seizures, decrease brain damage and enhance the survival rate compared to various controls. The enhancement of therapeutic outcomes is consistent with two key predicative suggestions underlying this study, namely that competitive engulfing ACh in CP6A cavity leads to an inhibition on hyperstimulation of ACh receptors and that rapid drug release is triggered by disordered ACh that characterizes POX poisoning. This work thus serves to highlight what may emerge as a feasible strategy for improving the emergency treatment effect of OPs poisoning. To the extent this proves true, it could win more time to carry out rescue and ultimately save more lives of victims.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the AAALAC and were approved by the Animal Care and Use Committee of the National Beijing Center for Drug Safety Evaluation and Research. The authors gratefully acknowledge the National Natural Science Foundation of China (21772118, 21971192, 81573354), the Shanghai Pujiang Program (16PJD024), the Shanghai Shuguang Program and the Military Medical Scientific and Technological Project (AWS17J007).Notes and references
- A. Verweij, H. L. Boter and C. E. A. M. Degenhardt, Science, 1979, 204, 616–618 CrossRef CAS PubMed.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- M. Eddleston, P. Eyr, F. Worek, F. Mohamed, L. Senarathna, L. V. Meyer, E. Juszczak, A. Hittarage, S. Azhar, W. Dissanayake, W. M. H. R. Sheriff, L. Szinicz, A. H. Dawson and N. A. Buckley, Lancet, 2005, 366, 1452–1459 CrossRef CAS.
- M. Eddleston, F. Mohamed, J. O. J. Davies, P. Eyer, F. Worek, M. H. R. Sheriff and N. A. Buckley, Q. J. Med., 2006, 99, 513–522 CrossRef CAS PubMed.
- D. A. Jett, Ann. Neurol., 2007, 61, 9–13 CrossRef CAS PubMed.
- T. Okumura, T. Hisaoka, T. Natio, H. Isonuma, S. Okumura, K. Miura, H. Maekawa, S. Ishimatsu, N. Takasu and K. Suzuki, Environ. Toxicol. Pharmacol., 2005, 19, 447–450 CrossRef CAS PubMed.
- E. Dolgin, Nat. Med., 2013, 19, 1194–1195 CrossRef PubMed.
- A. Karunarathne, D. Gunnell, F. Konradsen and M. Eddleston, Clin. Toxicol., 2019, 58, 227–232 CrossRef PubMed.
- D. Gunnell, M. Eddleston, M. R. Phillips and F. Konradsen, BMC Public Health, 2007, 7, 357–371 CrossRef PubMed.
- M. B. Čolović, D. Z. Kristić, T. D. Lazarević-Pašti, A. M. Bondžić and V. M. Vasić, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef PubMed.
- A. R. Main, Pharmacol. Ther., 1979, 6, 579–628 CrossRef CAS.
- S. Ehrenpreis, Nature, 1964, 4922, 887–893 CrossRef PubMed.
- D. M. Quinn, Chem. Rev., 1987, 87, 955–979 CrossRef CAS.
- M. D. Nimal Senanayake and M. B. B. S. Lakshman Karalliedde, N. Engl. J. Med., 1987, 316, 761–763 CrossRef PubMed.
- C. B. Millard, G. Koellner, A. Ordentlich, A. Shafferman, I. Silman and J. L. Sussman, J. Am. Chem. Soc., 1999, 121, 9883–9884 CrossRef CAS.
- B. Sanson, F. Nachon, J. P. Colletier, M. T. Froment, L. Toker, H. M. Greenblatt, J. L. Sussman, Y. Ashani, P. Masson, I. Silman and M. Weik, J. Med. Chem., 2009, 52, 7593–7603 CrossRef CAS PubMed.
- E. Carletti, J. P. Colletier, F. Dupeux, M. Trovaslet, P. Masson and F. Nachon, J. Med. Chem., 2010, 53, 4002–4008 CrossRef CAS PubMed.
- I. B. Wilson, J. Biol. Chem., 1951, 190, 111–117 CrossRef CAS.
- I. B. Wilson and E. K. Meislich, J. Am. Chem. Soc., 1953, 75, 4628–4629 CrossRef CAS.
- G. Mercey, T. Verdelet, J. Renou, M. Kliachyna, R. Baati, F. Nachon, L. Jean and P. Y. Renard, Acc. Chem. Res., 2012, 45, 756–766 CrossRef CAS PubMed.
- M. Eddleston and F. R. Chowdhury, Br. J. Clin. Pharmacol., 2015, 81, 462–470 CrossRef PubMed.
- M. Eddleston, A. Dawson, L. Karalliedde, W. Dissanayake, A. Hittarage, S. Azher and N. A. Buckley, Crit. Care, 2004, 8, 391–397 CrossRef PubMed.
- N. J. Connors, Z. H. Harnett and R. S. Hoffman, J. Med. Toxicol., 2014, 10, 143–147 CrossRef CAS PubMed.
- T. C. Marrs, Toxicol. Rev., 2003, 22, 75–81 CrossRef CAS PubMed.
- M. R. Murphy, D. W. Blick and M. A. Dunn, Aviat., Space Environ. Med., 1993, 64, 110–115 CAS.
- I. B. Wilson and B. Ginsburg, Biochim. Biophys. Acta, 1955, 18, 168–170 CrossRef CAS.
- (a) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed; (b) G. Yu, M. Xue, Z. Zhang, J. Li, C. Han and F. Huang, J. Am. Chem. Soc., 2012, 134, 13248–13251 CrossRef CAS PubMed.
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- H. Zhang, Z. Liu and Y. Zhao, Chem. Soc. Rev., 2018, 47, 5491–5528 RSC.
- M. Ueno, T. Tomita, H. Arakawa, T. Kakuta, T. Yamagishi, J. Terakawa, T. Daikoku, S. Horike, S. Si, K. Kurayoshi, C. Ito, A. Kasahara, Y. Tadokoro, M. Kobayashi, T. Fukuwatari, I. Tamai, A. Hirao and T. Ogoshi, Commun. Chem., 2020, 3, 183 CrossRef CAS.
- M. Kapoor, J. C. Cloyd and R. A. Siegel, J. Controlled Release, 2016, 237, 147–159 CrossRef CAS PubMed.
- A. R. Khan, M. Liu, M. W. Khan and G. Zhai, J. Controlled Release, 2017, 268, 364–389 CrossRef CAS PubMed.
- A. P. Silva and R. A. D. D. Rupasinghe, J. Chem. Soc., Chem. Commun., 1985, 1669–1670 RSC.
- J. Chen, Y. Zhang, Z. Meng, L. Guo, X. Yuan, Y. Zhang, Y. Chai, J. L. Sessler, Q. Meng and C. Li, Chem. Sci., 2020, 11, 6275–6282 RSC.
- H. Zhu, H. Wang, B. Shi, L. Shuangguan, W. Tong, G. Yu, Z. Mao and F. Huang, Nat. Commun., 2019, 10, 2412–2511 CrossRef PubMed.
- R. J. Racine, Electroencephalogr. Clin. Neurophysiol., 1972, 32, 281–294 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, and other materials. See DOI: 10.1039/d1sc00426c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |