
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne tool to bring them all: Au-catalyzed synthesis of B,O- and B,N-doped PAHs from boronic and borinic acids†
Omar
Ouadoudi‡
,
Tanja
Kaehler‡
,
Michael
Bolte
 ,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
Institut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt (Main), Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de;
First published on 17th March 2021
Abstract
The isoelectronic replacement of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds with −BN+ bonds in polycyclic aromatic hydrocarbons (PAHs) is a widely used tool to prepare novel optoelectronic materials. Far less well explored are corresponding B,O-doped PAHs, although they have a similarly high application potential. We herein report on the modular synthesis of B,N- and B,O-doped PAHs through the [Au(PPh3)NTf2]-catalyzed 6-endo-dig cyclization of BN–H and BO–H bonds across suitably positioned C
C bonds with −BN+ bonds in polycyclic aromatic hydrocarbons (PAHs) is a widely used tool to prepare novel optoelectronic materials. Far less well explored are corresponding B,O-doped PAHs, although they have a similarly high application potential. We herein report on the modular synthesis of B,N- and B,O-doped PAHs through the [Au(PPh3)NTf2]-catalyzed 6-endo-dig cyclization of BN–H and BO–H bonds across suitably positioned C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds in the key step. Readily available, easy-to-handle o-alkynylaryl boronic and borinic acids serve as starting materials, which are either cyclized directly or first converted into the corresponding aminoboranes and then cyclized. The reaction even tolerates bulky mesityl substituents on boron, which later kinetically protect the formed B,N/O-PAHs from hydrolysis or oxidation. Our approach is also applicable for the synthesis of rare doubly B,N/O-doped PAHs. Specifically, we prepared 1,2-B,E-naphthalenes and -anthracenes, 1,5-B2-2,6-E2-anthracenes (E = N, O) as well as B,O2-containing and unprecedented B,N,O-containing phenalenyls. Selected examples of these compounds have been structurally characterized by X-ray crystallography; their optoelectronic properties have been studied by cyclic voltammetry, electron spectroscopy, and quantum-chemical calculations. Using a new unsubstituted (B,O)2-perylene as the substrate for late-stage functionalization, we finally show that the introduction of two pinacolatoboryl (Bpin) substituents is possible in high yield and with perfect regioselectivity via an Ir-catalyzed C–H borylation approach.
C bonds in the key step. Readily available, easy-to-handle o-alkynylaryl boronic and borinic acids serve as starting materials, which are either cyclized directly or first converted into the corresponding aminoboranes and then cyclized. The reaction even tolerates bulky mesityl substituents on boron, which later kinetically protect the formed B,N/O-PAHs from hydrolysis or oxidation. Our approach is also applicable for the synthesis of rare doubly B,N/O-doped PAHs. Specifically, we prepared 1,2-B,E-naphthalenes and -anthracenes, 1,5-B2-2,6-E2-anthracenes (E = N, O) as well as B,O2-containing and unprecedented B,N,O-containing phenalenyls. Selected examples of these compounds have been structurally characterized by X-ray crystallography; their optoelectronic properties have been studied by cyclic voltammetry, electron spectroscopy, and quantum-chemical calculations. Using a new unsubstituted (B,O)2-perylene as the substrate for late-stage functionalization, we finally show that the introduction of two pinacolatoboryl (Bpin) substituents is possible in high yield and with perfect regioselectivity via an Ir-catalyzed C–H borylation approach.
Introduction
The targeted doping of polycyclic aromatic hydrocarbons (PAHs) with p-block atoms is a powerful tool to create new molecules for applications as diverse as drug development, materials science, and catalysis.1 Especially the isoelectronic replacement of a non-polar CC bond with a polar −BN+ bond can substantially influence the π-electron distribution and frontier-orbital energies of the resulting B,N-PAH and thus lead to chemical and physical properties that differ considerably from those of the corresponding carbonaceous congener (“BN/CC isosterism”).2 By varying the positions,3 orientations, and number of −BN+ units within the molecular framework, a greatly expanded structural and chemical space becomes accessible.4–15 In addition, breaking the symmetry of a molecular scaffold through the introduction of a −BN+ unit often allows late-stage functionalizations with higher regioselectivities than in the case of the parent PAH.16 Because of these appealing features, considerable research efforts are currently being made to develop novel B,N-PAHs. Although many synthesis approaches are still based on individual solutions of limited scope, the following more general strategies have already emerged. (i) B–N/B–C-bond formation cascades: the B–N bond is formed first, followed by an intramolecular electrophilic borylation reaction with a suitably positioned aryl, vinyl, or alkynyl group (Scheme 1a).17–21 (ii) C–C-bond formation/oxidation cascades: a ring-closing metathesis (RCM) is conducted on an already established B–N moiety and a subsequent oxidation step generates the conjugated π system (Scheme 1b).22,23 (iii) Photochemical or exciton-driven C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) B-bond formation: starting from B–N adducts with N-heteroaryl-benzyl backbones, the elimination of HR′ (R′ = alkyl, aryl), which occurs either upon UV/vis irradiation or upon application of an electric current to a corresponding electroluminescent device, closes the conjugation pathway (Scheme 1c).24,25
B-bond formation: starting from B–N adducts with N-heteroaryl-benzyl backbones, the elimination of HR′ (R′ = alkyl, aryl), which occurs either upon UV/vis irradiation or upon application of an electric current to a corresponding electroluminescent device, closes the conjugation pathway (Scheme 1c).24,25
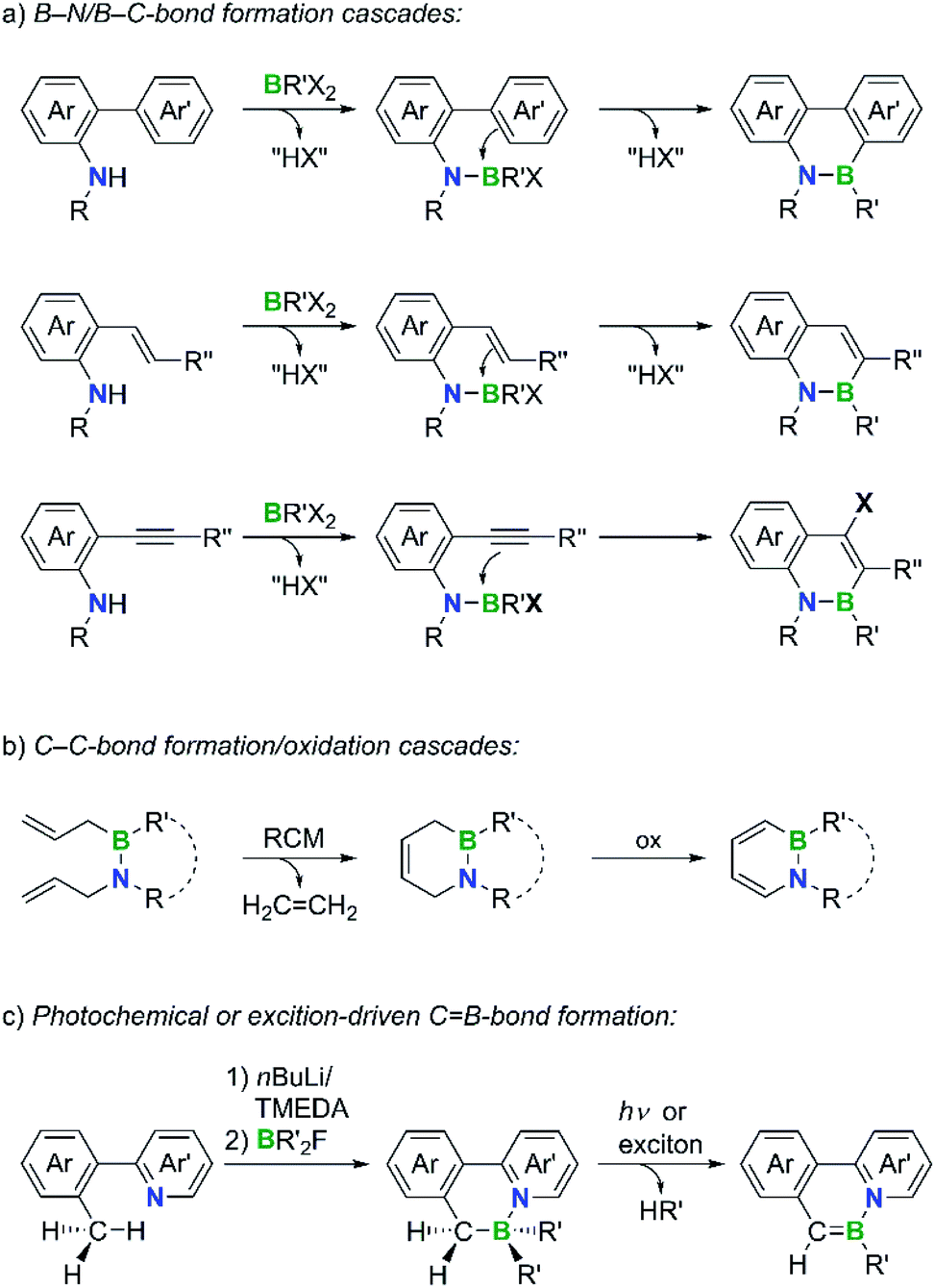 | ||
| Scheme 1 Common strategies for the synthesis of B,N-doped PAHs. (RCM = ring-closing metathesis). | ||
Compared to B,N-PAHs, analogous B,O-PAHs have been largely neglected, although a first derivative, 10-hydroxy-9,10-oxaboraphenanthrene, was published by Dewar et al. as early as in 1960.26 Still today, the majority of known B,O-PAHs contains the structural motif of a 9,10-oxaboraphenanthrene,27–42 likely due to its convenient accessibility via B–O-bond formation/electrophilic aromatic borylation sequences (Scheme 2a; compare the analogous B,N case shown in Scheme 1a).26–33
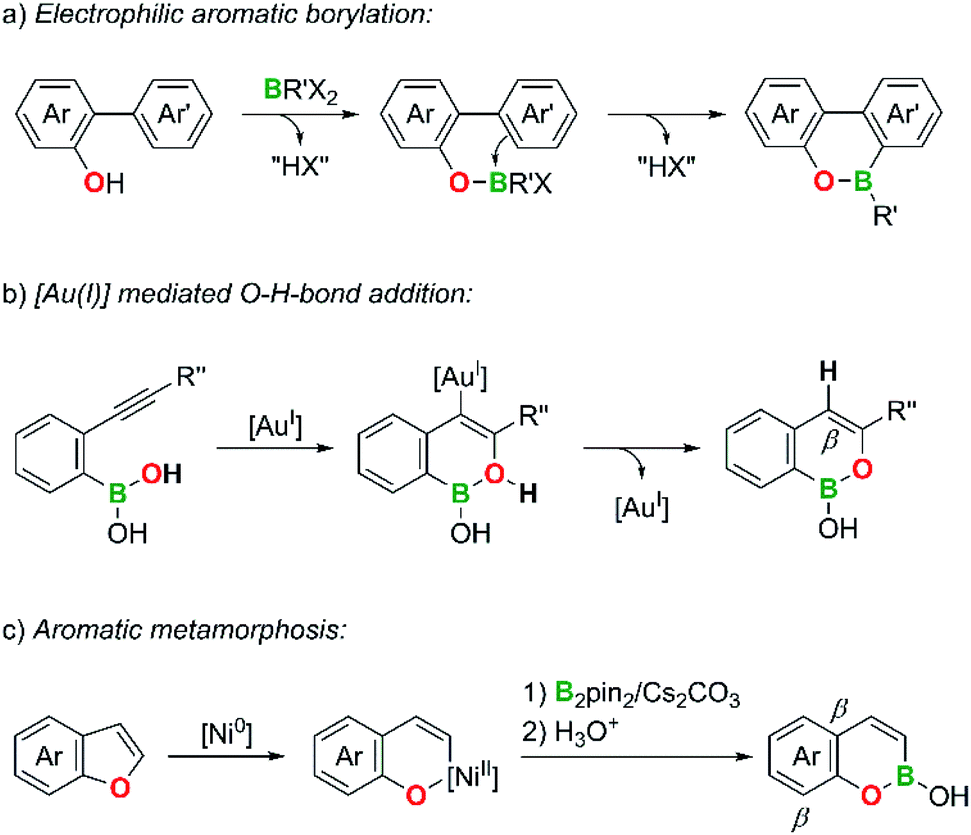 | ||
| Scheme 2 Common strategies for the synthesis of B,O-doped PAHs. | ||
A second class of B,O-PAHs with a reasonably large number of members are 1,2-B,O-naphthalenes. Sheppard et al. and Gong et al. selected o-alkynylphenyl boronic acids as the starting materials and used the Au- or Pd-catalyzed intramolecular addition of a BO–H bond to the CC bond in the final cyclization step (Scheme 2b).43–46 Yorimitsu et al. prepared an isomeric 2,1-B,O-naphthalene through Ni- or Mn-catalyzed O–C-bond activation of benzofurans and subsequent insertion of a [Bpin]− fragment generated by heterolysis of bis(pinacolato)diboron, pinB–Bpin (“aromatic metamorphosis”; Scheme 2c).47,48
Even the limited information available on the chemical properties of B,O-PAHs reveal some distinct patterns: (i) due to the higher electronegativity and weaker π-donor ability of O relative to the N atom, B,O-PAHs tend to be less aromatic and more Lewis acidic than corresponding B,N-PAHs.28,34,49–55 (ii) The 1,2-B,O-naphthalenes shown in Scheme 2b possess a relatively low stability due to the electron-rich C atom at the position β to the O atom (“boron enolates”).43–45 The associated peculiar reactivities of certain B,O-PAHs make them valuable synthetic intermediates for aldol reactions, aminations, Suzuki–Miyaura C–C couplings, Chan–Lam C–O couplings, and the formation of lactones by deborylative CO insertion.29,35–37,43,44,47,56
On the other hand, if the β position is embedded into an aromatic benzene ring (as in the cases of the isomeric 2,1-B,O-naphthalenes shown in Scheme 2c) and the B atom is equipped with an appropriate substituent,57 the resulting B,O-PAHs can become sufficiently inert to serve as luminescent materials for optoelectronic applications.31–33,38,39,41 It is also important to note in this context that the steric demand of an O atom is even lower than that of a CH or NH fragment in analogous B- or B,N-PAHs. This promotes a coplanar conformation between the respective B,O-heterocycle and, e.g., B-bonded phenyl substituents and can thus contribute to the increase of conjugation lengths and the extension of the frontier orbitals from the oxaborin moiety to the phenyl ring.38,47,52,58–60
If one proceeds further from B,E-PAHs to (B,E)n-PAHs with E = N or O and n > 1, the available information again becomes dramatically less,9,11,13,30,32,39,54,55,61–78 which is unfortunate, because some evidence has already been gathered that the number n of embedded −BE+ units significantly influences the chemical and physical properties of corresponding compounds.30,54,68,72,79 Thus, new atom-economic strategies for the construction of (B,E)n-PAHs, which ideally avoid the use of sensitive boron halides, sophisticated organometallic reagents, or forcing reaction conditions, are still in demand. The most time and cost-efficient approach would have to be sufficiently modular to implement both E = N and O via closely related reaction protocols. Some of these conditions are met by recently discovered ring-expansion reactions on five-membered ring precursors, which result in the insertion of one E atom into an endocyclic B–C bond. This way, boroles or 9-borafluorenes have been converted to 2,1-B,N/2,1-B,O-benzenes or 10,9-B,N/10,9-B,O-phenanthrenes, respectively.41,53,54,80–85
Inspired by the well-established oxypalladation of alkynes and Sheppard's above-mentioned work,43 our group herein discloses a straightforward protocol for the synthesis of (B,N)n- and (B,O)n-PAHs. The main structural motif present in all synthetic intermediates employed is that of an o-alkynyl-substituted aryl boronic or borinic acid (Scheme 3). These intermediates can either be cyclized directly with the help of a [AuI] catalyst or first transformed into the corresponding aminoboranes and then cyclized using the same [AuI] catalyst. Our approach offers the following advantages: (i) the chemistry of boronic and borinic acids is nowadays very well explored and most derivatives can easily be handled, purified, and stored.86 (ii) The possibility to decide in the last step before cyclization whether a B,N- or B,O-PAH should be prepared guarantees maximum efficiency. (iii) The protocol is applicable to the synthesis of both single and multiple B,E-doped PAHs. Our approach differs from all other strategies in that the cyclization step relies on the formation of C–N/O bonds.
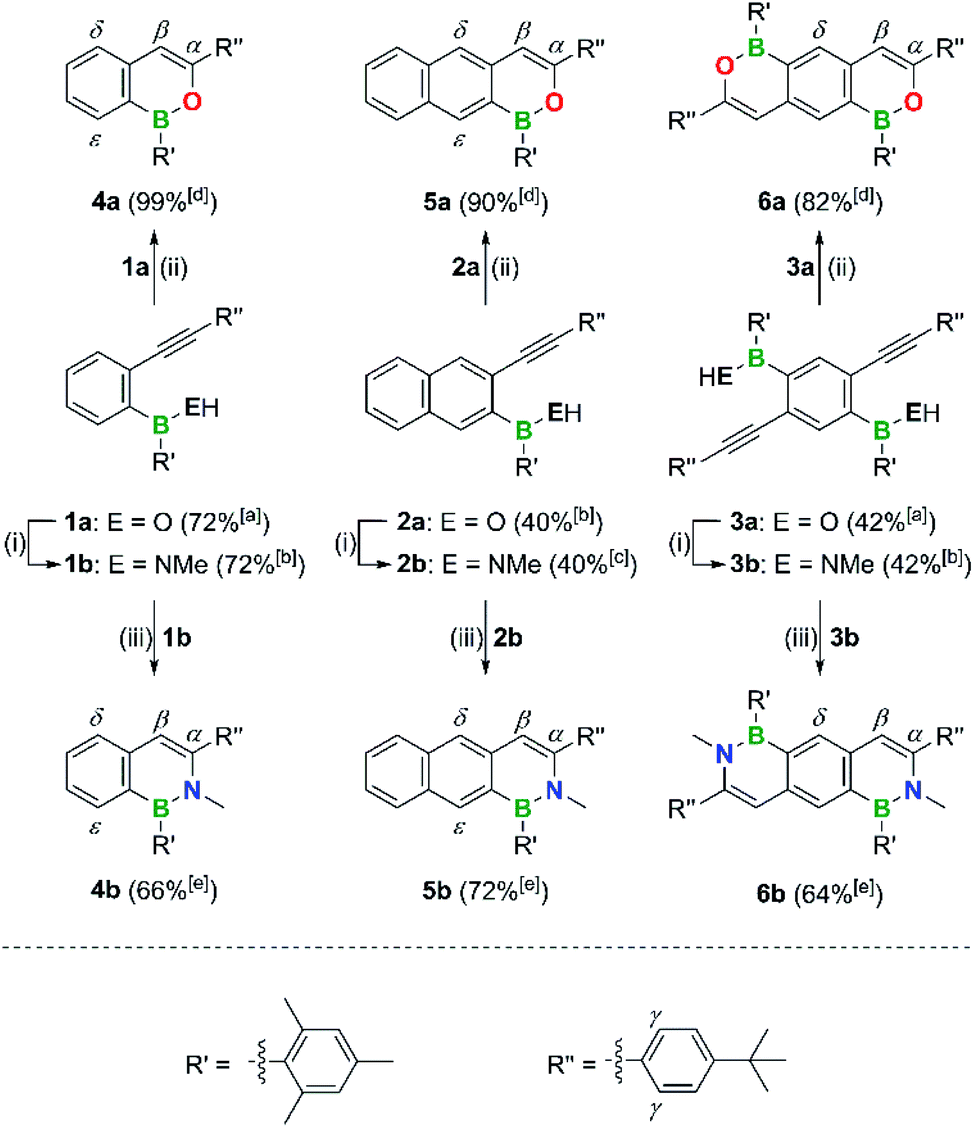 | ||
| Scheme 3 Synthesis of compounds 4a,b–6a,b. Reagents and conditions: (i) 1–2 equiv. (Me3Si)2NMe, C6D6, 120 °C, 2–3 d, sealed NMR tube. (ii) 1–2 mol% [Au(PPh3)NTf2], CHCl3, room temperature, 10 min. (iii) 5 mol% [Au(PPh3)NTf2], CDCl3, 60 °C, 4–48 h. [a] Overall yield over two steps from commercially available chemicals. [b] Overall yield over three steps from commercially available chemicals. [c] Overall yield over four steps from commercially available chemicals. [d] Yield of the final cyclization step. [e] Yield of the final cyclization step without isolation of 1b–3b. | ||
As a proof-of-principle, we have already produced (B,N)2- and (B,O)2-perylenes via our method.55 In our present work, we extend the scope of the reaction to (double) B,N/O-doped naphthalenes and anthracenes. It will also be shown that OBO- and NBO-doped phenalenyls are becoming accessible, the latter having no precedent in the literature.
Results and discussion
Syntheses
The o-alkynyl-substituted aryl borinic and boronic acid precursors 1a–3a and 7a required for the preparation of the (B,O)n- and (B,N)n-PAHs 4a,b–6a,b and 8a–c (Schemes 3 and 4) were synthesized starting from readily accessible mixed bromo(iodo)arenes. Negishi-coupling protocols were employed for the selective introduction of the alkynyl substituents at the iodinated C atoms. The subsequent borylation reactions were performed by Br/Li exchange with tBuLi, quenching of the resulting aryl lithium intermediates with MesB(OMe)2 or B(OMe)3, and aqueous acidic workup (see the ESI† for full details). The electronegative F3C group in 7a,b and 8a–c is a remnant of the synthesis of 2-bromo-1,3-diiodo-5-(trifluoromethyl)benzene from 1-bromo-4-(trifluoromethyl)benzene and served to direct the two I atoms to the ortho positions of the Br substituent during electrophilic aromatic iodination. F3C also proved to be a useful 19F NMR-spectroscopic handle. The tBuC6H4 moieties were chosen with the aim to increase the solubilities of the obtained intermediates and products in organic solvents. Moreover, these substituents offer some steric shielding to the otherwise fully exposed reactive β-CH sites (see above). Finally, the intense tBu signals in the sparsely populated alkyl regions of the 1H NMR spectra of 1a,b–8a–c effectively reveal the presence of possible side products and are thus a good measure of the selectivities of the desired conversions. | ||
| Scheme 4 Synthesis of compounds 8a–c. Reagents and conditions: (i) 1.5 equiv. (Me3Si)2NMe, C6D6, 120 °C, 2–3 d, sealed NMR tube. (ii) 1 mol% [Au(PPh3)NTf2], CHCl3, room temperature, 4–6 h. (iii) 2 mol% [Au(PPh3)NTf2], CDCl3, 60 °C, 4 h. (iv) 5 drops TfOH (approx. 1 equiv.), CH2Cl2, room temperature, 5 min. [a] Yield over three steps from commercially available chemicals. [b] Yield over four steps from commercially available chemicals. [c] Yield of the final cyclization step. [d] Yield of the final cyclization step without isolation of 7b. | ||
The B,O precursors 1a–3a were cyclized using the catalyst [Au(PPh3)NTf2] in CHCl3.43,55 Regardless of the presence of the bulky Mes substituents, which serve to protect the B atoms during workup, the reactions took place already at room temperature, required no more than a few minutes for completion, and gave yields in the range 82–99% (Scheme 3; all cyclizations were quantitative according to NMR).
The mono- or ditopic borinic acids 1a–3a were transformed into the corresponding (methylamino)boranes by heating with (Me3Si)2NMe in C6D6 (1–2 equiv., 120 °C, 2–3 d, sealed NMR tube). After the quantitative conversion was confirmed by NMR spectroscopy, the solvent was changed to CDCl3, [Au(PPh3)NTf2] was added and the mixture heated to 60 °C for 4–48 h while monitored by NMR spectroscopy. After workup, 4b–6b were isolated in yields of 64–72% over both steps (Scheme 3).
The boronic acid 7a turned out to be a special case: although a double cyclization reaction is possible and affords the B,O2-PAH 8a in 86% yield, it requires a reaction time of several hours rather than minutes at room temperature (Scheme 4).
Even upon heating of 7a with 2.5 equiv. of (Me3Si)2NMe, the amination reaction stopped at the stage of the methylamino(hydroxy)borane 7b (Scheme 4); for the subsequent targeted synthesis of 7b, 1.5 equiv. of (Me3Si)2NMe were used (C6D6, 120 °C, 2–3 d). Treatment of 7b with 2–25 mol% of [Au(PPh3)NTf2] resulted in a 6-endo-dig addition of the BN–H bond across the CC bond (60 °C, 4–48 h; 8c), whereas no B,O-doped ring was formed; the optimized protocol for the synthesis of 8c uses 2 mol% [Au(PPh3)NTf2] and a reaction time of 4 h. The complete cyclization of 8c to the B,N,O-phenalenyl 8ab was finally achieved by addition of F3CSO3H (TfOH).87,88
These results are remarkable in several respects: (i) 8ab is the first B,N,O-PAH available so far. (ii) When the reactions are independent, the ring closure to the (B,O)n-PAHs with the [Au(PPh3)NTf2] catalyst is generally faster than the formation of the corresponding (B,N)n-PAHs. However, for 7b, when there is now intramolecular competition between N–H and O–H, cyclization to the B,N heterocycle is preferred.89 (iii) TfOH can also be a suitable cyclization catalyst and in this particular example it is even superior to [Au(PPh3)NTf2].
The closest relatives of our B,O-naphthalene 4a are Sheppard's boron enolates shown in Scheme 2b.43 Some of these derivatives (e.g., R′′ = H,45nBu43) were characterized by NMR spectroscopy, IR spectroscopy, and high resolution mass spectrometry. The optoelectronic properties were not investigated. Yet, the presence of a strongly π-donating OH substituent at the B atom of Sheppard's enolates should lead to a lesser extent of double bonding in the endocyclic B–O bond than in the case of 4a, which carries an electronically more innocent Mes substituent.90 A doubly benzannulated congener of the B,O2-doped phenalenyl 8a was prepared by Hatakeyama via demethylative direct borylation of 2,2′′-dimethoxy-1,1′:3′,1′′-terphenyl (BBr3/TMP, 180 °C, 18 h; TMP = 2,2,6,6-tetramethylpiperidine; see Scheme 2a for a related reaction).31 No precedence exists for the B,O- and (B,O)2-anthracenes 5a and 6a.
Fig. 1 shows the sequence of relative thermodynamic stabilities of all conceivable parental B,N-naphthalene isomers according to quantum-chemical calculations (R1–R8 = H);91 substitution has been achieved at the indicated positions. We note that the energetically favorable 2,1-B,N- and 1,2-B,N-naphthalenes differ from the other known isomers by the presence of unperturbed aromatic C6 rings.3
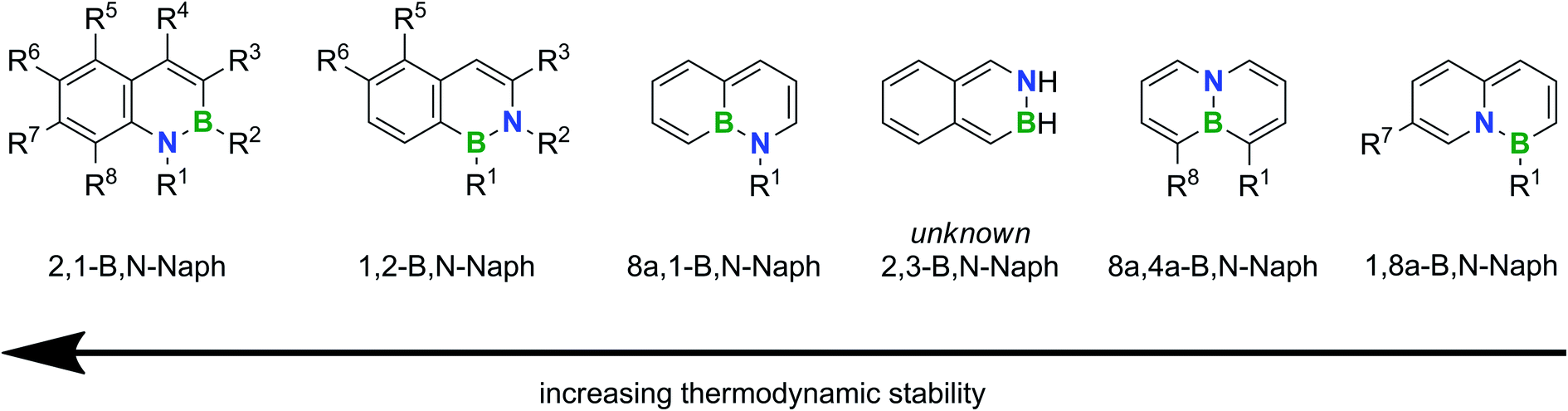 | ||
| Fig. 1 Relative thermodynamic stabilities of B,N isosters of naphthalene containing one B,N unit. Substitutions have been achieved at the indicated positions. | ||
The most stable 2,1-B,N-naphthalenes are also by far the most thoroughly studied isomers.18,19,92–103 The dominant synthesis strategies rely on ring-closing reactions between o-vinyl-/o-alkynylanilines and R′BCl2 or K[R′BF3]/SiCl4/NEt3 (cf.Scheme 1a; R′ = halogen, alkyl, alkenyl, alkynyl, (hetero)-aryl).18,19,92,93,95,99,102 An “aromatic metamorphosis” approach (cf.Scheme 2c) is based on reductive ring opening of indoles with excess Li metal, trapping of the dilithiated intermediate with RBpin, and aqueous workup.103
Apart from our current work on the second most stable 1,2-B,N-naphthalenes, the only other report comes from the Cui group.104 They used benzylic imines as key intermediates (3 synthesis steps) and converted them into the corresponding enamidyl dibromoboranes, which were cyclized via an Et3N-promoted intramolecular electrophilic aromatic borylation reaction at 80 °C (2 steps; yields: 30–51% with respect to the corresponding benzylic imine). The authors demonstrated a scope of their reaction sequence across the substituents R1 = alkynyl/(hetero)aryl, R2 = aryl, R3 = tBu/Ph, R5 = F and R6 = Me.
Apart from the B,N-anthracenes 5b and 6b, their orientational isomers 2,1-B,N-anthracene and 2,6-B2-1,5-N2-anthracene are known. The air-stable compounds are accessible through (double) borylative cyclization of 2-amino-3-vinylnaphthalene or 1,4-diamino-2,5-divinylbenzene with BCl3. Subsequent Cl/H exchange using LiAlH4 afforded the target molecules in about 80% yield over the last two steps.79
To conclude, our novel synthesis approach for (B,N)n- and (B,O)n-doped PAHs (i) provides previously inaccessible positional isomers in yields competitive with any alternative cyclization reaction developed to-date, (ii) is modular, as it allows the synthesis of (B,E)n derivatives with E = N and O starting from the same late precursors, and (iii) takes advantage of well-established borinic and boronic acid chemistry, avoiding the use of corrosive, air- and moisture-sensitive haloboranes.
NMR-spectroscopic and X-ray crystallographic characterization of 4a,b–6a,b and 8a–c
All spectra were run in CDCl3.105 In the 1H NMR spectra of 4a,b–6a,b and 8a–c, the successful cyclization reactions are indicated by the presence of singlet resonances in the aromatic regions, which are due to the newly generated Hβ protons (see Schemes 3 and 4 for the position labels). The 13C{1H} NMR spectra of 4a,b–6a,b no longer contain signals characteristic of C(sp) atoms; instead, new resonances appear that belong to Cβ (4a–6a: av. 106.2 ppm; 4b–6b: av. 113.4 ppm) and Cα (4a–6a: av. 151.8 ppm; 4b–6b: av. 145.7 ppm). Intense cross peaks between the NCH3 proton signals and the Cα signals are observed in all H,CHMBC spectra of 4b–6b. The 11B NMR shift values of 4a,b–6a,b are found in the expected region of 39–46 ppm.106The B,O2-phenalenyl 8a shows NMR features similar to those of the B,O-naphthalene 4a (8a: δ(Hβ) = 6.97, δ(Cβ) = 104.2, δ(Cα) = 154.2), but its 11B signal is significantly shifted to higher field as a result of better magnetic shielding by the two π-donating O atoms (8a: 29 ppm vs.4a: 46 ppm). For the singly ring-closed compound 8c, we observe simultaneously resonances attributable to CHβ and Cα units and those attributable to two alkynyl-C atoms. An H,CHMBC experiment proved the formation of a 1,2-B,N-naphthalene core with dangling OH substituent (δ(OH) = 7.14). The 1H NMR spectrum of the B,N,O-phenylene 8ab, in contrast, lacks an OH signal and instead shows two well-resolved Hβ resonances at 6.34 and 6.89 ppm. While two Cβ resonances appear at 104.2 and 108.1 ppm, alkynyl-C signals are not detectable in the 13C{1H} NMR spectrum of 8ab.
Further comparison of the NMR spectra of 4a–6a on the one hand and 4b–6b on the other reveals additional remarkable differences in the molecular and electronic structures as a function of B,O- vs. B,N-doping. While the 1H and 13C chemical shift values of the Mes substituents are largely the same, especially the CHγ values of the tBuC6H4 groups of 4a–6a (δ(Hγ) = av. 7.94 ppm; δ(Cγ) = av. 125.2 ppm) are significantly different from those of 4b–6b (δ(Hγ) = av. 7.39 ppm; δ(Cγ) = av. 129.1 ppm). The same is true for the B,O-half of 8ab (δ(Hγ) = 7.87 ppm; δ(Cγ) = 125.3 ppm) compared to its B,N-half (δ(Hγ) = 7.36 ppm; δ(Cγ) = 128.9 ppm). These differences can be explained by different conformations of the tBuC6H4 substituents with respect to the heterocyclic cores in 4a,b–6a,b: the small O atom allows an essentially coplanar arrangement in solution, whereas the larger NMe group enforces a twist between the two moieties (the bulky Mes rings are consistently orthogonally positioned). Different conformations between 4a–6a and 4b–6b are also observed in the solid state and are relevant for the interpretation of the optoelectronic properties of the B,O- vs. B,N-doped species (see below).
Looking at 4a,b/5a,b, we find that the Cε atoms are deshielded by about 10 ppm with respect to the Cδ atoms, regardless of whether the compounds contain O or N atoms (δ(Cδ) = 126.4 (4a), 126.1 (4b), 124.0 (5a), 123.4 ppm (5b); δ(Cε) = 136.5 (4a), 135.5 (4b), 138.9 (5a), 136.8 ppm (5b)). Given that the δ(Cδ) values are close to that of C6H6 (128.4 ppm), the +M effect of O and N does not seem to reach out to this position, whereas the π-charge density at Cε is apparently reduced due to the −M effect of B. In line with this interpretation, the two equivalent CH atoms of the benzene cores of 6a,b, which should experience the ±M effects of both dopant elements, are also considerably deshielded (134.7 (6a), 133.2 ppm (6b)). We also note that plots of the highest occupied molecular orbitals (HOMOs) of 4a,b/5a,b consistently indicate a larger contribution of the pz orbital located at Cδ than of the pz orbital located at Cε (see the ESI†).
The product molecules 4a, 6a, 8a and 4b, 5b have been characterized by X-ray crystallography (Fig. 2; the solid-state structures of numerous intermediates are compiled in the ESI†). As a general feature, the B–O distances (1.368(4)–1.388(2) Å) are significantly shorter than the B–N distances (1.417(5)–1.435(7) Å), and this is even true for the boronic acid ester 8a, in which two π-donating O atoms compete for the same pz(B) acceptor orbital. Apart from different grades of −BE+ double-bond character (E = O, N), other influencing factors are the smaller covalent radius of the O atom compared to the N atom (which also carries a Me substituent here) and a lower degree of intramolecular steric repulsion in the B,O-doped compounds. In agreement with this latter factor and the NMR data discussed above, we find much smaller dihedral angles between the B,O heterocycles and the tBuC6H4 substituents in 4a, 6a, 8a (14.4(2)–33.6(1)°) than between the B,N heterocycles and the tBuC6H4 ring in 4b, 5b (82.1(2)–83.0(1)°).
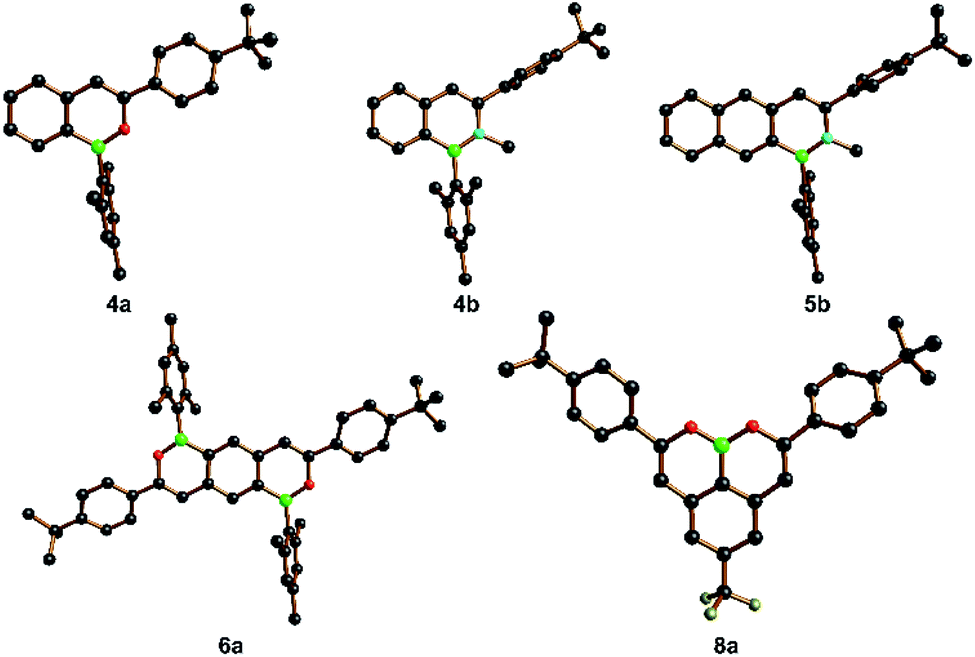 | ||
| Fig. 2 Crystallographically determined solid-state structures of 4a, 4b, 5b, 6a, and 8a; H atoms were omitted for clarity. Selected bond lengths [Å] and dihedral angles [°]: 4a: B–O = 1.388(2), C(α)–C(β) = 1.345(2); BO ring//mesityl = 83.1(1), BO ring//tBuC6H4 = 17.0(1). 4b: B–N = 1.417(5), C(α)–C(β) = 1.356(6); BN ring//mesityl = 78.0(1), BN ring//tBuC6H4 = 83.0(1). 5b: B–N = 1.435(7), C(α)–C(β) = 1.328(7); BN ring//mesityl = 85.3(2), BN ring//tBuC6H4 = 82.1(2). 6a: B–O = 1.374(3), C(α)–C(β) = 1.340(3); BO ring//mesityl = 75.7(1), BO ring//tBuC6H4 = 28.3(1). 8a: B–O = 1.368(4)/1.381(5), C(α)–C(β) = 1.344(5)/1.345(4); BO ring//tBuC6H4 = 14.4(2)/33.6(1). | ||
A comparison of the C–C distances in 4a, 6a, 8a and 4b, 5b also provides insight into the degree of cyclic π-delocalization along the molecules' cores: all Cα–Cβ distances fall in the range 1.328(7)–1.356(6) Å, which is indicative for largely isolated CαCβ double bonds (ideal value: 1.34 Å).107 The adjacent Cβ–Ar distances, in turn, are consistently longer (1.437(6)–1.447(7) Å) than CαCβ and also longer than all C–C distances in the annulated benzene rings. Taken together, these data point toward Clar's sextets in the carbonaceous six-membered rings and attached –BE–CC– heterobutadiene fragments. We finally note that the −BN+ bond lengths found in our 1,2-B,N-naphthalene 4b and 1,2-B,N-anthracene 5b do not differ from those in corresponding 2,1-B,N-naphthalenes/anthracenes and are thus not influenced by the orientation of the B,N pairs.95,96,108
Optoelectronic properties of 4a,b–6a,b and 8a, 8ab
Of the newly synthesized B,O- and B,N-PAHs compiled in Schemes 3 and 4, only the B,E-anthracenes 5a,b and the (B,N)2-anthracene 6b show (quasi)reversible redox waves in their cyclic voltammograms (vs. FcH/FcH+; THF, room temperature, supporting electrolyte: 0.1 M [nBu4N][PF6]; Table 1). 5a (E1/2 = −2.58 V) is easier to reduce by 220 mV than 5b (E1/2 = −2.80 V), likely due to the higher electronegativity of the O atom compared to the N atom and the extension of the conjugation pathway into the coplanar tBuC6H4 ring (cf. the conformations adopted by 4avs.4b in the solid state; Fig. 2). For a comparable 2,1-B,N-anthracene bearing a mesityl group on boron as the sole substituent, a peak cathodic potential of Ep,c = −2.59 V (DMF) was reported.108 These data indicate that the electrochemical properties of the B,E-anthracenes are not only influenced by the choice of E = N or O but also by subtle effects resulting from the orientation of the introduced B,E pairs.12 Reduction of the (B,N)2-anthracene 6b occurs at E1/2 = −3.00 V and is thus somewhat harder to achieve than the reduction of the B,N-anthracene 5b (E1/2 = −2.80 V).| λ abs [nm] (ε [M−1 cm−1]) | λ onset [nm] | λ em [nm] | Φ PL [%] | E optG [eV] | E′DFTGe [eV] | E 1/2 [V] | E CVLUMO [eV] | E′DFTLUMOg [eV] | E′DFTHOMOh [eV] | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Each onset wavelength (λonset) was determined by constructing a tangent on the point of inflection of the bathochromic slope of the most red-shifted absorption maximum. b Resolved vibrational fine structure. c Quantum yields were determined by using a calibrated integrating sphere. d Optical band gap EoptG = 1240/λonset. e For better comparability with the EoptG values, the computed energy gaps (EDFTG) have been scaled according to the following linear equation: E′DFTG = 0.65 × EDFTG + 0.48. f E CVLUMO = −4.8 eV − E1/2 (FcH/FcH+ = −4.8 eV vs. vacuum level). g For better comparability with the ECVLUMO values, the computed LUMO energies (EDFTLUMO) have been scaled according to the following linear equation: E′DFTLUMO = 0.65 × EDFTLUMO −1.20. h Scaled HOMO energies (E′DFTHOMO) were calculated from the scaled LUMO energies (E′DFTLUMO) and the scaled HOMO–LUMO energy gaps (E′DFTHOMO = E′DFTLUMO − E′DFTG). i Quantum yields measured in c-hexane. sh = shoulder. | ||||||||||
| 5a | 290 (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 925) 925) |
419 | 426 (sh) | 20 | 2.96 | 2.87 | −2.58 | −2.22 | −2.23 | −5.10 |
| 297 (45316) |
445 | 31i | ||||||||
| 328 (sh) | ||||||||||
| 339 (34508) |
||||||||||
| 351 (20644) |
||||||||||
| 379 (sh) | ||||||||||
| 395 (sh) | ||||||||||
| 5b | 268 (44824) |
422 | 428 (sh) | 32 | 2.94 | 2.93 | −2.80 | −2.00 | −2.03 | −4.96 |
| 326 (9621) | 448 | 51i | ||||||||
| 341 (12571) |
470 (sh) | |||||||||
| 379 (sh) | ||||||||||
| 405 (sh) | ||||||||||
| 6a | 312 (sh) | 447 | 452 | 41 | 2.77 | 2.73 | — | — | −2.27 | −5.00 |
| 326 (sh) | 475 | 49i | ||||||||
| 343 (sh) | 507 (sh) | |||||||||
| 360 (75215) |
||||||||||
| 373 (56253) |
||||||||||
| 404 (13108) |
||||||||||
| 425 (8628) | ||||||||||
| 6b | 347 (27027) |
422 | 426 | 16 | 2.94 | 2.98 | −3.00 | −1.80 | −1.80 | −4.78 |
| 388 (sh) | 446 | 52i | ||||||||
| 8a | 270 (13015) |
399 | 397 | 43 | 3.11 | 3.02 | — | — | −2.20 | −5.22 |
| 314 (25351) |
419 | 52i | ||||||||
| 329 (sh) | 443 | |||||||||
| 366 (5963) | 468 | |||||||||
| 386 (4312) | 510 (sh) | |||||||||
| 8ab | 307 (30228) |
412 | 398 (sh) | 34 | 3.01 | 2.98 | — | — | −2.03 | −5.01 |
| 315 (sh) | 415 | 44i | ||||||||
| 329 (27393) |
434 | |||||||||
| 374 (10481) |
461 (sh) | |||||||||
| 393 (sh) | 491 (sh) | |||||||||
UV/vis spectra were recorded for 4a,b–6a,b and 8a, 8ab (CHCl3); photoluminescence spectra of these compounds were measured both in CHCl3 and c-hexane (Fig. 3 and Table 1). The naphthalene derivatives 4a,b show low photoluminescence quantum efficiencies of ΦPL < 10% and will therefore not be discussed further. We first focus on the onsets of absorbance (λonset), since these are directly correlated with the HOMO–LUMO energy gaps (EoptG). The λonset values of 5a/5b (419/422 nm) are almost identical, which is also true for the emission wavelengths λem (426/428 nm). Thus, despite their different LUMO levels, EoptG is the same for the two B,E-anthracenes. In contrast, both the onset of absorbance and the emission wavelength are bathochromically shifted for 6a compared to 6b (λonset = 447 vs. 422 nm; λem = 452 vs. 426 nm). Here, the impact of two vs. zero coplanar tBuC6H4 substituents and an associated enlarged π-electron system in 6a may play a role.
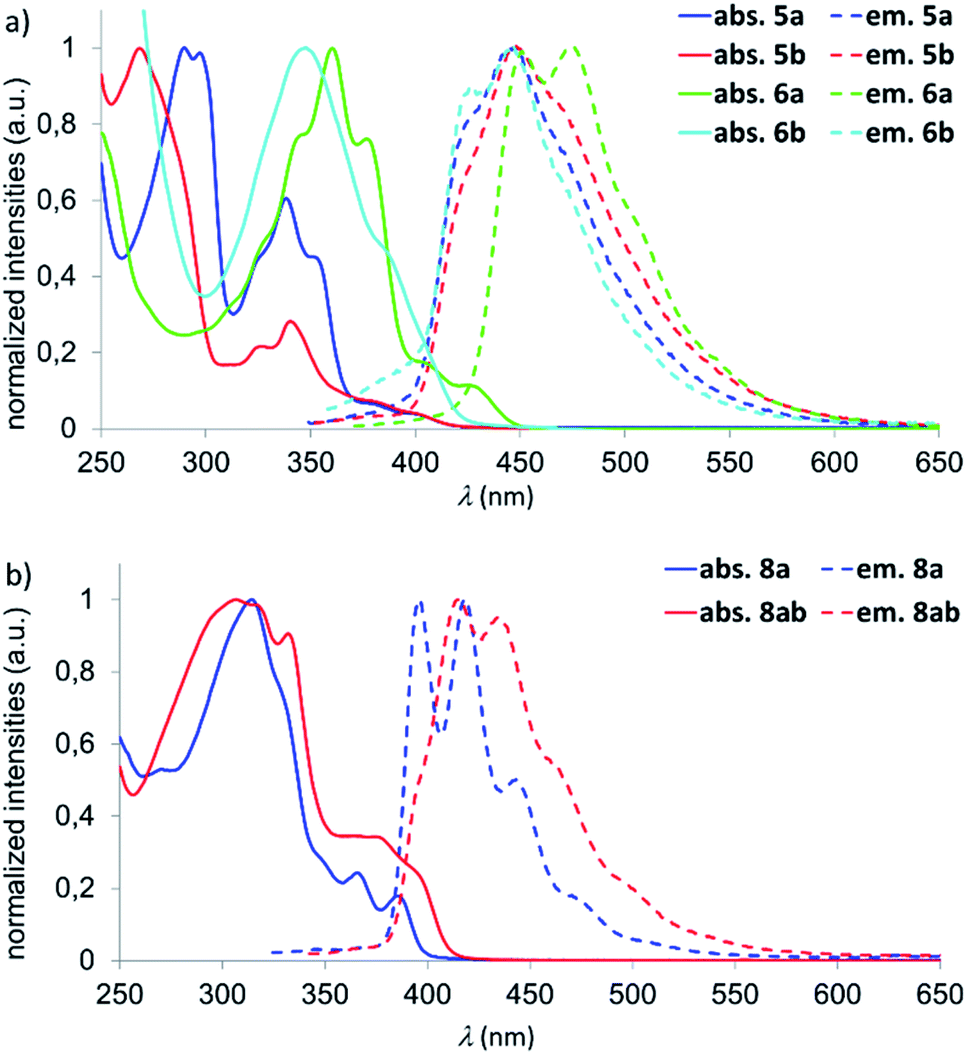 | ||
| Fig. 3 (a) Normalized UV/vis absorption (solid lines) and emission (dashed lines) spectra in CHCl3 of compounds 5a,b–6a,b. (b) Normalized UV/vis absorption (solid lines) and emission (dashed lines) spectra in CHCl3 of compounds 8a and 8ab. | ||
If recorded in c-hexane, all emissions are detected at somewhat shorter wavelengths compared to measurements in CHCl3 (Δ(λem) = 260–920 cm−1), the vibrational fine structures are even better resolved, and quantum efficiencies of ΦPL = 31% (5a), 51% (5b), 49% (6a), and 52% (6b) are obtained. The trends in the experimentally determined optical band gaps EoptG(5a) ≈ EoptG(5b) ≈ EoptG(6b) > EoptG(6a) are well reproduced by quantum-chemical calculations (cf. the scaled values E′DFTG in Table 1). The calculations also confirm a less cathodic reduction potential of 5a compared to 5b, reflected by the lower computed LUMO energy of 5a.
The gas-phase structures and frontier-orbital configurations of 6a/6b and their carbonaceous congener 6 are exemplarily shown in Fig. 4. Orthogonally positioned Mes rings are seen throughout, while the tBuC6H4 substituents in 6a are less twisted with respect to the heterocyclic core than in 6b, which agrees well with the X-ray crystallography results (Fig. 2). Compound 6 possesses a smaller energy gap E′DFTG = 2.63 eV than 6a (2.73 eV) and 6b (2.98 eV). As an important difference between our B,N-anthracenes 5b/6b and Liu's positional isomers 2,1-B,N-anthracene and 2,6-B2-1,5-N2-anthracene, the π systems of the CαCβ bonds contribute strongly to the HOMOs of the former compounds (Fig. 4) but only negligibly to those of the latter.79
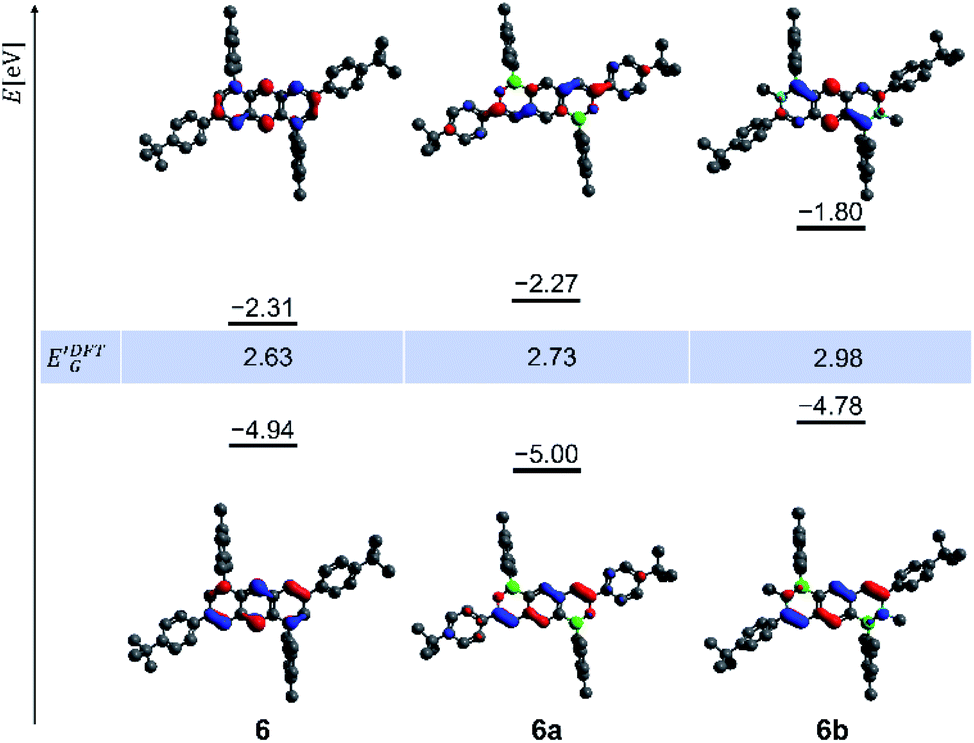 | ||
| Fig. 4 DFT-calculated nodal structures and scaled energy levels of the frontier orbitals of the parental 1,5-dimesityl-3,7-di(tBuC6H4)anthracene (6), 6a, and 6b (HOMOs bottom, LUMOs top; isovalue of the isosurface plots: 0.05 a0−3/2; B3LYP/6-31G*). | ||
The optical properties of the B,O2- and B,N,O-phenalenyls are similar and therefore do not require further discussion (Table 1 and Fig. 3b). However, we note pleasingly high photoluminescence quantum efficiencies of ΦPL = 52% (8a) and 44% (8ab) in c-hexane.
Late-stage derivatization of the (B,O)2-perylene 9
With increasing knowledge about B,N-PAHs it becomes more and more obvious that functional groups do not necessarily have to be introduced before the cyclization step. Late stage derivatization, a widely used tool in PAH chemistry, is also possible and often remarkably regioselective (see corresponding reactions on 2,1-B,N-benzenes and 2,1-B,N-naphthalenes).16 Given the higher reactivity of B,O- compared to B,N-PAHs (see above), we were interested in exploring whether late-stage functionalizations of the former are also possible, while maintaining the structural integrity of the heterocyclic scaffold. For this purpose, we selected the parent (B,O)2-perylene 9 (Scheme 5), which allows a direct comparison with its highly symmetric carbonaceous counterpart. As functional group to be introduced, the pinacolatoboryl (Bpin) substituent was chosen.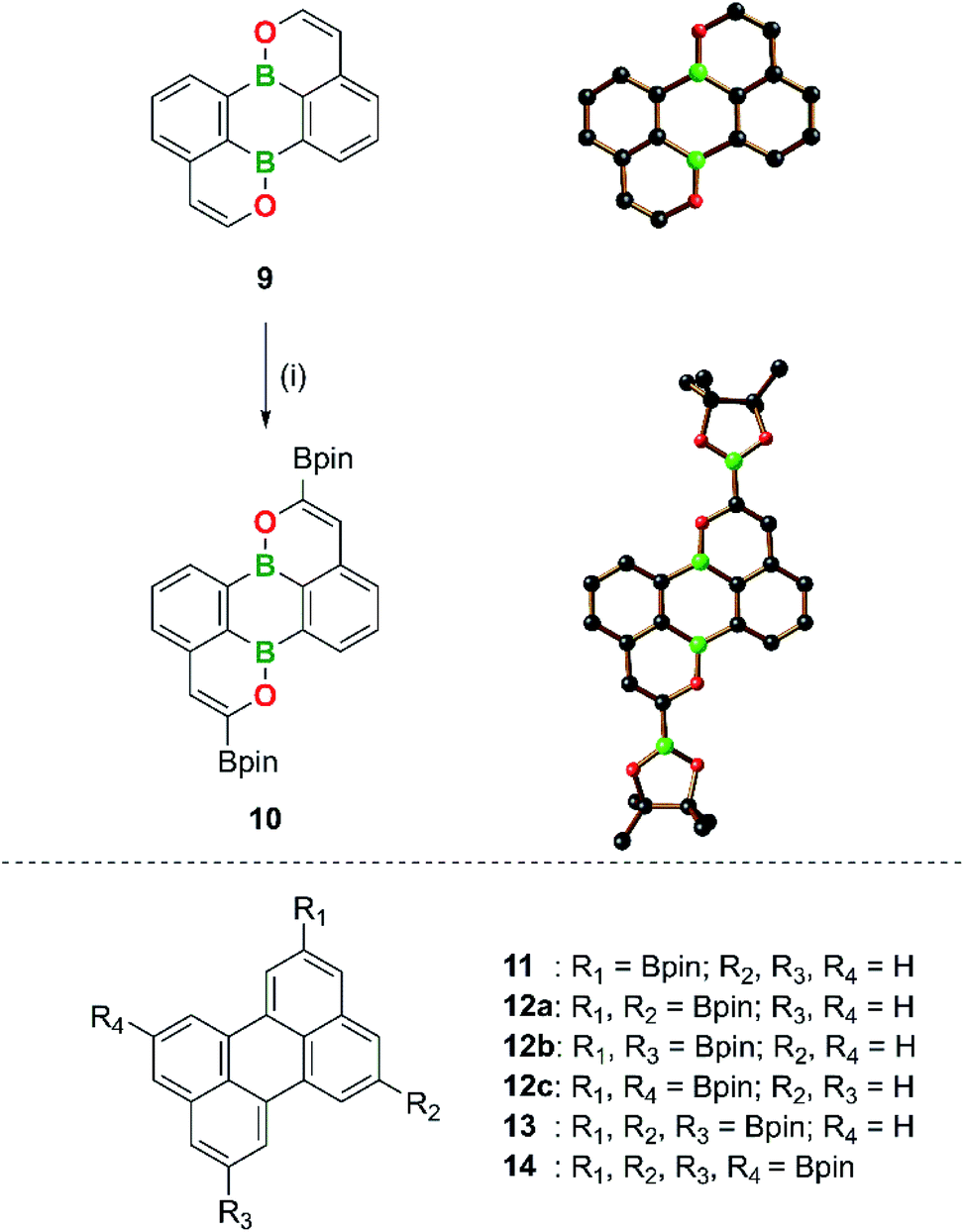 | ||
| Scheme 5 Transformation of (B,O)2-perylene 9 to the doubly Bpin-substituted (B,O)2-perylene 10 by Ir-catalyzed borylation and their structures in the solid state. Reagents and conditions: (i) 3.1 equiv. B2pin2, 3 mol% [Ir(COD)(μ-OMe)]2, 6 mol% dtbpy, THF, 80 °C, 42 h. | ||
Similar to other (B,O)2-perylenes,55 compound 9 was synthesized by double cyclization of the diethynylated 9,10-dihydroxy-9,10-dihydro-9,10-diboraanthracene derivative using the [Au(PPh3)NTf2] catalyst (Scheme 5).109 The successful preparation of 9 thus demonstrates the applicability of our method to substrates carrying terminal alkynes (see the ESI† for full details). Treatment of 9 with 3.1 equiv. of B2pin2 in the presence of the catalyst system 1/2 [Ir(COD)(μ-OMe)]2/dtbpy110–112 led to a quantitative (TLC) double C–H borylation at the two C atoms in α-position to the O atoms and gave the corresponding product 10 in 63% yield after workup (Scheme 5; COD = 1,5-cyclooctadiene, dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine). No mono-, tri-, or tetraborylated derivatives were observed; an increase in the amount of added B2pin2 to 5 equiv. did not lead to higher borylation levels either. The regioselectivity of the functionalization reaction was confirmed by X-ray crystallography (Scheme 5) and NMR spectroscopy. 10 gives rise to two resonances in the 11B NMR spectrum and four resonances in the aromatic region of the 1H NMR spectrum. Three doublets of doublets (each integrating 2H) can be assigned to the C6H3 fragments and the remaining singlet (2H) to the protons at the positions β to the O atoms (H,CHMBC experiment).
The regioselectivity of the borylation of 9 is based on two factors: (i) the key intermediate of the catalytic cycle, fac-[Ir(dtbpy)(Bpin)3], is a rather bulky metal complex that avoids performing the rate-determining C–H-activation step at sterically encumbered positions ortho to ring junctions or other substituents.113,114 Activation of the peri-C–H bonds of 9 (or perylene) is therefore unfavorable. (ii) Of the four remaining C–H bonds, the two adjacent to the electronegative O atom, which is also smaller than a neighboring C–H unit, react preferentially.115 In contrast to the selective diborylation of 9, the corresponding reaction between carbonaceous perylene and 2.2 equiv. of B2pin2 furnished a mixture of mono- (11), di- (three isomers, 12a–c), tri- (13), and tetraborylated (14) products (Scheme 5). While the perylenes carrying different numbers of Bpin substituents have been separated by HPLC, it has been found impossible to separate 12a–c. On the other hand, the 2,5,8,11-tetraborylated perylene 14, which does not (yet) have a (B,O)2-analogue, was accessible in 83% yield by using 4.4 equiv. of B2pin2 and the Ir catalyst (Scheme 5).114,116–118
Conclusions
We have shown that the mild Au(I)-mediated cyclization of aryl(amino)boranes or aryl boronic and borinic acids with ortho-positioned CC bonds is an ideal tool for the synthesis of singly and doubly B,N- or B,O-doped polycyclic aromatic hydrocarbons (PAHs). We see the following advantages of this new method over many of the existing protocols: (i) boronic and borinic acids, which are nowadays standard reagents in every synthesis-oriented laboratory, serve as starting materials. (ii) The use of sophisticated organometallic reagents or corrosive boron halides is avoided. (iii) Modularity is achieved by the fact that the aminoboranes required for the synthesis of the B,N-PAHs are accessible in one step from the boronic and borinic acids required for the fabrication of the B,O-PAHs. The cyclization protocols presented herein thus represent a significant addition to the currently available toolbox for doping PAHs with main-group elements. Moreover, by using an unsubstituted (B,O)2-perylene as the substrate for Ir-catalyzed C–H borylation, we (i) present a rare example of late-stage functionalization of a B,O-PAH and (ii) show that the exchange of two CC for −BO+ bonds leads to dramatically improved product selectivity compared to the case of the carbonaceous perylene.
Author contributions
T. K. performed the late-stage derivatization of compound 9; O. O. synthesized and characterized all other compounds. M. B. performed the X-ray crystal structure analyses. H.-W. L. and M. W. supervised the project. The manuscript was written by M. W. and O. O. and edited by all the co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Aleksandar Lucic for his assistance with manuscript preparation.Notes and references
- (a) Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons, Ltd., Chichester, UK, 1st edn, 2017 Search PubMed; (b) S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363 DOI:10.1002/adsc.202001356; (c) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS; (d) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (e) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (f) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (g) S. Yamaguchi, A. Fukazawa and M. Taki, J. Synth. Org. Chem., 2017, 75, 1179–1187 CrossRef CAS; (h) P. Zhao, D. O. Nettleton, R. G. Karki, F. J. Zécri and S. Y. Liu, ChemMedChem, 2017, 12, 358–361 CrossRef CAS PubMed; (i) L. Schweighauser and H. A. Wegner, Chem.–Eur. J., 2016, 22, 14094–14103 CrossRef CAS PubMed; (j) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (k) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS; (l) J. Kahlert, C. J. D. Austin, M. Kassiou and L. M. Rendina, Aust. J. Chem., 2013, 66, 1118–1123 CrossRef CAS; (m) B. C. Das, P. Thapa, R. Karki, C. Schinke, S. Das, S. Kambhampati, S. K. Banerjee, P. van Veldhuizen, A. Verma, L. M. Weiss and T. Evans, Future Med. Chem., 2013, 5, 653–676 CrossRef CAS PubMed; (n) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (o) S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed.
-
(a) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed;
(b) J. Huang and Y. Li, Front. Chem., 2018, 6, 1–22 CrossRef PubMed;
(c) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 2017, 4353–4368 CrossRef;
(d) H. Helten, Chem.–Eur. J., 2016, 22, 12972–12982 CrossRef CAS PubMed;
(e) X.-Y. Wang, J.-Y. Wang and J. Pei, Chem.–Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed;
(f) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed;
(g) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef;
(h) Z. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed. For selected B- and N-doped polycyclic aromatic hydrocarbons that are devoid of direct −BN+ bonds and show exciting optoelectronic properties, see:
(i) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed;
(j) S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087–5090 CrossRef CAS PubMed;
(k) X. Liang, Z.-P. Yan, H.-B. Han, Z.-G. Wu, Y.-X. Zheng, H. Meng, J.-L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed;
(l) K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed;
(m) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Various numbering schemes have been used in the literature to indicate the positions (x,y) and orientations of the B,E pairs in x,y-B,E-doped PAHs (E = N, O). In this publication, we use the conventional IUPAC scheme for numbering the C atoms in the parental carbonaceous PAHs. We will also always name the B atom first and adapt the sequence of the numbers x and y in the compounds' names accordingly (see Fig. 1).
- C. A. Jaska, D. J. H. Emslie, M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, J. Am. Chem. Soc., 2006, 128, 10885–10896 CrossRef CAS PubMed.
- M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, Angew. Chem., Int. Ed., 2007, 46, 4940–4943 CrossRef CAS PubMed.
- M. J. D. Bosdet, C. A. Jaska, W. E. Piers, T. S. Sorensen and M. Parvez, Org. Lett., 2007, 9, 1395–1398 CrossRef CAS PubMed.
- A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS PubMed.
- A. Chrostowska, S. Xu, A. N. Lamm, A. Mazière, C. D. Weber, A. Dargelos, P. Baylère, A. Graciaa and S.-Y. Liu, J. Am. Chem. Soc., 2012, 134, 10279–10285 CrossRef CAS PubMed.
- B. Neue, J. F. Araneda, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2013, 52, 9966–9969 CrossRef CAS PubMed.
- E. R. Abbey, A. N. Lamm, A. W. Baggett, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2013, 135, 12908–12913 CrossRef CAS PubMed.
- X. Y. Wang, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 7668–7671 CrossRef CAS PubMed.
- J. S. A. Ishibashi, A. Dargelos, C. Darrigan, A. Chrostowska and S.-Y. Liu, Organometallics, 2017, 36, 2494–2497 CrossRef CAS.
- H. Lin, C. R. McConnell, B. Jilus, S.-Y. Liu and F. Jäkle, Macromolecules, 2019, 52, 4500–4509 CrossRef CAS.
- A. S. Scholz, J. G. Massoth, M. Bursch, J.-M. Mewes, T. Hetzke, B. Wolf, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, J. Am. Chem. Soc., 2020, 142, 11072–11083 CrossRef CAS PubMed.
- M. Chen, K. S. Unikela, R. Ramalakshmi, B. Li, C. Darrigan, A. Chrostowska and S.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 1556–1560 CrossRef CAS PubMed.
- C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC.
- T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS PubMed.
- S. R. Wisniewski, C. L. Guenther, O. A. Argintaru and G. A. Molander, J. Org. Chem., 2014, 79, 365–378 CrossRef CAS PubMed.
- F.-D. Zhuang, J.-M. Han, S. Tang, J.-H. Yang, Q.-R. Chen, J.-Y. Wang and J. Pei, Organometallics, 2017, 36, 2479–2482 CrossRef CAS.
- For further examples of aromatic borylations, see: (a) M. J. S. Dewar, V. P. Kubba and R. Pettit, J. Chem. Soc., 1958, 3073–3076 RSC; (b) X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed; (c) X.-Y. Wang, F.-D. Zhuang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, Chem. Commun., 2015, 51, 4368–4371 RSC; (d) X.-Y. Wang, D.-C. Yang, F.-D. Zhuang, J.-J. Liu, J.-Y. Wang and J. Pei, Chem.–Eur. J., 2015, 21, 8867–8873 CrossRef CAS PubMed; (e) A. John, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 5588–5592 CrossRef CAS PubMed; (f) J. S. A. Ishibashi, C. Darrigan, A. Chrostowska, B. Li and S.-Y. Liu, Dalton Trans., 2019, 48, 2807–2812 RSC.
- For related examples, in which an N-bonded boron electrophile attacks a dangling CC or CC bond rather than an aryl ring, see:
(a) H. Wei, Y. Liu, T. Y. Gopalakrishna, H. Phan, X. Huang, L. Bao, J. Guo, J. Zhou, S. Luo, J. Wu and Z. Zeng, J. Am. Chem. Soc., 2017, 139, 15760–15767 CrossRef CAS PubMed;
(b) A. Abengózar, P. García-García, D. Sucunza, L. M. Frutos, O. Castano, D. Sampedro, A. Pérez-Redondo and J. J. Vaquero, Org. Lett., 2017, 19, 3458–3461 CrossRef PubMed;
(c) A. Abengózar, D. Sucunza, P. García-García, D. Sampedro, A. Pérez-Redondo and J. J. Vaquero, J. Org. Chem., 2019, 84, 7113–7122 CrossRef PubMed;
(d) L. Zi, J. Zhang, C. Li, Y. Qu, B. Zhen, X. Liu and L. Zhang, Org. Lett., 2020, 22, 1499–1503 CrossRef CAS PubMed;
(e) A. Abengózar, I. Valencia, G. G. Otárola, D. Sucunza, P. García-García, A. Pérez-Redondo, F. Mendicuti and J. J. Vaquero, Chem. Commun., 2020, 56, 3669–3672 RSC.
- A. J. V. Marwitz, E. R. Abbey, J. T. Jenkins, L. N. Zakharov and S.-Y. Liu, Org. Lett., 2007, 9, 4905–4908 CrossRef CAS PubMed.
- For further examples, see: (a) A. J. Ashe and X. Fang, Org. Lett., 2000, 2, 2089–2091 CrossRef CAS PubMed; (b) E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2010, 132, 16340–16342 CrossRef CAS PubMed; (c) A. D. Rohr, J. W. Kampf and A. J. Ashe, Organometallics, 2014, 33, 1318–1321 CrossRef CAS; (d) F. Sun, L. Lv, M. Huang, Z. Zhou and X. Fang, Org. Lett., 2014, 16, 5024–5027 CrossRef CAS PubMed; (e) A. N. Brown, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2015, 137, 8932–8935 CrossRef CAS PubMed; (f) A. Abengózar, P. García-García, D. Sucunza, A. Pérez-Redondo and J. J. Vaquero, Chem. Commun., 2018, 54, 2467–2470 RSC.
- J.-S. Lu, S.-B. Ko, N. R. Walters, Y. Kang, F. Sauriol and S. Wang, Angew. Chem., Int. Ed., 2013, 52, 4544–4548 CrossRef CAS PubMed.
- For further examples, see: (a) S. Wang, D. T. Yang, J. Lu, H. Shimogawa, S. Gong, X. Wang, S. K. Mellerup, A. Wakamiya, Y. L. Chang, C. Yang and Z. H. Lu, Angew. Chem., Int. Ed., 2015, 54, 15074–15078 CrossRef CAS PubMed; (b) D. T. Yang, S. K. Mellerup, X. Wang, J. S. Lu and S. Wang, Angew. Chem., Int. Ed., 2015, 54, 5498–5501 CrossRef CAS PubMed; (c) S. B. Ko, J. S. Lu and S. Wang, Org. Lett., 2014, 16, 616–619 CrossRef CAS PubMed; (d) Y. G. Shi, D. T. Yang, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Org. Lett., 2016, 18, 1626–1629 CrossRef CAS PubMed; (e) D. T. Yang, Y. Shi, T. Peng and S. Wang, Organometallics, 2017, 36, 2654–2660 CrossRef CAS; (f) Y. G. Shi, X. Wang, N. Wang, T. Peng and S. Wang, Organometallics, 2017, 36, 2677–2684 CrossRef CAS; (g) W. J. Wu, Q. S. Li and Z. S. Li, J. Phys. Chem. A, 2017, 121, 753–761 CrossRef CAS PubMed.
- M. J. S. Dewar and R. Dietz, J. Chem. Soc., 1960, 1344–1347 RSC.
- W. Maringgele, A. Meller, M. Noltemeyer and G. M. Sheldrick, Z. Anorg. Allg. Chem., 1986, 536, 24–34 CrossRef CAS.
- L. M. Greig, B. M. Kariuki, S. Habershon, N. Spencer, R. L. Johnston, K. D. M. Harris and D. Philp, New J. Chem., 2002, 26, 701–710 RSC.
- Q. J. Zhou, K. Worm and R. E. Dolle, J. Org. Chem., 2004, 69, 5147–5149 CrossRef CAS PubMed.
- L. M. Greig, A. M. Z. Slawin, M. H. Smith and D. Philp, Tetrahedron, 2007, 63, 2391–2403 CrossRef CAS.
- M. Numano, N. Nagami, S. Nakatsuka, T. Katayama, K. Nakajima, S. Tatsumi, N. Yasuda and T. Hatakeyama, Chem.–Eur. J., 2016, 22, 11574–11577 CrossRef CAS PubMed.
- X.-Y. Wang, A. Narita, W. Zhang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 9021–9024 CrossRef CAS PubMed.
- K. Shigemori, M. Watanabe, J. Kong, K. Mitsudo, A. Wakamiya, H. Mandai and S. Suga, Org. Lett., 2019, 21, 2171–2175 CrossRef CAS PubMed.
- P. J. Grisdale and J. L. R. Williams, J. Org. Chem., 1969, 34, 1675–1677 CrossRef CAS.
- J. He, J. L. Crase, S. H. Wadumethrige, K. Thakur, L. Dai, S. Zou, R. Rathore and C. S. Hartley, J. Am. Chem. Soc., 2010, 132, 13848–13857 CrossRef CAS PubMed.
- S. Mathew, L. A. Crandall, C. J. Ziegler and C. S. Hartley, J. Am. Chem. Soc., 2014, 136, 16666–16675 CrossRef CAS PubMed.
- Y. Sumida, R. Harada, T. Kato-Sumida, K. Johmoto, H. Uekusa and T. Hosoya, Org. Lett., 2014, 16, 6240–6243 CrossRef CAS PubMed.
- A. Budanow, E. von Grotthuss, M. Bolte, M. Wagner and H.-W. Lerner, Tetrahedron, 2016, 72, 1477–1484 CrossRef CAS.
- T. Katayama, S. Nakatsuka, H. Hirai, N. Yasuda, J. Kumar, T. Kawai and T. Hatakeyama, J. Am. Chem. Soc., 2016, 138, 5210–5213 CrossRef CAS PubMed.
- H. Saito, K. Nogi and H. Yorimitsu, Chem. Lett., 2017, 46, 1122–1125 CrossRef CAS.
- S. Kirschner, S. S. Bao, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Org. Biomol. Chem., 2019, 17, 5060–5065 RSC.
- For corresponding 9,10-thiaboraphenanthrenes, see: F. A. Davis and M. J. S. Dewar, J. Am. Chem. Soc., 1968, 90, 3511–3515 CrossRef CAS.
- C. Körner, P. Starkov and T. D. Sheppard, J. Am. Chem. Soc., 2010, 132, 5968–5969 CrossRef PubMed.
- R. Guo, K.-N. Li, B. Liu, H.-J. Zhu, Y.-M. Fan and L.-Z. Gong, Chem. Commun., 2014, 50, 5451–5454 RSC.
- L. Benhamou, D. W. Walker, D. K. Bučar, A. E. Aliev and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 8039–8043 RSC.
- The Hg(II)-mediated addition of Ph2BO–H to HCCOEt has also been reported: M. Murakami and T. Mukaiyama, Chem. Lett., 1982, 11, 241–244 CrossRef.
- H. Saito, S. Otsuka, K. Nogi and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 15315–15318 CrossRef CAS PubMed.
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Org. Lett., 2017, 19, 5557–5560 CrossRef CAS.
- F. A. Davis and M. J. S. Dewar, J. Org. Chem., 1968, 33, 3324–3326 CrossRef CAS.
- F. A. Davis, M. J. S. Dewar and R. Jones, J. Am. Chem. Soc., 1968, 90, 706–708 CrossRef CAS.
- B. M. Mikhailov and M. E. Kuimova, J. Organomet. Chem., 1976, 116, 123–133 CrossRef CAS.
- J. Chen, Z. Bajko, J. W. Kampf and A. J. Ashe, Organometallics, 2007, 26, 1563–1564 CrossRef CAS.
- S. Yruegas, D. C. Patterson and C. D. Martin, Chem. Commun., 2016, 52, 6658–6661 RSC.
- W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
- T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2019, 58, 11379–11384 CrossRef CAS PubMed.
- S. M. Mathew and C. S. Hartley, Macromolecules, 2011, 44, 8425–8432 CrossRef CAS.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- X. Wang, F. Zhang, J. Gao, Y. Fu, W. Zhao, R. Tang, W. Zhang, X. Zhuang and X. Feng, J. Org. Chem., 2015, 80, 10127–10133 CrossRef CAS PubMed.
- W. Zhang, F. Zhang, R. Tang, Y. Fu, X. Wang, X. Zhuang, G. He and X. Feng, Org. Lett., 2016, 18, 3618–3621 CrossRef CAS PubMed.
- C. Zhang, L. Zhang, C. Sun, W. Sun and X. Liu, Org. Lett., 2019, 21, 3476–3480 CrossRef CAS PubMed.
- S. S. Chissick, M. J. S. Dewar and P. M. Maitlis, Tetrahedron Lett., 1960, 1, 8–10 CrossRef.
- G. C. Culling, M. J. S. Dewar and P. A. Marr, J. Am. Chem. Soc., 1964, 86, 1125–1127 CrossRef CAS.
- P. J. Comina, D. Philp, B. M. Kariuki and K. D. M. Harris, Chem. Commun., 1999, 2279–2280 RSC.
- P. R. Ashton, K. D. M. Harris, B. M. Kariuki, D. Philp, J. M. A. Robinson and N. Spencer, J. Chem. Soc., Perkin Trans. 2, 2001, 11, 2166–2173 RSC.
- D. J. H. Emslie, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2003, 42, 1252–1255 CrossRef CAS PubMed.
- M. Lepeltier, O. Lukoyanova, A. Jacobson, S. Jeeva and D. F. Perepichka, Chem. Commun., 2010, 46, 7007–7009 RSC.
- D. Carnevale, V. del Amo, D. Philp and S. E. Ashbrook, Tetrahedron, 2010, 66, 6238–6250 CrossRef CAS.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed.
- X. Wang, F. Zhang, J. Liu, R. Tang, Y. Fu, D. Wu, Q. Xu, X. Zhuang, G. He and X. Feng, Org. Lett., 2013, 15, 5714–5717 CrossRef CAS PubMed.
- X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed.
- G. Li, W. W. Xiong, P. Y. Gu, J. Cao, J. Zhu, R. Ganguly, Y. Li, A. C. Grimsdale and Q. Zhang, Org. Lett., 2015, 17, 560–563 CrossRef CAS PubMed.
- A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS PubMed.
- Z. Lu, H. Quanz, O. Burghaus, J. Hofmann, C. Logemann, S. Beeck, P. R. Schreiner and H. A. Wegner, J. Am. Chem. Soc., 2017, 139, 18488–18491 CrossRef CAS PubMed.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562–13565 CrossRef CAS PubMed.
- F. Liu, H. Zhang, J. Dong, Y. Wu and W. Li, Asian J. Org. Chem., 2018, 7, 465–470 CrossRef CAS.
- J. Zhang, F. Liu, Z. Sun, C. Li, Q. Zhang, C. Zhang, Z. Liu and X. Liu, Chem. Commun., 2018, 54, 8178–8181 RSC.
- H. Huang, Y. Zhou, M. Wang, J. Zhang, X. Cao, S. Wang, D. Cao and C. Cui, Angew. Chem., Int. Ed., 2019, 58, 10132–10137 CrossRef CAS PubMed.
- Y. Appiarius, T. Stauch, E. Lork, P. Rusch, N. C. Bigall and A. Staubitz, Org. Chem. Front., 2021, 8, 10–17 RSC.
- J. S. A. Ishibashi, J. L. Marshall, A. Mazière, G. J. Lovinger, B. Li, L. N. Zakharov, A. Dargelos, A. Graciaa, A. Chrostowska and S. Y. Liu, J. Am. Chem. Soc., 2014, 136, 15414–15421 CrossRef CAS PubMed.
- S. Biswas, C. Maichle-Mössmer and H. F. Bettinger, Chem. Commun., 2012, 48, 4564–4566 RSC.
- M. Müller, C. Maichle-Mössmer and H. F. Bettinger, Angew. Chem., Int. Ed., 2014, 53, 9380–9383 CrossRef PubMed.
- H. F. Bettinger and M. Müller, J. Phys. Org. Chem., 2015, 28, 97–103 CrossRef CAS.
- S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC.
- X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC.
- In a conceptually related fashion, 1,2-dihydro-1,2-azaborines and 1,2-dihydro-1,2-oxaborines have been synthesized through carbene insertion into 1,2-azaborolide and 1,2-oxaborolide anions, respectively: (a) A. J. Ashe, X. X. Fang, X. X. Fang and J. W. Kampf, Organometallics, 2001, 20, 5413–5418 CrossRef CAS; (b) J. Pan, J. W. Kampf and A. J. Ashe, Organometallics, 2004, 23, 5626–5629 CrossRef CAS. The single or double carbene insertion into 9-borafluorene, which affords six- and seven-membered rings has also been reported: (c) T. A. Bartholome, K. R. Bluer and C. D. Martin, Dalton Trans., 2019, 48, 6319–6322 RSC.
- Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2nd edn, 2011 Search PubMed.
- L. Hintermann and A. Labonne, Synthesis, 2007, 2007, 1121–1150 CrossRef.
- W. Liu, H. Wang and C.-J. Li, Org. Lett., 2016, 18, 2184–2187 CrossRef CAS PubMed.
- We are grateful to one anonymous referee, who brought the following to our attention: in a simplified picture, it can be assumed that there are two particularly important reaction steps in the catalytic cycle: step (1) is the irreversible attack of the B-bonded N/O atom on an η2-alkynyl–[Au] pre-complex to form vinylic N/O–CC–[Au] intermediates as resting states (see Scheme 2b). Step (2) is the irreversible proto-demetalation of N/O–CC–[Au] to give N/O–CC–H with regeneration of the active catalyst. Step (1) is faster for the BN(Me)–H substrates. Step (2) is faster for the more acidic BO–H substrates and is generally rate-determining. In the cases of independent reactions, this results in a more rapid formation of B,O-PAHs. The situation changes in an intramolecular competition, where most of the catalyst is arrested in the N–CC–[Au] state, ultimately leading to the B,N-PAH as the main product (here: 8c). For a related case, see: S. Guo, J. C. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 15976–15984 CrossRef CAS PubMed.
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS.
- Z. Liu, J. S. A. Ishibashi, C. Darrigan, A. Dargelos, A. Chrostowska, B. Li, M. Vasiliu, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2017, 139, 6082–6085 CrossRef CAS PubMed.
- M. J. S. Dewar and R. Dietz, J. Chem. Soc., 1959, 2728–2730 RSC.
- M. J. S. Dewar and R. Dietz, J. Org. Chem., 1961, 26, 3253–3256 CrossRef CAS.
- M. J. S. Dewar, J. Hashmall and V. P. Kubba, J. Org. Chem., 1964, 29, 1755–1757 CrossRef CAS.
- P. Paetzold, C. Stanescu, J. R. Stubenrauch, M. Bienmüller and U. Englert, Z. Anorg. Allg. Chem., 2004, 630, 2632–2640 CrossRef CAS.
- J. Pan, J. W. Kampf and A. J. Ashe, Organometallics, 2009, 28, 506–511 CrossRef CAS.
- G. E. Rudebusch, L. N. Zakharov and S.-Y. Liu, Angew. Chem., Int. Ed., 2013, 52, 9316–9319 CrossRef CAS PubMed.
- G. A. Molander, S. R. Wisniewski and K. M. Traister, Org. Lett., 2014, 16, 3692–3695 CrossRef CAS PubMed.
- G. A. Molander, S. R. Wisniewski and J. Amani, Org. Lett., 2014, 16, 5636–5639 CrossRef CAS PubMed.
- G. A. Molander and S. R. Wisniewski, J. Org. Chem., 2014, 79, 6663–6678 CrossRef CAS PubMed.
- G. A. Molander, S. R. Wisniewski and E. Etemadi-Davan, J. Org. Chem., 2014, 79, 11199–11204 CrossRef CAS PubMed.
- F. J. R. Rombouts, F. Tovar, N. Austin, G. Tresadern and A. A. Trabanco, J. Med. Chem., 2015, 58, 9287–9295 CrossRef CAS PubMed.
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 3855–3860 CrossRef CAS PubMed.
- X. Liu, P. Wu, J. Li and C. Cui, J. Org. Chem., 2015, 80, 3737–3744 CrossRef CAS PubMed.
- In order to increase the readability of this chapter, we have averaged all NMR chemical shift values of nuclei at the same positions in 4a–6a on the one hand and 4b–6b on the other. In the cases of multiplet signals, we have taken the midpoint of each multiplet and averaged those. In all cases the spread of the shift values within the subsets 4a–6a or 4b–6b was significantly smaller than the differences in the average values between 4a–6a and 4b–6b.
- H. Nöth and B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer, Berlin, Heidelberg, New York, 1978 Search PubMed.
- M. A. Fox and J. K. Whitesell, Organic Chemistry, Jones and Bartlett Publisher, Sudbury, Mass., 3rd edn, 2004 Search PubMed.
- H. L. van de Wouw, J. Y. Lee, M. A. Siegler and R. S. Klausen, Org. Biomol. Chem., 2016, 14, 3256–3263 RSC.
- The corresponding methylamino borane did not undergo the cyclization reaction to furnish a parent (B,N)2-perylene devoid of C-bonded substituents.
- T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056–3058 CrossRef CAS PubMed.
- T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924–2925 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114–16126 CrossRef CAS PubMed.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- For B-monosubstituted 2,1-B,N-benzenes and suitable 2,1-B,N-naphthalenes, Liu et al. and Molander et al. have shown that the Ir-catalyzed C–H borylation occurs preferentially at the most acidic hydrogen atoms and therefore as close as sterically possible to the embedded electronegative N atoms: (a) G. H. M. Davies, M. Jouffroy, F. Sherafat, B. Saeednia, C. Howshall and G. A. Molander, J. Org. Chem., 2017, 82, 8072–8084 CrossRef CAS PubMed; (b) A. W. Baggett, M. Vasiliu, B. Li, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2015, 137, 5536–5541 CrossRef CAS PubMed; (c) A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and F. Jäkle, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS PubMed; (d) C. R. McConnell, F. Haeffner, A. W. Baggett and S. Y. Liu, J. Am. Chem. Soc., 2019, 141, 9072–9078 CrossRef CAS PubMed. Based on an investigation of various quinoline and nonsymmetrical 1,2-disubstituted benzene substrates, Marder, Steel et al. have found that preferential borylation occurs at the site of the most deshielded H/C atom in the 1H/13C{1H} NMR spectrum (provided that it is sterically accessible): (e) H. Tajuddin, P. Harrisson, B. Bitterlich, J. C. Collings, N. Sim, A. S. Batsanov, M. S. Cheung, S. Kawamorita, A. C. Maxwell, L. Shukla, J. Morris, Z. Lin, T. B. Marder and P. G. Steel, Chem. Sci., 2012, 3, 3505–3515 RSCIn the case of 9 (and also of pristine 2,1-B,N-benzene), the 1H-NMR tool failed to provide a correct prediction, whereas the 13C-NMR tool worked faithfully (cf. the NMR data given in the ESI†).
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem. Sci., 2019, 10, 7516–7534 RSC.
- J. Merz, L. Dietrich, J. Nitsch, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem.–Eur. J., 2020, 26, 12050–12059 CrossRef CAS PubMed.
- Shinokubo et al. succeeded in the Ir-catalyzed direct tetraborylation of perylene bisimides, which took place at the same sites as in the case of 9 despite the presence of ortho-substituents: T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532–2535 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2058862–2058872, 2068160, and 2068161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00543j |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |