
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically driven stereoselective approach to syn-1,2-diol derivatives from vinylarenes and DMF†
Da Sol
Chung‡
,
Steve H
Park‡
,
Sang-gi
Lee
* and
Hyunwoo
Kim
 *
*
Department of Chemistry and Nanoscience, Ewha Womans University, 03760 Seoul, Korea. E-mail: sanggi@ewha.ac.kr; khw7373@ewha.ac.kr
First published on 23rd March 2021
Abstract
We have developed an electrochemically driven strategy for the stereoselective synthesis of protected syn-1,2-diols from vinylarenes with N,N-dimethylformamide (DMF). The newly developed system obviates the need for transition metal catalysts or external oxidizing agents, thus providing an operationally simple and efficient route to an array of protected syn-1,2-diols in a single step. This reaction proceeds via an electrooxidation of olefin, followed by a nucleophilic attack of DMF. Subsequent oxidation and nucleophilic capture of the generated carbocation with a trifluoroacetate ion is proposed, which gives rise predominantly to a syn-diastereoselectivity upon the second nucleophilic attack of DMF.
Introduction
The dihydroxylation of alkenes is a fundamental and straightforward transformation for the preparation of 1,2-diols, which is widely used in the preparation of key intermediates in fragrances, pharmaceuticals and functional materials.1 There are several classical methods that are capable of achieving this goal, including the Woodward–Prevost reaction2a–c and the epoxidation followed by ring-opening,2d in which both reactions proceed via cyclic intermediates that define the stereochemistry of the product (eqn (1) in Scheme 1A). A concerted syn-dihydroxylation mediated by OsO4 along with its asymmetric version, the Sharpless dihydroxylation, has also been recognized as the most widely used synthetic method for 1,2-diols (eqn (2) in Scheme 1A).3 Despite these landmark achievements, the replacement of the use of an expensive and highly toxic osmium catalyst led to the dioxygenation of alkenes employing other metal-based catalysts which has successfully been developed in the presence of stoichiometric chemical oxidants such as PhI(OAc)2 or dioxygen (eqn (3) in Scheme 1A).4 More recently, transition-metal free approaches such as peroxide5 or radical-mediated protocols6 have emerged as the alternative synthetic routes to 1,2-diols, although it is sometimes difficult to predict stereochemical outcome (eqn (4) in Scheme 1A).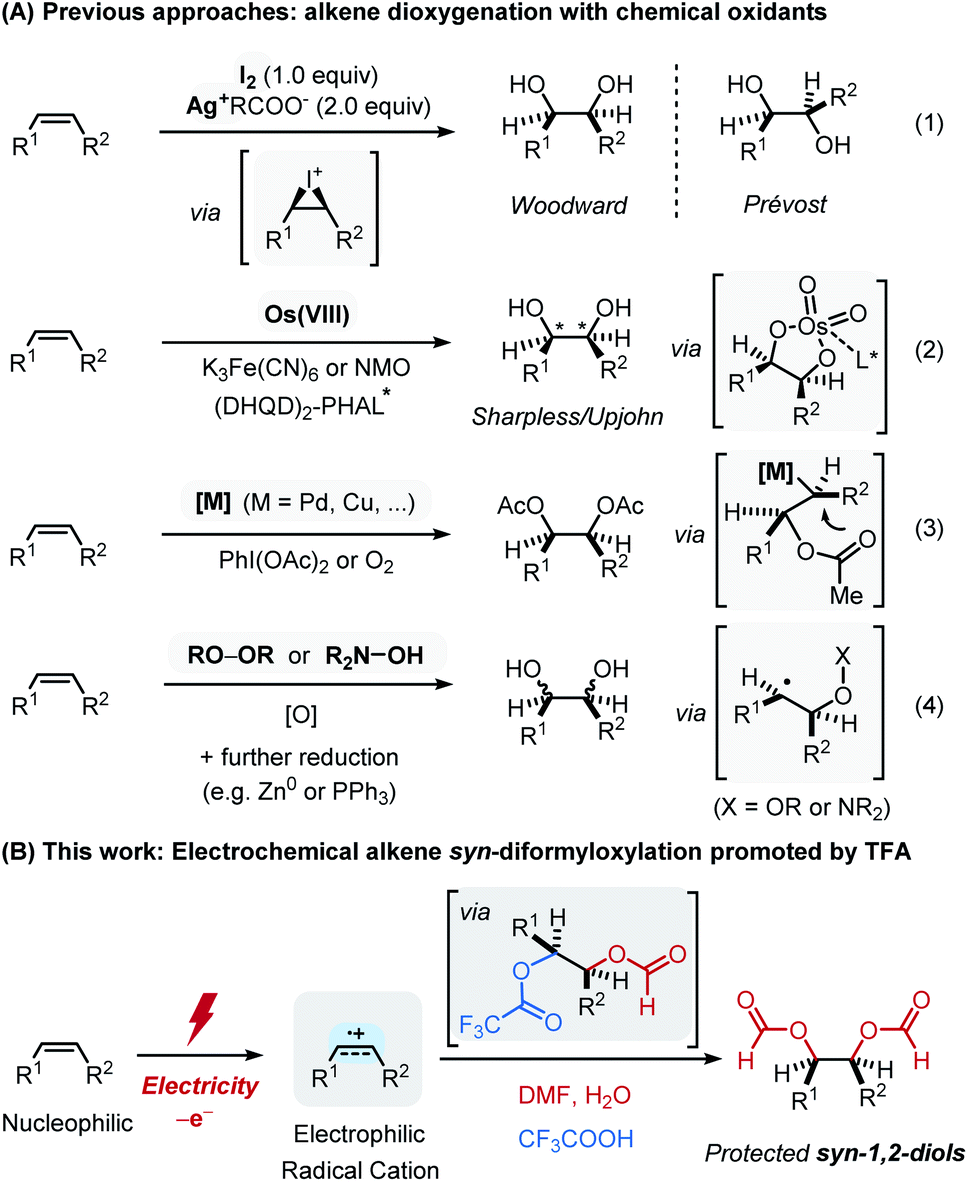 | ||
| Scheme 1 Synthetic approaches to 1,2-diols from alkenes. | ||
Most of these precedent examples however still rely on the use of transition metals for the requisite redox process or otherwise require additional synthetic steps for the preparation of reaction mediators. In addition, an employment of a chemical oxidant system (e.g. hypervalent iodines) often leads to limited functional group compatibility. In this regard, we envisioned an electrochemical alkene oxidation as an ideal approach. Electrocatalytically driven organic synthesis allows to precisely select the redox potential for use, which circumvents selectivity and compatibility issue that often arise from its purely chemical counterparts.7 Despite recent efforts in the development of electrochemical dialkoxylation,8 it is highly desirable to develop a conventional and stereoselective approach for 1,2-diol derivatives using feedstock chemicals.
Herein, we report an electrochemical syn-diformyloxylation of vinylarenes using DMF as an oxygen source (Scheme 1B). This newly developed catalyst-free approach provides a straightforward and efficient route to a wide range of protected syn-1,2-diols in a single step. Electrochemical oxidation of alkenes to its radical cation enabled the direct addition of DMF, in which the formyloxy group derived from DMF can readily be converted into hydroxyl group.9 Subsequent oxidation and a facile nucleophilic capture of the generated carbocation with a trifluoroacetate ion is proposed to grant high syn-diastereoselectivity upon second nucleophilic attack of DMF.
Results and discussion
We set out to investigate our proposed olefin diformyloxylation by choosing 4-tert-butylstyrene (1) as the model substrate (Table 1). After optimization, we observed that the application of a constant cell voltage of 2.5 V (corresponding to an anodic potential of 1.3 V vs. SCE) enabled the formation of a formyl protected diol 2 in 90% yield (entry 1). The optimal conditions employed trifluoroacetic acid (TFA, 5.0 equiv.) and water (3.0 equiv.) as additives, TBABF4 as the electrolyte, carbon felt and platinum plate as the anode and cathode respectively, in DMF. A control experiment without applied potential revealed that electric current is necessary for reactivity (entry 2). The reaction efficiency was significantly diminished when the reaction was conducted in the presence of molecular sieves, suggesting that stoichiometric amount of water is requisite for the reaction (entry 3). We found that the employment of acids weaker than TFA were detrimental to the reaction (entries 4 and 5). On the other hand, acids stronger than TFA were productive, albeit with slightly reduced efficiencies (entries 6 and 7). Switching the solvent from DMF into CH3CN significantly hampered the reactivity, even under high excess amount of DMF (entries 8 and 9). We have also found that the reactivity was not significantly affected when electrolysis was conducted under a constant current of 2 mA (entry 10). Notably, the reaction under O2 atmosphere was not beneficial for the desired transformation, showing a significant drop in reaction efficiency (entry 11). Similarly, the reactivity was found to be slightly diminished when the reaction was conducted open to air (entry 12).|
|
||
|---|---|---|
| Entry | Variation from “standard conditions” | Yield of 2 (%) |
| a 1 (0.2 mmol, 1.0 equiv.), CF3COOH (1.0 mmol, 5.0 equiv.), H2O (0.6 mmol, 3.0 equiv.), TBABF4 (0.1 M), DMF (3 mL); cell voltage (Ucell = 2.5 V); yields determined by 1H NMR (isolated yields in parenthesis). | ||
| 1 | None | 95 (90) |
| 2 | No applied voltage | <5 |
| 3 | With 4 Å molecular sieves | 33 |
| 4 | CH3COOH instead of TFA | <5 |
| 5 | HCOOH instead of TFA | 31 |
| 6 | HOTf instead of TFA | 60 |
| 7 | HNTf2 instead of TFA | 50 |
| 8 | DMF (20 equiv.) in CH3CN (0.05 M) | 25 |
| 9 | DMF (10 equiv.) in CH3CN (0.05 M) | 15 |
| 10 | Under constant current (Icell = 2.0 mA) | 75 |
| 11 | Under O2 atmosphere (balloon, 1 atm) | 45 |
| 12 | Under air atmosphere | 70 |

To investigate the scope and functional group compatibility of the current diformyloxylation protocol, an array of terminal vinylarenes were initially examined (Table 2). Vinylarenes with different functional groups such as simple alkyls (2–4), acetoxy (5), ester (6) and halogen (7–9) groups at para-position were well tolerated. An ortho-substituted vinylarene (10), 2-vinylnaphthalene (11) and 1,1-disubstituted olefin (12) were also smoothly participated in the reaction without difficulties. Importantly, vinyl heterocycles derived from dibenzofuran (13) and carbazole (14) were also applicable to the current protocol, while pyridine, quinolone and thiophene derived vinyl heteroarenes were reluctant to participate in the reaction mainly due to the formation of polymeric side products. Interestingly, 1,2-diacetoxylation (15) product was obtained when N,N-dimethylacetamide (DMA) was employed as a solvent. Moreover, the reactivity toward biorelevant structures was examined to illustrate the installation of protected 1,2-diol group as a late-stage synthesis, furnishing estrone (16) and tyrosine (17) derivatives. It was also notable to see that a readily oxidizable electron rich alkene such as phenyl vinyl ether,10 could also be engaged in this reaction (18).
| a Isolated yields are reported. Optimal conditions from Table 1 used. Diastereoselectivity was determined by 1H NMR spectra of crude reaction mixture. b Yield determined by 1H NMR using 1,2-dimethoxyethane as an internal standard. c N,N-Dimethylacetamide (DMA) was used as a solvent. d 10 equiv. of TFA was used. e 5% of 33 was also obtained along with 32. |
|---|

|
To explore the stereoselectivity of the current procedure, we next set out to examine the scope of various internal alkenes. As summarized in Table 2, diformyloxylation of a wide range of internal alkenes granted access to products bearing vicinal stereogenic centers with good diastereocontrol. Dioxygenation of acyclic 1,2-disubstituted alkenes provided formyl-protected syn-diol products in good to excellent diastereomeric ratios (19–24, >7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Notably, substrates having labile groups on allylic positions underwent desired transformations without dissociation of the leaving groups (21–23). The cyclic alkenes such as dibenzosuberone and indene were viable substrates as well, albeit in somewhat diminished diastereoselectivity (25–26, >4:1, diastereomeric ratios). Interestingly, the reaction was found to be highly chemoselective towards a more readily oxidizable, electronically rich alkene when a substrate bearing multiple alkenes was tested. For example, cinnamyl cinnamate gave a mono-dioxygenated product 27 with high chemo- and diasteroselectivity. Trisubstituted alkenes were also reacted efficiently to afford the corresponding products in good diasteroselectivity (28–29). However, tetrasubstituted or unactivated alkenes derived from simple hydrocarbons were found to be recalcitrant to the current dioxygenation method (see Scheme S1 in ESI† for unsuccessful substrates). The electrochemical diformyloxylation tested positive in the radical clock experiment with cycloproyl-substituted alkene 30, implying the intermediacy of a benzylic radical during the reaction.
:1). Notably, substrates having labile groups on allylic positions underwent desired transformations without dissociation of the leaving groups (21–23). The cyclic alkenes such as dibenzosuberone and indene were viable substrates as well, albeit in somewhat diminished diastereoselectivity (25–26, >4:1, diastereomeric ratios). Interestingly, the reaction was found to be highly chemoselective towards a more readily oxidizable, electronically rich alkene when a substrate bearing multiple alkenes was tested. For example, cinnamyl cinnamate gave a mono-dioxygenated product 27 with high chemo- and diasteroselectivity. Trisubstituted alkenes were also reacted efficiently to afford the corresponding products in good diasteroselectivity (28–29). However, tetrasubstituted or unactivated alkenes derived from simple hydrocarbons were found to be recalcitrant to the current dioxygenation method (see Scheme S1 in ESI† for unsuccessful substrates). The electrochemical diformyloxylation tested positive in the radical clock experiment with cycloproyl-substituted alkene 30, implying the intermediacy of a benzylic radical during the reaction.
We have also found that the reaction took place selectively at the terminal position when 1,3-diene was employed as a substrate (32). Different from conventional vinylarene substrates, however, the dioxygenation product 33 possessing a trifluoroacetoxy group at 3-position was obtained as the major product (45%) along with 5% of the desired product (34). This observation led us to further investigate the possible intermediacy of our reaction. As hypothesized, we were able to observe the formation of desired diformyloxylation product 34 upon treatment of 33 with DMF in the presence of water.
The uncommon diastereoselectivity trend observed in this catalyst-free approach piqued our interest in elucidating its mechanism. The reaction under deuterated DMF-d7 solvent revealed that the formyl groups in the product originate from DMF, as we envisioned at the outset (Fig. 1A). In contrast, we found that the reaction with deuterated formic acid did not result in incorporation of deuterium on both formyl groups (Fig. 1B). These findings suggested that the engagement of formic acid with the olefin radical cation is less conceivable, in which the formic acid is initially generated by hydrolysis of DMF in the presence of strong acid.9a,11 Notably, the stereo-outcome of this reaction was dependent on the alkenylic geometry of the starting materials, with the major isomers irrespectively arising from syn-diformyloxylation. For example, trans- and cis-β-methylstyrene (trans- and cis-35) both gave corresponding syn-diformyloxylation products with 10:1 and 3:1 of diastereomeric ratios respectively (Fig. 1C). We note that this retention of diastereoselectivity upon choice of the stereoisomeric starting materials is unusual compared to previous examples that employ different alkene oxidation strategies.12
 | ||
| Fig. 1 Mechanistic investigations. (A) Deuterium labeling experiment with DMF-d7. (B) Deuterium labeling experiment with formic acid-d2. (C) Diastereoselectivity test with isomeric vinylarene substrates. (D) O18-labeling experiments. (E) Cyclic voltammetric experiments. | ||
To elucidate the origin of the oxygen atoms, we performed an isotopic labeling study with indene (36) as a substrate, using 97% O18 enriched water as an additive (Fig. 1D). The relative amounts of doubly-labeled diformyloxylation products (>69.3%) with singly-labeled (<26.9%) and unlabeled (<4%) products were determined for both diastereomers (syn- and anti-26), using high-resolution mass spectroscopy (HRMS). On the other hand, hydrolysis of the formyloxy groups resulted in significant removal of the oxygen 18-label, leading to the formation of unlabeled diol as the major product for both diastereomers (syn- and anti-37). These results suggest that both oxygens for the hydroxyl group originate from DMF during the reaction, not from water.
Cyclic voltammetry (CV) data showed that the oxidation of alkene 1 to the corresponding alkene radical cation results in a feature at around Ep/2 = 1.7 V (vs. SCE; Fig. 1E, black line). The addition of TFA, DMF, or both did not cause significant anodic peak shift of this redox event in thermodynamically more feasible way, implying that the alkene substrate is directly oxidized on the carbon anode during the reaction. We also recognized the possibility of an oxidation of DMF prior to the alkene. However, such mechanism is proved to be difficult because of high oxidation potential of DMF (Ep/2 = 2.1 V, dashed black lines), even in the presence of TFA (dashed blue lines).
On the basis of these experimental results and precedent literature,13 a mechanistic rationale is shown in Fig. 2. First, electrochemical oxidation of the alkene substrate A generates the alkene radical cation B (the oxidation potentials Eox for the different vinylarenes tested range from 1.15 V to 1.75 V vs. SCE).14 The low potential threshold for such electron transfer is reported to be 0.5 V lower than the thermodynamic potential of the substrate,15 which supports that our measured initial anodic potential (Ea,i = 1.3 V vs. SCE) is above the onset potential of alkene oxidation. As an additional note, the anodic potential was maintained throughout the reaction, showing 1.22 V of the final anodic potential (Ea,f). This result suggests that the desired reactivity can be achieved at the low potential threshold of the alkene oxidation. The nucleophilic trapping of B with DMF produces the carbon-centered radical C, which is concurrently oxidized into the dication D on a carbon anode. Indeed, the second anodic oxidation is calculated to be thermodynamically more feasible than the first oxidation (Eox = 0.69 V vs. SCE, when Ar = Ph and R = Me).16 A facile nucleophilic capture of sterically biased dication D by trifluroacetate ion would result in the predominant formation of an anti-dioxygenated intermediate F, upon hydrolysis of iminium intermediate E. A nucleophilic displacement of trifluoroacetate group17 from an isolable intermediate F by DMF eventually furnishes a syn-diformyloxylation product H followed by the second hydrolysis. It should be noted that the high diastereoselectivity observed in linear alkene substrates suggests that the formation of E occurs prior to the erosion of diastereocontrol caused by C–C bond rotation. The reactivity trend upon the choice of acid observed in Table 1 is likely related to the final nucleophilic displacement step. Moreover, the role of TFA in affecting diastereoselectivity was also verified by a series of control experiments with internal alkene substrate, where TFA is replaced by other acids.18 We also recognized the possibility of a nucleophilic attack of D directly from DMF to give anti-diformyloxylation product (H′). However, this pathway is considered to be less likely because trifluoroacetate is presumably a better nucleophile than DMF to capture dicationic intermediate D mainly due to its anionic character. This is also consistent to the predominant syn-diastereoselectivity of the current diformyloxylation protocol.
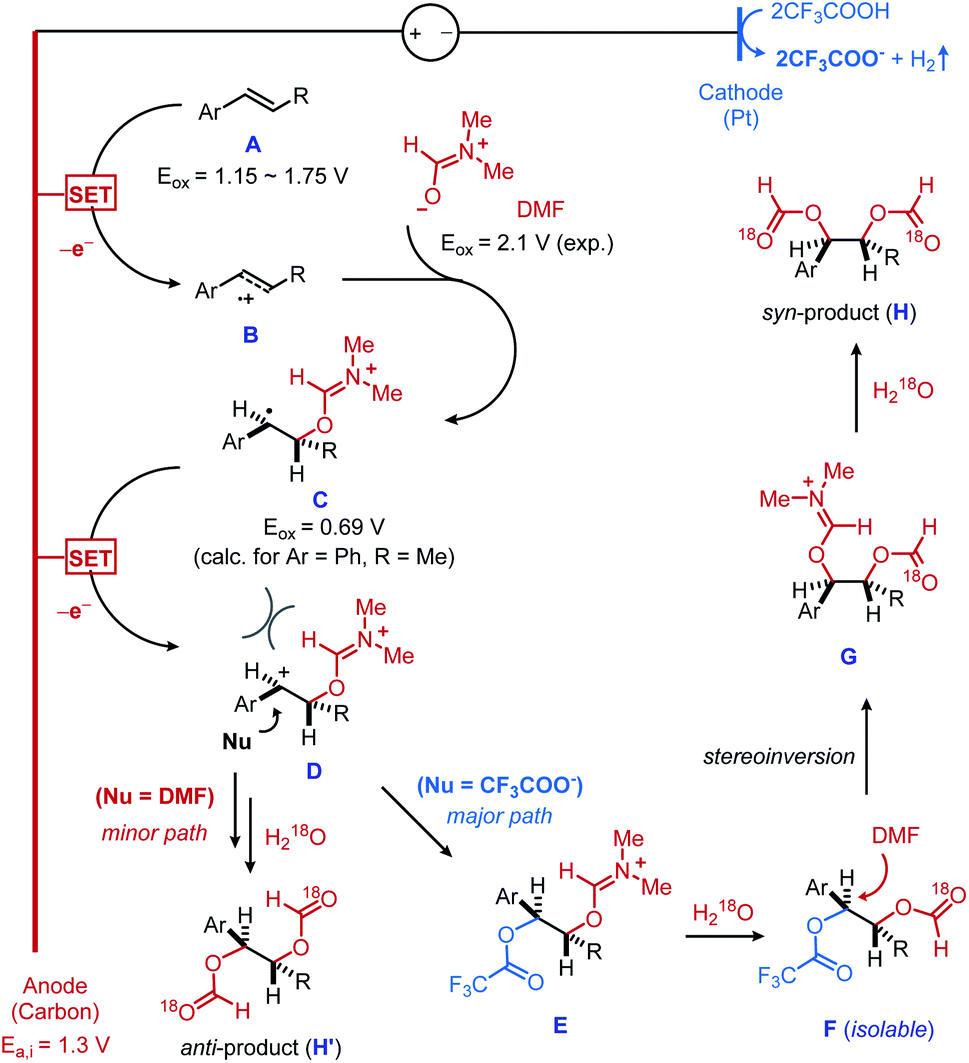 | ||
| Fig. 2 Mechanistic rationale. | ||
Conclusions
In conclusion, we devised an electrooxidative strategy that grants access to formyl-protected syn-1,2-diols from vinylarenes and DMF. This reaction is initiated by the electrochemical oxidation of the alkene substrates followed by the nucleophile attack of DMF. Mechanistic studies imply that trifluoroacetate ion is presumably engaged in the nucleophilic capture of the carbocation intermediate, which gives rise to high syn-diastereoselectivity. A simple deprotection of formyl protecting groups from the dioxygenated product was also presented, highlighting synthetic utility of this electrochemical method toward a variety of 1,2-diols. We anticipate this electrochemical synthetic approach promoted by trifluoroacetic acid will be broadly applicable in further development of nucleophilic olefin functionalization reactions.Author contributions
S. H. P. and D. S. C. performed the experiments and analysed the data. H. K. conceived the project, analysed experimental data and performed the calculations. The manuscript was written by H. K. and S.-g. L. and S. H. P. contributed to the editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [NRF-2021R1C1C1004605 (H. Kim)]. D. S. Chung acknowledges the National Research Foundation (NRF-2019R1A6A3A13096586). This study made use of the NMR facility supported by Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (NRF-2020R1A6C101B194).Notes and references
-
(a)
J. I. Kroscwitz and M. Howe-Grant, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 1991 Search PubMed
; (b) K. Kulkaand and J. W. Dittrick, Cosmet. Perfum., 1975, 90, 90–95 Search PubMed
-
(a) R. B. Woodward and F. V. Brutcher, J. Am. Chem. Soc., 1958, 80, 209–211 CrossRef CAS
-
(a) M. Schroeder, Chem. Rev., 1980, 80, 187–213 CrossRef CAS
-
(a) Y. Zhang and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 3076–3077 CrossRef CAS PubMed
-
(a) Y. Usui, K. Sato and M. Tanaka, Angew. Chem., Int. Ed., 2003, 42, 5623–5625 CrossRef CAS PubMed
-
(a) B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320–13322 CrossRef CAS PubMed
- For representative articles on electrochemically driven organic synthesis, see:
(a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
-
(a) C.-Y. Cai and H.-C. Xu, Nat. Commun., 2018, 9, 3551 CrossRef PubMed
-
(a) M. M. Heravi, M. Ghavidel and L. Mohammadkhani, RSC Adv., 2018, 8, 27832–27862 RSC
- B. M. Peterson, S. Lin and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 2076–2079 CrossRef CAS PubMed
- T. Cottineau, M. Richard-Plouet, J.-Y. Mevellec and L. Brohan, J. Phys. Chem. C, 2011, 115, 12269–12274 CrossRef CAS
-
(a) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed
-
(a) A. J. J. Lennox, J. E. Nutting and S. S. Stahl, Chem. Sci., 2018, 9, 356–361 RSC
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC
- Oxidation potential of C (when Ar = Ph and R = Me) was calculated using quantum chemical simulations and referenced to the absolute oxidation potential of NHE (−4.430 V). See ESI† (page S4–6) for details. For related references, see:
(a) D. G. Truhlar, C. J. Cramer, A. Lewis and J. A. Bumpus, J. Chem. Educ., 2004, 81, 596 CrossRef
-
(a) A. Abad, C. Agulló, A. C. Cuñat and I. Navarro, Synthesis, 2005, 2005, 3355–3361 CrossRef
- A set of control experiments was conducted where TFA is replaced by other acids using an internal alkene (trans-35) as a substrate. Both reactivity and syn-selectivity was decreased upon treatment of other acids, supporting that the mechanistic role of TFA is important in the final nucleophilic displacement step. See ESI† page S12 for details.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00760b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |