
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriarylamine-based porous coordination polymers performing both hydrogen atom transfer and photoredox catalysis for regioselective α-amino C(sp3)–H arylation†
Hanning
Li
,
Yang
Yang
,
Xu
Jing
 *,
Cheng
He
and
Chunying
Duan
*
*,
Cheng
He
and
Chunying
Duan
*
State Key Laboratory of Fine Chemicals, Zhang Dayu College of Chemistry, Dalian University of Technology, 116024, P. R. China. E-mail: xjing@dlut.edu.cn; cyduan@dlut.edu.cn
First published on 18th May 2021
Abstract
Direct functionalization of C(sp3)–H bonds in a predictable, selective and recyclable manner has become a central challenge in modern organic chemistry. Through incorporating different triarylamine-containing ligands into one coordination polymer, we present herein a heterogeneous approach to the combination of hydrogen atom transfer (HAT) and photoredox catalysis for regioselective C–H arylation of benzylamines. The different molecular sizes and coordination modes of the ligands, tricarboxytriphenylamine (H3TCA) and tris(4-(pyridinyl)phenyl)amine (NPy3), in one coordination polymer consolidate the triarylamine (Ar3N) moiety into a special structural intermediate, which enhances the chemical and thermal stability of the polymers and diminishes structural relaxation during the catalytic process. The inherent redox potentials of Ar3N moieties prohibit the in situ formed Ar3N˙+ to earn an electron from C(sp3)–H nucleophiles, but allow the abstraction of a hydrogen atom from C(sp3)–H nucleophiles, enabling the formation of the C(sp3)˙ radical and the cross-coupling reaction to proceed at the most electron-rich sites with excellent regioselectivity. The new heterogeneous photoredox HAT approach skips several interactions between transient species during the typical synergistic SET/HAT cycles, demonstrating a promising redox-economical and reagent-economical heterogeneous platform that has not been reported for α-amino C–H arylation to form benzylamine derivatives. Control experiments based on monoligand coordination polymers suggested that the mixed-ligand approach improved the photochemical and photophysical properties, providing important insight into rational design and optimization of recyclable photocatalysts for rapid access to complex bioactive molecules and late-stage functionalized pharmaceuticals.
Introduction
The use of C(sp3)–H bonds, which are ubiquitous in organic molecules, as latent nucleophile equivalents for cross-coupling reactions has the potential to substantially streamline synthetic efforts in organic chemistry.1,2 Visible-light photocatalysis has led to a paradigm shift in organic synthesis that encompasses the activation of small molecules,3–7 fostering the discovery of promising tools to activate aliphatic C–H bonds; however, functionalization of C(sp3)–H bonds in a predictable, selective, and efficient manner remains a central challenge.8,9 Hydrogen atom transfer (HAT),10–12 when involved in photocatalysis, can activate organic substrates without the limitation of redox potentials, allowing kinetically controlled functionalization of hydridic C–H bonds in the presence of other weaker, polarity-mismatched ones. However, besides the limits of photocatalysts capable of performing single electron transfer and hydrogen atom transfer (SET/HAT) synergistic catalysis in a homogeneous fashion,13,14 developing efficient heterogeneous photocatalytic systems15,16 to expand the real applications in organic syntheses and the fine chemicals industry remains largely unexplored, partly due to the difficulties in controlling the kinetics of the multistep electron transfer or hydrogen atom transfer catalytic cycles in a heterogeneous fashion.17Porous coordination polymers with ordered photoactive organic ligands and inorganic nodes are well-known for their promising catalytic performance.18–22 The regular distribution of photocatalytic sites in confined environments benefits the fixation and stabilization of the active radicals formed under light-irradiation to overcome the restrictions in a homogeneous system.23–25 Taking advantage of Ar3N-based coordination polymers for photocatalysis with excellent photocatalytic performance and recyclability26–28 and the tertiary amine radical cation (i.e., quinuclidine radical) capable of engendering a selective HAT at the most electron-rich site of C(sp3)–H nucleophiles,29 we envisioned that incorporating triarylamine (Ar3N) moieties into one coordination polymer would combine HAT and photoredox catalysis for regioselective C(sp3)–H functionalization at electron-rich C–H sites.
It is noted that organic dyes with Ar3N have never been used as HAT catalysts in a homogeneous fashion, partly due to the heavy structural relaxation30 from Ar3N˙+ to Ar3NH+ limiting the kinetics of H-atom exchange in solution. To avoid the potential structural relaxation during the HAT process, tertiary amine HAT catalysts, such as quinuclidine,29,31,32 were always constrained in a robust sp3 conformation. Alternatively, the efficiency of the photocatalytic conversions using Ar3N-based chromophores is dependent on the planar conformation of the N-center in Ar3N chromophores essentially. Our approach thus focused on the modification of the N-center conformation in the Ar3N-based coordination polymers to balance the efficiency of photosensitization with that of HAT catalysis. We envisioned that incorporating both tricarboxytriphenylamine (H3TCA)33 and tris(4-(pyridinyl)phenyl)amine (NPy3)34 into one coordination polymer would provide the possibility of optimizing the conformation of Ar3N moieties into a special intermediate structure that mixed the characteristics of both N(sp3) and N(sp2), decreasing the potential structural relaxation during the HAT process. Meanwhile, electron-rich C–H bonds adjacent to amine functionalities could serve as useful precursors to form the C-centered radical C(sp3)˙ via a HAT event when interacting with the aforementioned catalytically generated amine radical cation.35,36
The proposed catalytic strategy was evaluated by the activation of an N-benzylic C(sp3)–H bond, as benzylic amines are important pharmaceutical structural motifs that are difficult to access via simple substrates. The mechanistic details are outlined in Fig. 1. Irradiation of polymers containing Ar3N moieties produces a strongly reductive Ar3N* (Ered = −2.04 V, vs. Ag/AgCl) that initially undergoes SET with an electron-deficient radical precursor, 1,4-dicyanobenzene (1,4-DCB, Ered1/2 = −1.61 V, vs. SCE),37 to afford the corresponding anionic radical (sub1˙−) and the oxidized photosensitizer Ar3N˙+. The latter selectively abstracts a H atom from the substrate that contains a C(sp3)–H bond with a lower C–H bond dissociation energy (BDE) to form C(sp3)˙, as the inherent moderate oxidization potential of Ar3N˙+ prohibits the direct oxidation of substrates via SET. The direct coupling of the two generated organic radicals predominates the desired product at the most electron-rich site of the C–H nucleophile and completes the SET/HAT catalytic cycles. This redox-economical heterogeneous platform skipped several interactions between transient species during the reactions, allowing rapid access to complex bioactive molecules and late-stage functionalized pharmaceuticals in a recyclable manner.
 | ||
| Fig. 1 (a) The mixed-ligand strategy for the construction of coordination polymers showing the structures of the two ligands mentioned and the measured oxidative potentials (vs. Ag/AgCl) (in parentheses) of the coordination polymers. (b) Schematic illustration of the heterogeneous SET/HAT photocatalytic approach in which the in situ formed oxidized Ar3N˙+ in mixed-ligand Cd-MIX acts as a HAT catalyst for regioselective C(sp3)–H arylation. | ||
Results and discussion
The solvothermal reaction of tricarboxytriphenylamine (H3TCA, 0.05 mmol) and tris(4-(pyridinyl)phenyl)amine (NPy3, 0.05 mmol) with Cd(NO3)2·4H2O (0.25 mmol) gave crystals of Cd-MIX in a yield of approximately 50%. 1H NMR of Cd-MIX in DMSO-d6/DCl gave a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of TCA3− and NPy3 ligands in the structure (Fig. S16†). The similar powder X-ray diffraction (PXRD) patterns based on the single-crystal and as-synthesized Cd-MIX indicated that the bulk Cd-MIX consisted of a pure phase (Fig. S17†). Changing the molar ratio of the two ligands does not affect the structure of Cd-MIX, but affects its purity and yield. Single-crystal analysis revealed that Cd-MIX crystallized in the C2/c space group, and the asymmetric unit of Cd-MIX comprised three independent Cd ions, one NPy3, and two TCA3− ligands. Both Cd1 and Cd3 were six-coordinated by one N atom from NPy3, four O atoms from carboxylates, and one O atom from H2O molecules (Fig. 2a). However, Cd2 was seven-coordinated by one N atom from NPy3, four O atoms from two bidentate carboxylates, and two O atoms from bridged carboxylates. The carboxylates connected Cd1 and Cd2 together to form tetranuclear clusters. The propeller-like TCA3− ligands coordinated to the clusters and Cd3 to generate a 3D coordination polymer with channel openings of 8.6 × 13.0 Å2 (Fig. 2c). The NPy3 ligand coordinated with three different Cd ions and was positioned in a conformation consolidated by the TCA3−-linked Cd nodes (Fig. 2b). Obvious π⋯π stacking interactions between the two ligands were also found to stabilize the mixed-ligand coordination polymer with the interplanar separation reaching 3.62 Å (Fig. S4†).
:1 molar ratio of TCA3− and NPy3 ligands in the structure (Fig. S16†). The similar powder X-ray diffraction (PXRD) patterns based on the single-crystal and as-synthesized Cd-MIX indicated that the bulk Cd-MIX consisted of a pure phase (Fig. S17†). Changing the molar ratio of the two ligands does not affect the structure of Cd-MIX, but affects its purity and yield. Single-crystal analysis revealed that Cd-MIX crystallized in the C2/c space group, and the asymmetric unit of Cd-MIX comprised three independent Cd ions, one NPy3, and two TCA3− ligands. Both Cd1 and Cd3 were six-coordinated by one N atom from NPy3, four O atoms from carboxylates, and one O atom from H2O molecules (Fig. 2a). However, Cd2 was seven-coordinated by one N atom from NPy3, four O atoms from two bidentate carboxylates, and two O atoms from bridged carboxylates. The carboxylates connected Cd1 and Cd2 together to form tetranuclear clusters. The propeller-like TCA3− ligands coordinated to the clusters and Cd3 to generate a 3D coordination polymer with channel openings of 8.6 × 13.0 Å2 (Fig. 2c). The NPy3 ligand coordinated with three different Cd ions and was positioned in a conformation consolidated by the TCA3−-linked Cd nodes (Fig. 2b). Obvious π⋯π stacking interactions between the two ligands were also found to stabilize the mixed-ligand coordination polymer with the interplanar separation reaching 3.62 Å (Fig. S4†).
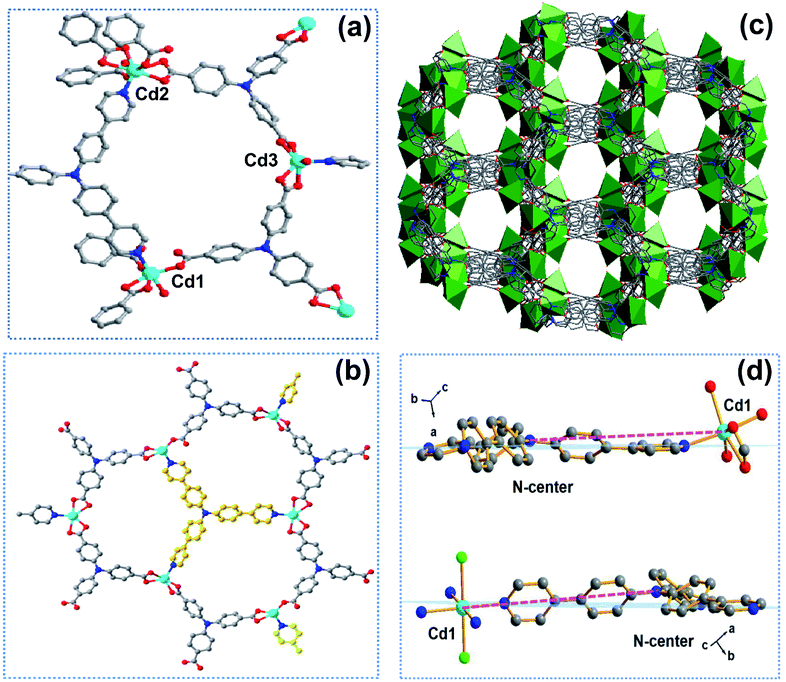 | ||
| Fig. 2 (a) Coordination environments of Cd ions with related TCA3− and NPy3 ligands in Cd-MIX. (b) The structural constraints of NPy3 ligands consolidated by the coordination of TCA3− with Cd ions in Cd-MIX. (c) Structure of Cd-MIX showing the 3D framework with channel openings of 8.6 × 13.0 Å2. (d) The conformations of the NPy3 ligand in Cd-MIX (top) and Cd-NPy3 (bottom). Hydrogen atoms and solvent molecules are omitted for clarity. Cyan, Cd; red, O; blue, N; gray, C; green, Cl. | ||
Detailed structural analysis revealed that the included angles around the central nitrogen atom N2 of the NPy3 ligands in Cd-MIX were 121.3, 123.4, and 115.0°. The structural constraints consolidated the central N2 atom which was positioned 0.36 Å above the plane defined by the other three N atoms (Fig. S10†). The conformation of NPy3 thus was fixed as a special structural intermediate that mixed both N(sp3) and N(sp2) characteristics of the central N atom, which is beneficial for the transformation from Ar3N˙+ to Ar3NH+ to abstract a hydrogen atom at the most electron-rich site of C(sp3)–H nucleophiles. The average distance between the central N2 atom and NPy3 ligand-bridged Cd ions (Cd⋯N2 distance ca. 10.7 Å) in Cd-MIX was approximately 0.2 Å shorter than that of the monoligand coordination polymer Cd-NPy3, which is assembled from only the NPy3 ligand (Fig. 2d), further confirming a distorted conformation in the mixed-ligand coordination polymer.38,39
Moreover, the d10 structure of the Cd centers as well as the variable bridging mode between the two kinds of ligands and the metal nodes brought some flexible elements, allowing the inherent structural relaxation to occur in the solid state in a controllable way during the hydrogen atom transfer process. The free volume in the desolvated coordination polymer Cd-MIX is approximately 43.5%, as determined by PLATON.40 Dye uptake experiments performed by soaking Cd-MIX crystals in a methylene blue solution exhibited 8.0% uptake based on crystal weight. The uniform distribution of methylene blue throughout the crystals suggested that these dye molecules penetrated deeply into these channels (Fig. S22 and S23†), revealing the porosity of Cd-MIX for guest accommodation.41
The solid-state ultraviolet-visible (UV-vis) absorption spectrum of Cd-MIX exhibited broader absorption that spanned the visible light range than those of the monoligand coordination polymers Cd-TCA,42 which is assembled from ligand H3TCA, and Cd-NPy3, respectively (Fig. 3a). Similar to that in the reported mixed-ligand coordination polymers, the stacking π⋯π interactions between ligands and the distorting coordination geometry of the metal centers in the mixed-ligand coordination polymers resulted in an obvious redshift of the absorption band, providing the possibility of photocatalytic conversion under visible light irradiation.43 Solid-state cyclic voltammetry revealed that Cd-MIX exhibited simultaneous two oxidation couples at 0.56 V and 0.76 V (vs. Ag/AgCl), corresponding to the oxidation of ligands TCA3− and NPy3, respectively (Fig. 3b). The inherent moderate oxidization potential of Ar3N in the coordination polymer prohibited the oxidized state of Cd-MIX from earning an electron from the C(sp3)–H nucleophiles, i.e., N-phenylpyrrolidine (1a) (Fig. 3b).44
 | ||
| Fig. 3 (a) Solid-state UV-vis spectra of Cd-MIX, Cd-TCA, and Cd-NPy3. (b) Cyclic voltammetry of Cd-MIX in the solid state and amines examined in solution. Fluorescence decay curves (c) and EPR spectra (d) of Cd-MIX suspension with and without 1a, 1c, and 1,4-dicyanobenzene, respectively. | ||
When excited at 420 nm, the suspension of Cd-MIX in acetonitrile solution exhibited a broad emission band at 508 nm (Fig. S24†). In combination with the free energy change (E0–0)45,46 between the ground and excited states, which was obtained from the absorption and emission spectra of the triarylamine-based coordination polymer Cd-MIX, the excited state potentials were calculated to be −2.04 and −1.84 V (vs. Ag/AgCl) for TCA3− and NPy3 (Fig. S19†), respectively. These potentials demonstrated that the excited state of Cd-MIX was able to reduce the radical precursor 1,4-dicyanobenzene (−1.61 V, vs. SCE)37 to form the anionic radical sub1˙− and cation radical Ar3N˙+.
1,4-Dicyanobenzene is able to quench the emission of the coordination polymer Cd-MIX in an acetonitrile suspension with a Stern–Volmer constant47 of about 1.0 × 103 M−1, demonstrating a SET from the excited state of Cd-MIX to the radical precursor 1,4-dicyanobenzene (Fig. S24†). No obvious emission variation could be detected when C(sp3)–H nucleophiles N-phenylpyrrolidine (1a) and N-benzylpyrrolidine (1c) were added (Fig. S27 and S28†). Time-dependent emission decay experiments estimated a significant decrease of the emission lifetime of Cd-MIX from 1.40 to 1.15 ns when 1,4-dicyanobenzene was added, but no lifetime decrease could be found when N-phenylpyrrolidine (1a) and N-benzylpyrrolidine (1c) were present (Fig. 3c). The IR spectrum of solid-state Cd-MIX immersed in a 1,4-dicyanobenzene solution revealed a CN stretching vibration at 2233 cm−1 (Fig. S29†), suggesting the direct absorption of 1,4-dicyanobenzene within the pores of Cd-MIX. Direct irradiation of the solid-state Cd-MIX adsorbed with 1,4-dicyanobenzene led to a radical signal with a g value of approximately 2.004 only after a minute (Fig. 3d), whereas irradiation of Cd-MIX itself did not cause any obvious radical signals under identical conditions in electron paramagnetic resonance experiments. We infer that the excited state of Cd-MIX cannot oxidize 1a or 1c but is able to reduce the substrate 1,4-dicyanobenzene to generate the aromatic radical for further coupling reactions.
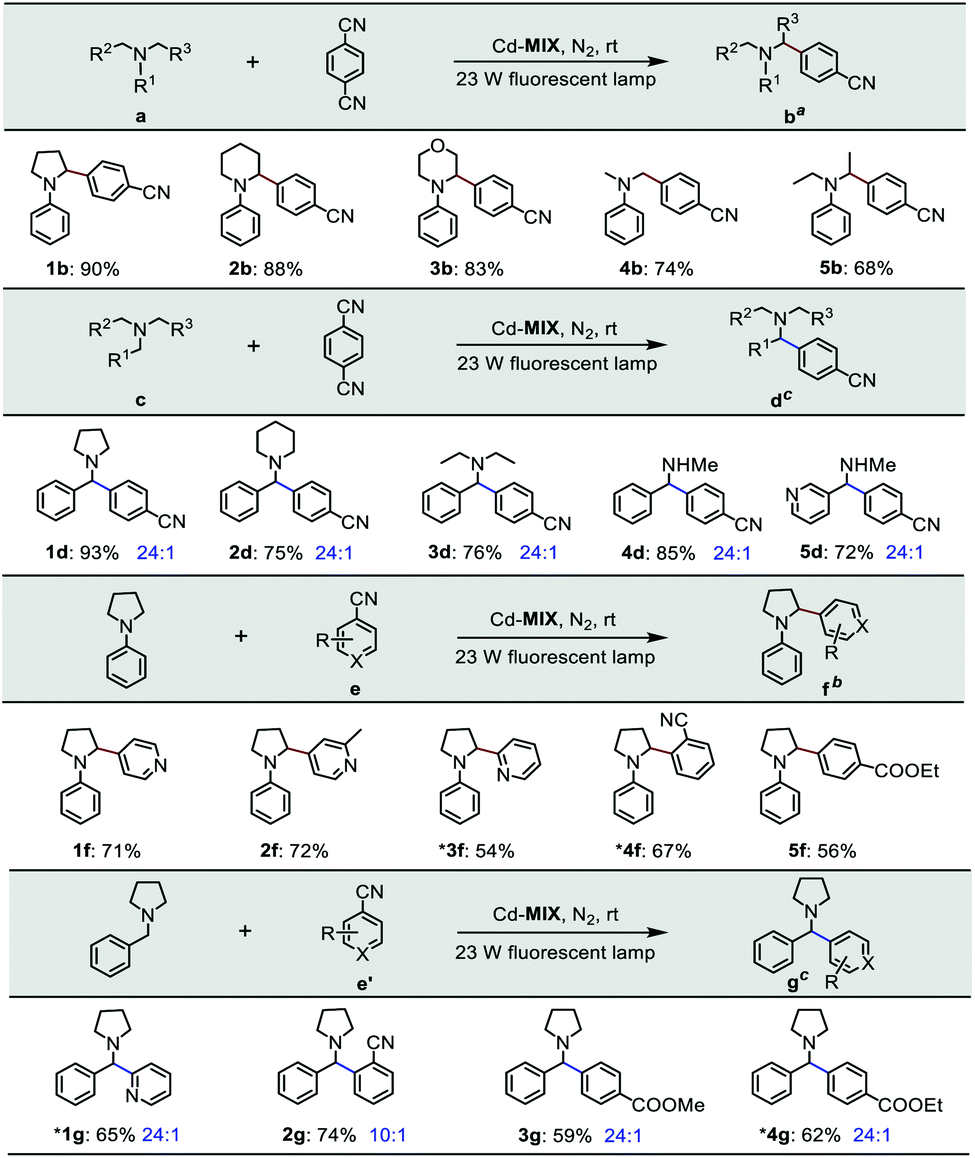
The photocatalytic experiment was initiated with N-phenyl-pyrrolidine (1a) and 1,4-dicyanobenzene as starting substrates. With a 23 W household fluorescent lamp as the light source, the photocatalytic transformation of an amine gave an α-amino cyanobenzene product (1b) with a conversion of 90% upon 0.5 mol% (based on the Ar3N moiety in the mixed-ligand coordination polymer) loading of Cd-MIX at room temperature for 12 h (Table 2). In the reaction of N-benzylpyrrolidine (1c) with 1,4-dicyanobenzene, arylation at the N-benzylic position gave a coupling product (1d) with a conversion of 93% and regioselectivity >96% under the standard reaction conditions, and almost no arylation products at the cyclic N-methylene position were detected (Table 1). Meanwhile, no additional activation of products 1b and 1d was detected, as only electron-deficient benzonitriles could be activated in this photocatalytic system.48 In addition, no further arylation of the resulting amine products could be observed, and benzylic amine substrates were added in twice the amount to ensure the selectivity.44 Control experiments exhibited negligible conversion in the absence of Cd-MIX, while no products were detected in the dark, which suggested that both the light source and Cd-MIX were indispensable (Tables S4 and S5,† entries 2 and 14). Cd salt, or the simple mixing of Cd salt and ligands (H3TCA and NPy3), resulted in negative activity under identical conditions (Tables S4 and S5,† entries 3–5), verifying the high photocatalytic activity of Cd-MIX for the C–H arylation and that the heavy structural relaxation from Ar3N˙+ to Ar3NH+ in a homogeneous fashion indeed limited the kinetics of H-atom exchange. With the decrease of NaOAc content, the conversion of the reaction sharply declined, whereas the replacement of NaOAc by K2HPO4 (with a similar pKb in aqueous solution) hardly affected the conversion (Tables S4 and S5,† entries 12 and 13). We estimate that an appropriate weak base environment is important for neutralizing the acidic Ar3NH+ or HCN produced in the reaction system to obtain an efficient reaction.
|
|
||
|---|---|---|
| Entry | Photocatalysts | Conversion (%) |
| a Catalyst (0.5 mol%, based on the Ar3N moiety, Cd salt is based on the molar ratio to Ar3N in Cd-MIX), 1,4-DCB (1.0 mmol), NaOAc (1.0 mmol), amine 1c (2.0 mmol), and 4.0 mL of DMSO for 10 h. Conversions were determined by GC-MS. b Isolated yield. | ||
| 1 | Cd-MIX | 98 (93b) |
| 2 | None | No reaction |
| 3 | Cd(NO3)2·4H2O | No reaction |
| 4 | H3TCA + NPy3 | 27 |
| 5 | H3TCA + NPy3 + Cd(NO3)2·4H2O | 32 |
| 6 | Cd-TCA | 16 |
| 7 | Cd-NPy3 | 49 |
| 8 | Cd-TCA + NPy3 | 19 |
| 9 | Cd-NPy3 + H3TCA | 48 |
| 10 | Cd-TCA + Cd-NPy3 | 53 |

Meanwhile, when triphenylamine was used as the photocatalyst with 1a or 1c as coupling substrates, almost no products were obtained because the free rotation of the three arms in the free triphenylamine molecules consumes the energy at the excited state and diminishes the photosensitization efficiency (Tables S4 and S5,† entries 11), in addition to the heavy structural relaxation from Ar3N˙+ to Ar3NH+ limiting the kinetics of H-atom exchange in solution. As the oxidative state of Ar3N moieties in Cd-MIX cannot oxidize the substrates 1a and 1cvia the SET process, and the BDE of Ar3N+–H, i.e., the quinuclidine radical cation (N+–H 100 kcal mol−1),49 is larger than that of the N-benzylic C–H bond (85 kcal mol−1),50 we deduced that the regioselectivity at the N-benzylic position was well consistent with the idea of a mechanistic switchover from SET to HAT catalysis.44 Deuterium labelling experiments were conducted by treating N-((phenyl-D5)methyl-D2)pyrrolidine (1cD) and 1c with 1,4-dicyanobenzene in two different vessels or in one vessel under optimal conditions (Fig. 4a), resulting in kH/kD values of 3.26 and 2.78, respectively. The kinetic isotope effect indicated that the C–H cleavage was indeed a hydrogen atom transfer process and might occur at the rate-determining step of the reaction.51
 | ||
| Fig. 4 (a) Kinetics curves of the C–H arylation using N-benzylpyrrolidine or N-((phenyl-D5)methyl-D2)pyrrolidine with 1,4-dicyanobenzene under optimal conditions. (b) The turnover number in scale-up experiments for C–H arylation of N-benzylpyrrolidine with 1,4-dicyanobenzene (0.64 g) within 50 h under optimal conditions. | ||
Compared to typical direct HAT photocatalysis in which the activated photocatalyst acts as a HAT catalyst to abstract a H atom from a substrate,12 our approach, in which the in situ formed oxidized Ar3N˙+ acts as a HAT catalyst, possibly avoids the use of a strong oxidizer to abstract the H-atom from the substrate and diminishes the direct oxidization of the substrate to form a byproduct. It is noted that the H-atom abstraction agents in previously reported SET/HAT synergistic photocatalysis have always been transient radical species generated either through electron transfer or energy transfer by photocatalysts.52 Our approach thus simplified the transformation steps by using the oxidized state of the photosensitizer itself for the H-atom abstraction, representing a new HAT strategy parallel to direct HAT catalysis in which the excited state of the photocatalyst behaves as a HAT catalyst for H-atom abstraction.53
Besides the advantages of fewer constituents and decreased reaction steps, the new heterogeneous photoredox-mediated HAT approach exhibits recyclability of the heterogeneous photocatalyst.54 We adopted a model fluidized bed reactor55 for C–H arylation of amine compounds to investigate the recycling properties of Cd-MIX: after one round of reaction, the reaction solution was removed, and then fresh reaction solution containing substrates was added for another round of reaction. No significant decrease in conversion was observed after three cycles (Fig. S36 and S37†). The similar powder X-ray diffraction patterns of the synthesized and recovered Cd-MIX confirmed that the polymer structure was maintained (Fig. S40†). By adding excess substrates, 5.0 mmol of 1,4-dicyanobenzene and 10.0 mmol of N-benzylpyrrolidine (1c), the loading of 0.1 mol% Cd-MIX (based on the Ar3N moiety) gave a TON value of about 900 per mole of catalyst after 50 hours of irradiation. The total pseudo-first order kinetics behavior (rate constant k = 0.048 ± 0.004 h−1) demonstrated that the catalytic efficiency was maintained after 50 hours of constant irradiation (Fig. 4b and S39†). There was no obvious variation in catalytic efficiency when the size of Cd-MIX was reduced from 200 to 1.0 μm by mechanical grinding. We infer that the surface area of the catalyst is not a crucial factor for the arylation reaction (Fig. S41†). Therefore, Cd-MIX is a real heterogeneous photocatalyst that allows the catalytic reaction to occur in the channels of the coordination polymer.
As functional benzylic amines are important building blocks in pharmaceuticals,56 scope exploration began with a change in amines (Table 2). Piperidines and morpholines provided desirable results (88% and 83%). Acyclic amines were converted with moderate conversions (74% and 68%). Meanwhile, amines with benzyl groups resulted in good conversions to N-benzylic arylation products with high regioselectivity. Several electron-deficient arenes or heteroaromatic benzonitriles as coupling substrates could also undergo coupling with amines (1a and 1c), forming the corresponding products when 1.0 mol% Cd-MIX was used. The results indicated that Cd-MIX as a new heterogeneous photoredox HAT catalyst can achieve efficient conversion via photocatalysis and excellent regioselectivity via HAT catalysis, which has not been reported for regioselective functionalization of C(sp3)–H bonds.
| a Reaction conditions: catalyst (0.5 mol%, based on the Ar3N moiety), 1,4-DCB (1.0 mmol), NaOAc (1.0 mmol), amine (2.0 mmol), and 4.0 mL of DMAC for 12 h. b Catalyst (1.0 mol%, based on the Ar3N moiety), arenes (0.5 mmol), NaOAc (0.5 mmol), amine (1.0 mmol), and 2.0 mL of DMAC for 12 h. c DMSO for 10 h. *0.2 mmol scale. Conversions were determined by 1H NMR. The regioselectivity was determined by GC-MS. |
|---|
|
To further explore the potential factors affecting the new direct HAT strategy, monoligand coordination polymers Cd-TCA and Cd-NPy3 were used as reference photocatalysts (Fig. 5). Cd-NPy3 has a 2D layered structure, while Cd-TCA is a 3D framework consolidated by pentanuclear Cd-clusters with channel openings of 7.7 × 8.1 Å2 (Fig. S6†). PXRD analyses indicated that the bulk samples of Cd-TCA and Cd-NPy3 consisted of a pure phase (Fig. S18†). Dye adsorption experiments on the two monoligand coordination polymers revealed that the layered Cd-NPy3 has a poor adsorption effect, while the porous Cd-TCA has comparable adsorption ability to Cd-MIX (Fig. S22†). Control experiments demonstrated that conversions of 20% and 35% with 1a as the substrate, or 16% and 49% with 1c as the substrate were obtained using Cd-TCA and Cd-NPy3 as catalysts, respectively (Tables S4 and S5,† entries 6 and 7). The higher catalytic efficiency of Cd-NPy3 than that of Cd-TCA demonstrated that the different porous behaviors of the coordination polymers were not the important factors dominating the catalytic behavior directly.
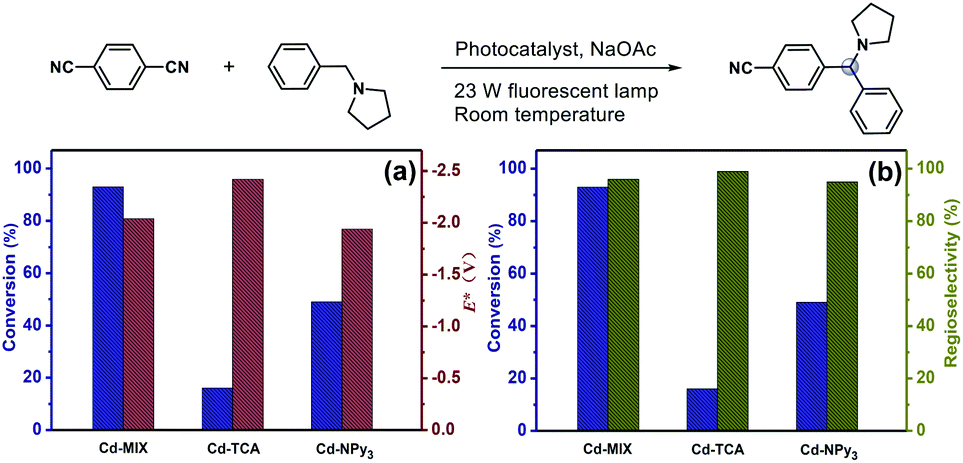 | ||
| Fig. 5 (a) Conversions of the C–H arylation of 1c using Cd-MIX, Cd-TCA, and Cd-NPy3 as catalysts and the excited state potentials of the three coordination polymers under optimal conditions. (b) Conversions of the C–H arylation of 1c using Cd-MIX, Cd-TCA, and Cd-NPy3 as catalysts and the regioselectivity of N-benzyl arylation products under optimal conditions. | ||
When one monoligand polymer was mixed with another free ligand as a photocatalyst, such as Cd-TCA with NPy3 or Cd-NPy3 with H3TCA, the conversions of the reaction were not ideal under the same conditions (Tables S4 and S5,† entries 8 and 9). The photocatalytic efficiency of the mixture of Cd-TCA and Cd-NPy3 was still lower than that of the mixed-ligand Cd-MIX (Tables S4 and S5,† entries 10). Solid-state cyclic voltammetry of Cd-TCA (0.53 V) and Cd-NPy3 (0.75 V) (vs. Ag/AgCl) (Fig. S20 and S21†) revealed that the oxidation potentials of both were less positive than those of 1a and 1c (Fig. 3b), and the excited state potentials of Cd-TCA (−2.42 V) and Cd-NPy3 (−1.94 V) were both sufficient to reduce 1,4-dicyanobenzene (Fig. 5a). We concluded that it is not the redox potential but the special structural intermediate that mixed both N(sp3) and N(sp2) characteristics of the central nitrogen atom, which decreased the structural relaxation from Ar3N˙+ to Ar3NH+ in the mixed-ligand coordination polymer Cd-MIX, dominates the differentiation of catalytic performance of the aforementioned coordination polymers, which is consistent well with the idea of a mechanistic switchover from SET to HAT catalysis. The proton-coupled electron transfer mechanism,57,58 in which a proton and an electron are exchanged in a concerted elementary step, which would require a deprotonation step of 1a and 1c ahead of the oxidation event, was precluded.
Considering the difference in spectral properties of Cd-MIX, Cd-TCA, and Cd-NPy3 (Fig. 3a), a comparison of the photocatalytic efficiency of the three catalysts was conducted under LED irradiation with three single wavelengths (365 nm, 395 nm, and 420 nm). As expected, when irradiated by a 365 nm LED with 1a or 1c as the substrate, all three catalysts were able to achieve complete transformation of 1,4-dicyanobenzene (Tables S4 and S5,† entries 15), but byproducts were present owing to excessive activation by the strong irradiation (Fig. S42 and S43†). When irradiated by a 395 nm (or 420 nm) LED with 1a or 1c as the substrate, Cd-MIX gave high conversions (>95%) based on 1,4-dicyanobenzene, but the catalytic conversions by Cd-TCA and Cd-NPy3 were both lower than those of Cd-MIX (Tables S4 and S5,† entries 16 and 17). When using a household fluorescent lamp that closely matches natural light as the light source, the gap in the catalytic efficiency of the three photocatalysts gradually widened (Tables S4 and S5,† entries 1, 6, and 7). We infer that the mixed-ligand approach was of great significance to extend the absorption spectra of the catalysts, in addition to the favourable photosensitivity and the HAT performance, which is beneficial for the synergistic SET/HAT catalysis to proceed under visible light irradiation.
Transient photocurrent/time tests revealed that Cd-MIX produces a higher photocurrent response than Cd-TCA and Cd-NPy3 (Fig. 6a), indicating that Cd-MIX has a higher electron–hole (e–h) separation efficiency. As photoinduced excitons are active only after effective charge separation,59 the higher photocurrent response of Cd-MIX indicates the higher photocatalytic activity.60 Electrochemical impedance spectroscopy plots showed that Cd-MIX has lower transport resistance than Cd-TCA and Cd-NPy3 (Fig. 6b). The faster electron transfer rate in the mixed-ligand polymer Cd-MIX would be another factor endowing higher catalytic efficiency.21,61 We concluded that the mixed-ligand approach first improved the photochemical and photophysical properties over two monoligand polymers, and then improved the photocatalytic efficiency of the C–H arylation reaction, which provided insights into the rational design and optimization of recyclable photocatalysts for rapid access to complex bioactive molecules and late-stage functionalized pharmaceuticals.
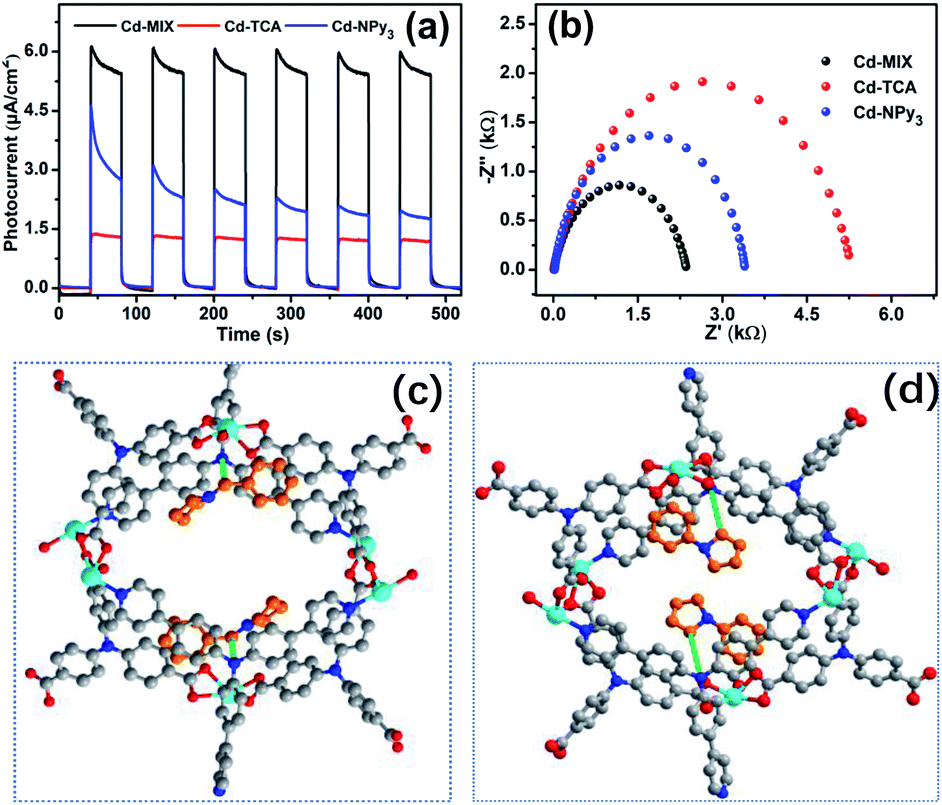 | ||
| Fig. 6 Photocurrent tests (a) and EIS plots (b) of Cd-MIX, Cd-TCA, and Cd-NPy3 under the same conditions, showing the superiority of Cd-MIX over the others. The interaction patterns between the embedded substrates (1c and 1a) and active sites in the channels of 1c@Cd-MIX (c) and 1a@Cd-MIX (d). | ||
1H NMR of the desolvated Cd-MIX immersed in a methanol solution of 1a and 1c revealed that Cd-MIX could adsorb these substrates within its pores (in DMSO-d6/DCl, Fig. S30a and 31a†). The IR spectra of Cd-MIX adsorbed with 1a and 1c showed characteristic signals at 2973–2869 cm−1 and 2961–2732 cm−1, demonstrating the adsorption and activation of the substrates within the pores of Cd-MIX (Fig. S30b and 31b†). The crystal structures of the regular crystals of 1a- and 1c-impregnated Cd-MIX (denoted as 1a@Cd-MIX and 1c@Cd-MIX) provided activation information for elucidating the mechanism details.28,62
As shown in Table S1,† the uptake of substrates 1a or 1c did not influence the main structure of the framework, and the cell dimensions and pores were retained. In 1a@Cd-MIX, the amines were located close to the NPy3 moiety; they were inserted between the layers of molecular sheets and interacted with the phenyl rings of NPy3via π⋯π stacking interactions with an interplanar separation of approximately 3.75 Å (Fig. 6d and S12†). The α-position of 1a was positioned near the central N atom of NPy3, reflecting that the well-positioned NPy3 ligands in Cd-MIX should be the active sites for the HAT event. When the trapped substrates contained an N-benzyl group (1c), the embedded amine molecules were adjusted into a suitable conformation to allow the phenyl rings of NPy3 and 1c to stack with an interplanar separation of 3.66 Å (Fig. 6c and S13†). In addition, there is also a weak interaction between the phenyl rings of 1c and the TCA3− ligand, and the vertical π⋯π stacking distance is approximately 3.72 Å (Fig. S13†). Such double π⋯π stacking interactions provided a driving force for the amine to approach the active sites. Substrate 1c can be docked near the central N atom of NPy3 by the C–H⋯N interactions between N-alkyl and central N as well as between N-benzyl and central N (ca. 3.87 Å and 3.40 Å), which facilitates the in situ formed oxidized NPy3˙+ to abstract a H atom from the benzyl C–H bond of 1c. These results support our hypothesis that the strong interaction between the amines and photocatalyst is an important factor in promoting catalytic conversion. The interactions between the amines and active sites in the channels of the polymer accelerate the capture of the substrate amines and lock the amines around the catalytic sites,24 facilitating the generation of amine radicals through the activation of N-alkyl and N-benzyl C–H bonds by the HAT process.
Conclusions
In summary, we have reported a mixed-ligand approach to the construction of dual functional coordination polymers which enables the arylation of α-amino C(sp3)–H bonds in a predictable, regioselective and recyclable manner. The different molecular sizes and coordination modes of the two Ar3N-containing ligands consolidate and optimize the Ar3N moieties into special structural intermediates, facilitating the oxidized Ar3N˙+ to abstract a hydrogen atom from C(sp3)–H nucleophiles, which is not capable of proceeding on a homogeneous triarylamine-based photocatalyst. The new direct SET/HAT strategy endows the electron transfers via a synergistic effect and skips several interactions between transient species during the typical synergistic SET/HAT cycles, demonstrating a promising redox-economical and reagent-economical heterogeneous photocatalytic platform that has not been reported for α-amino C–H arylation to form benzylamine derivatives. The mixed-ligand approach improves the photochemical and photophysical properties over two monoligand coordination polymers, providing deep insights into the rational design and optimization of recyclable photocatalysts for rapid access to complex bioactive molecules and late-stage functionalized pharmaceuticals.Experimental section
Materials and measurements
Unless stated otherwise, all chemicals and solvents were of reagent grade quality obtained from commercial sources and used without further purification. H3TCA and NPy3 were synthesized using previously reported procedures.27,63 Cd-TCA was synthesized according to a published procedure.42Elemental analyses of C, N, and H were performed on a Vario EL III elemental analyzer. Powder X-ray diffraction (PXRD) tests were performed on a SmartLab 9kW X-ray diffractometer instrument with a Cu sealed tube. Fourier transform infrared spectroscopy (IR) spectra were recorded using KBr pellets on a JASCO FT/IR-430 instrument. 1H NMR and 13C NMR spectra were recorded on a Varian INOVA 400 M spectrometer. Solution and solid-state UV-vis spectra were obtained on TU-1900 and Hitachi U-4100 spectrophotometers. Thermogravimetric analyses (TGA) were carried out at a ramp rate of 10 °C min−1 up to 800 °C under a nitrogen flow with a Mettler-Toledo TGA/SDTA851 instrument.
Scanning electron microscopy (SEM) images were taken using a HITACHI UHR FE-SEM SU8200 microscope. Fluorescence spectra were collected on an EDINBURGH F900 instrument. GC analyses were performed on an Agilent Technologies 6890 N GC system. Electron paramagnetic resonance (EPR) experiments were performed on a Bruker E500 instrument, and the intensity was recorded at 100 K under an Ar atmosphere. Cyclic voltammograms (CV) and electrochemical impedance spectroscopy (EIS) plots were measured on a ZAHNER ENNIUM electrochemical workstation. Transient photocurrent/time measurements were performed on a CHI 650E electrochemical workstation. Confocal laser scanning microscopy micrographs were collected using an Olympus Fluoview FV1000 instrument.
Preparation of coordination polymers
Single crystal X-ray crystallography
Data of Cd-MIX, 1a@Cd-MIX, 1c@Cd-MIX, and Cd-NPy3 were measured on a Bruker SMART APEX CCD diffractometer with Mo-Kα radiation (λ = 0.71073 Å) using the SMART and SAINT programs.64,65 The crystal structures were solved by direct methods and then refined by full-matrix least-squares refinements on F2 using SHELXL-97 software. And an absorption correction was performed using the SADABS program.66 The remaining atoms were found from successive full-matrix least-squares refinements on F2 and Fourier syntheses. Non-H atoms were refined with anisotropic displacement parameters. H atoms in the backbones were fixed geometrically at calculated distances and allowed to ride on the parent non-hydrogen atoms, whereas some of the disordered solvent molecules were not treated during the structural refinements. To help the stability of the refinement for the impregnated related substrate molecules, the bond distances between several atoms were fixed, and thermal parameters on adjacent atoms in two molecules were restrained to be similar. The SQUEEZE program was carried out for Cd-MIX, 1a@Cd-MIX, and 1c@Cd-MIX. The crystallographic data for Cd-MIX, 1a@Cd-MIX, 1c@Cd-MIX, and Cd-NPy3 are summarized in Table S1.†Catalysis details
Before catalysis experiments, solvent exchange experiments of catalysts were carried out to activate the catalysts. First, fresh catalysts were immersed in a methanol solution for 48 h and then fully dried out in a vacuum oven (373 K) for 10 h to obtain the activated catalysts.Arylation of amines with 1,4-dicyanobenzene. A glass tube was filled with 1,4-dicyanobenzene (1.0 mmol), photocatalyst (0.5 mol%, based on the Ar3N moiety), NaOAc (1.0 mmol), amine (2.0 mmol), and solvent (4.0 mL). Then the reaction mixture was cooled to −78 °C, degassed and backfilled with N2 three times. The resulting mixture was stirred and irradiated with a 23 W fluorescent lamp at room temperature. The conversions were determined by 1H NMR analyses, and the regioselectivity was determined by GC-MS. The conversions of the optimization process of experimental conditions were determined by GC-MS.
Arylation of amines with heteroaromatic benzonitriles and electron-deficient arenes. A glass tube was filled with heteroaromatic benzonitriles and electron-deficient arenes (0.5 mmol), photocatalyst (1.0 mol%, based on the Ar3N moiety), NaOAc (0.5 mmol), amine (1.0 mmol), and solvent (2.0 mL). Then the reaction mixture was cooled to −78 °C, degassed and backfilled with N2 three times. The resulting mixture was stirred and irradiated with a 23 W fluorescent lamp at room temperature. The conversions were determined by 1H NMR analyses, and the regioselectivity was determined by GC-MS.
Author contributions
C. Duan conceived the project, designed the experiments and supervised the work. H. Li and X. Jing carried out the main experiments, collected and interpreted the data. Y. Yang and C. He solved and refined the X-ray single crystal structures. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21890381 and 21971030).Notes and references
- Y. Qin, L. H. Zhu and S. Z. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- A. Hossiain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. Le, Y. F. Liang, R. W. Evans, X. M. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- A. H. Hu, J. J. Guo, H. Pan and Z. W. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS PubMed.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- Y. Y. Shen, Y. T. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- C. Gao, J. Wang, H. X. Xu and Y. J. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- C. Yang, R. Li, K. A. I. Zhang, W. Lin, K. Landfester and X. C. Wang, Nat. Commun., 2020, 11, 1239 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC.
- X. Yu and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 12320–12323 CrossRef CAS PubMed.
- X. J. Yang, T. Liang, J. X. Sun, M. J. Zaworotko, Y. Chen, P. Cheng and Z. J. Zhang, ACS Catal., 2019, 9, 7486–7493 CrossRef CAS.
- C. Y. Xu, Y. T. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- A. Dhakshinamoorthy, Z. H. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- L. Zeng, T. Liu, C. He, D. Y. Shi, F. L. Zhang and C. Y. Duan, J. Am. Chem. Soc., 2016, 138, 3958–3961 CrossRef CAS PubMed.
- Z. Q. Xia, C. He, X. G. Wang and C. Y. Duan, Nat. Commun., 2017, 8, 361 CrossRef PubMed.
- B. Z. Lü, Y. F. Chen, P. Y. Li, B. Wang, K. Müllen and M. Z. Yin, Nat. Commun., 2019, 10, 767 CrossRef PubMed.
- (a) Y. X. Tan, Y. P. He, D. Q. Yuan and J. Zhang, Appl. Catal., B, 2018, 221, 664–669 CrossRef CAS; (b) X. K. Wang, J. Liu, L. Zhang, L. Z. Dong, S. L. Li, Y. H. Kan, D. S. Li and Y. Q. Lan, ACS Catal., 2019, 9, 1726–1732 CrossRef CAS.
- P. Y. Wu, C. He, J. Wang, X. J. Peng, X. Z. Li, Y. L. An and C. Y. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- T. X. Zhang, X. Y. Guo, Y. S. Shi, C. He and C. Y. Duan, Nat. Commun., 2018, 9, 4024 CrossRef PubMed.
- X. H. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed.
- B. Adelizzi, I. A. W. Filot, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2017, 23, 6103–6110 CrossRef CAS PubMed.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed.
- H. K. Chae, J. Kim, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2003, 42, 3907–3909 CrossRef CAS PubMed.
- Z. Q. Yao, J. Xu, B. Zou, Z. P. Hu, K. Wang, Y. J. Yuan, Y. P. Chen, R. Feng, J. B. Xiong, J. L. Hao and X. H. Bu, Angew. Chem., Int. Ed., 2019, 58, 5614–5618 CrossRef CAS PubMed.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed.
- L. Shi and W. J. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- Y. Mori, Y. Sakaguchi and H. Hayashi, J. Phys. Chem. A, 2000, 104, 4896–4905 CrossRef CAS.
- Y. C. Qiu, S. Yuan, X. X. Li, D. Y. Du, C. Wang, J. S. Qin, H. F. Drake, Y. Q. Lan, L. Jiang and H. C. Zhou, J. Am. Chem. Soc., 2019, 141, 13841–13848 CrossRef CAS PubMed.
- H. Jiang, J. t Jia, A. Shkurenko, Z. J. Chen, K. Adil, Y. Belmabkhout, L. J. Weselinski, A. H. Assen, D. Xu Xue, M. O'Keeffe and M. Eddaoudi, J. Am. Chem. Soc., 2018, 140, 8858–8867 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- T. L. Easun, F. Moreau, Y. Yan, S. H. Yang and M. Schröder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- Y. Shen, X. F. Yang, H. B. Zhu, Y. Zhao and W. S. Li, Dalton Trans., 2015, 44, 14741–14746 RSC.
- (a) M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijna and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC; (b) Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, VCH, New York, 1993 Search PubMed.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 1999 Search PubMed.
- (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (b) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- W. Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778–4783 CrossRef CAS PubMed.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed.
- D. M. Yan, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- M. M. Bello, A. A. Abdul Raman and M. Purushothaman, J. Cleaner Prod., 2017, 141, 1492–1514 CrossRef CAS.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS PubMed.
- T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118–3123 RSC.
- H. Liu, C. Y. Xu, D. D. Li and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- D. Y. Shi, R. Zheng, M. J. Sun, X. R. Cao, C. X. Sun, C. J. Cui, C. S. Liu, J. W. Zhao and M. Du, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- T. Song, L. Zhang, P. Y. Zhang, J. Zeng, T. T. Wang, A. Alia and H. P. Zeng, J. Mater. Chem. A, 2017, 5, 6013–6018 RSC.
- T. Kawamichi, T. Haneda, M. Kawano and M. Fujita, Nature, 2009, 461, 633–635 CrossRef CAS PubMed.
- C. Hua, A. Baldansuren, F. Tuna, D. Collison and D. M. D'Alessandro, Inorg. Chem., 2016, 55, 7270–7280 CrossRef CAS PubMed.
- SMART, Data collection software (version 5.629), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- SAINT, Data reduction software (version 6.54), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, SHELXTL97, Program for crystal structure solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1970513, 1970515, 1970516 and 1970538. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00828e |
| This journal is © The Royal Society of Chemistry 2021 |