DOI:
10.1039/D1SC00694K
(Edge Article)
Chem. Sci., 2021,
12, 5825-5833
Highly efficient visible-light photocatalytic ethane oxidation into ethyl hydroperoxide as a radical reservoir†
Received
4th February 2021
, Accepted 17th March 2021
First published on 18th March 2021
Abstract
Photocatalytic ethane conversion into value-added chemicals is a great challenge especially under visible light irradiation. The production of ethyl hydroperoxide (CH3CH2OOH), which is a promising radical reservoir for regulating the oxidative stress in cells, is even more challenging due to its facile decomposition. Here, we demonstrated a design of a highly efficient visible-light-responsive photocatalyst, Au/WO3, for ethane oxidation into CH3CH2OOH, achieving an impressive yield of 1887 μmol gcat−1 in two hours under visible light irradiation at room temperature for the first time. Furthermore, thermal energy was introduced into the photocatalytic system to increase the driving force for ethane oxidation, enhancing CH3CH2OOH production by six times to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 233 μmol gcat−1 at 100 °C and achieving a significant apparent quantum efficiency of 17.9% at 450 nm. In addition, trapping active species and isotope-labeling reactants revealed the reaction pathway. These findings pave the way for scalable ethane conversion into CH3CH2OOH as a potential anticancer drug.
233 μmol gcat−1 at 100 °C and achieving a significant apparent quantum efficiency of 17.9% at 450 nm. In addition, trapping active species and isotope-labeling reactants revealed the reaction pathway. These findings pave the way for scalable ethane conversion into CH3CH2OOH as a potential anticancer drug.
Introduction
Ethane, as the second abundant constituent of natural gas (∼10%) and one of the major pollutants emitted from both biogenic and anthropogenic sources (∼9.57 Tg per year), can be utilized to produce value-added chemical substances such as ethanol, acetaldehyde, acetic acid, ethylene, and hydrochloric ether.2–4 Although ethyl hydroperoxide (CH3CH2OOH) could be generated in ethane oxidation, it is generally regarded as an intermediate species due to its facile decomposition.5,6 However, the synthesis of CH3CH2OOH is very attractive because it is a reservoir for both reactive oxygen species (ROS) such as HOx radicals and 1O2, and some carbon-containing free radicals such as alkyl, alkyloxy, and alkylperoxyl radicals.6–9 In the presence of a trace amount of enzyme, hematin, or some metal ions, these active species can be released by CH3CH2OOH to regulate the oxidative stress in the cell, as a promising approach for cancer therapy.7–13 This will even outperform the present photodynamic therapy techniques that are highly dependent on the oxygen and external light.13–15 Thus, achieving the stable conversion of ethane into CH3CH2OOH is highly desirable.
Compared with traditional catalytic processes, photocatalytic ethane conversion is much more intriguing because it can take place under a milder condition with a lower cost.1,16–25 So far, TiO2, ZnO, CeO2, MoO3, and V2O5 have been explored as the photocatalysts for ethane oxidation,22–26 but only UV light can be absorbed due to their large band gaps. However, in the solar irradiation spectrum, UV light accounts for only 3%, while visible light constitutes 44%.27,28 Thus, it is highly expected to develop visible-light-responsive photocatalysts for ethane oxidation. Here, WO3 is a promising candidate with a narrow band gap (2.4–2.8 eV) and a high resistance to photo-corrosion.29,30 Nevertheless, pure WO3 generally shows a high charge carrier recombination rate, requiring modifications such as loading cocatalysts, introducing dopants, and adjusting morphologies.31–33 Modified WO3 has been successfully applied in water splitting, pollutant degradation, and solar energy conversion,34,35 and is desirable to achieve efficient ethane oxidation under visible light irradiation. This is the first hypothesis of the present work.
Being able to absorb visible light cannot ensure the effective utilization of visible light to drive corresponding redox reactions. For example, Pt-loaded black TiO2 could absorb visible light, but it exhibited a negligible activity for water splitting under visible light irradiation.36 Here, as revealed in our previous work, though a narrow band gap of the photocatalyst allows visible light absorption, it meanwhile causes the insufficient driving force for the redox reactions, resulting in a poor catalytic performance.37 The introduction of thermal energy into the photocatalytic system, as a novel thermo-photo catalytic approach, can effectively address this issue. This is because the kinetic energy of the reactants increased by the temperature rise can compensate the potential driving force for corresponding redox reactions. As a result, the visible-light driven photocatalytic activities were significantly improved in a series of chemical processes including water splitting,37–39 CO2 reforming of CH4,40 steam reforming of CH4,41 CO hydrogenation,42 and CO2 hydrogenation.43 Based on our previous successes, introducing thermal energy is expected to enhance the efficiency of ethane oxidation under visible light irradiation, which constitutes the second hypothesis of this work.
On top of these two hypotheses, revealing the reaction pathway of photocatalytic ethane oxidation is even more essential. On the one hand, multiple products with various oxidation degrees might be generated in ethane oxidation. Elucidating the reaction pathway will help to exploit the strategies to enhance the selectivity for the desirable product. On the other hand, identifying the roles of each reactants (e.g., oxygen and water) will contribute to the exploration of approaches to accelerate the reaction. So far, only a few efforts have been made, mainly based on the results of nuclear magnetic resonance spectroscopy,44in situ infrared spectroscopy and trapping active species,25,45 whereas the solid and comprehensive investigation into the reaction mechanism has not been conducted yet.
In this work, Au/WO3 was designed and synthesized as a visible-light-responsive photocatalyst for visible-light driven photocatalytic ethane oxidation in the presence of oxygen and water. An impressive yield of CH3CH2OOH (1887 μmol gcat−1) was obtained in the two-hour visible-light driven photocatalytic process at room temperature for the first time, which was further enhanced by six times via elevating the reaction temperature to 100 °C, with a high selectivity of 63.6% and an apparent quantum efficiency up to 17.9% at 450 nm. More importantly, the synergistic roles of the Au/WO3 catalyst, visible light irradiation, and thermal energy in efficient ethane oxidation into CH3CH2OOH, as well as the comprehensive reaction mechanism of ethane conversion, were uncovered.
Results and discussion
Design and synthesis of Au/WO3 as a visible-light-responsive catalyst
There are three requirements for designing an efficient photocatalyst. First, the band structure of the catalyst must match the redox potentials of the desirable reaction. Second, it can absorb as much light as possible. Third, photo-generated electrons and holes can be effectively separated to drive corresponding redox reactions. In this light, to create an ideal catalyst for ethane oxidation, we selected WO3 as an active component due to its narrow band gap for visible light response and Au as a promoter since it can not only assist charge transfer but also enhance light harvest due to the surface plasmon resonance (SPR) effect.30,46
Au/WO3 was prepared by photo-depositing Au onto monoclinic WO3 nanoparticles. The Au loading amount was 0.33 wt% as determined by ICP-MS. In spite of the small loading amount, a weak peak corresponding to metallic Au (at 38.337°) was observed in the XRD pattern (Fig. S1†).47 The morphological structure of Au/WO3 is shown in Fig. 1a, where Au nanoparticles with an average size of ∼16 nm dispersed on the surface of WO3 (see Fig. S2† for the size distribution of Au). Moreover, the d-spacings of lattice fringes of 0.260, 0.230, and 0.202 nm were recognized (Fig. 1b and c), attributed to WO3(202), Au(111), and Au(200), respectively.47,48 Furthermore, elemental mapping (Fig. 1d–g) revealed the uniform distribution of Au, W, and O. The BET surface area and the BJH average pore diameter and volume of this catalyst were determined as 4.51 m2 g−1, 262 Å, and 0.0368 cm3 g−1, respectively (Fig. S3†). In addition, both lattice oxygen (O2−) and adsorbed oxygen (O− and O2−) were observed in the O 1s XPS spectrum (Fig. S4a†),48 while W existed in the +6 state with the peaks for W 4f7/2 and W 4f5/2 shifted to higher values compared with those for pure WO3, indicating the strong interaction between Au and WO3 (Fig. S4b†). The Au 4f XPS spectrum of the catalyst was just the same as the standard one of metallic Au (Fig. S4c†).47
 |
| | Fig. 1 TEM images of Au/WO3. (a) TEM image. (b) and (c) HRTEM images. (d) HAADF-STEM image. (e) Au element EDX map. (f) W element EDX map. (g) O element EDX map. | |
The UV-vis absorption spectra of Au/WO3 and WO3 are shown in Fig. 2a. Compared with WO3, Au/WO3 showed an enhanced light absorption from 450 to 700 nm with an absorption peak at 550 nm, owing to the SPR effect of Au nanoparticles.46 The band gap of Au/WO3 was then calculated to be 2.43 eV, enabling a visible-light driven photocatalytic process. To ensure that the thermodynamic requirements are satisfied, the band structure of Au/WO3 was analysed as following. The Mott–Schottky plots (Fig. S5†) indicated that the Fermi level locates at 0.71 V (vs. NHE) while the valence-band XPS spectrum (Fig. S4d†) showed a main absorption onset at 1.51 eV, by which the valence band maximum was estimated to be 2.22 V (vs. NHE). Considering the band gap of 2.43 eV, the conduction band minimum was then obtained as −0.21 V (vs. NHE). Thereby, the band structure of Au/WO3 was depicted in Fig. 2b, consistent with reported ones.46,49
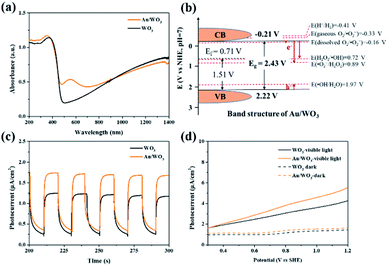 |
| | Fig. 2 Optical and photoelectric properties of the catalyst. (a) UV-visible absorption spectra of Au/WO3 and WO3. (b) Band structure of Au/WO3 and redox potentials of possible reactions. (c) Photocurrent–time curves of Au/WO3 and WO3. (d) Linear sweep voltammetry curves of Au/WO3 and WO3. | |
Here, a series of reduction reactions would take place with photo-generated electrons, forming ˙O2−, H2O2, and ˙OH. The reduction of gas-phase O2 cannot happen since its redox potential (−0.33 V vs. NHE) is above the conduction band minimum, but dissolved O2 can be reduced with a favorable redox potential of −0.16 V vs. NHE.50–52 This implies the vital importance of carrying out ethane oxidation in an aqueous condition. Moreover, owing to this unique band structure, the reduction of H+ into H2 is inhibited, effectively enhancing the production of above oxidizing agents. Meanwhile, photo-generated holes could oxidize water with ˙OH as the byproduct. The formation of these ROS would make a great contribution to ethane oxidation.
Moreover, Au/WO3 showed a high efficiency for separating photo-generated electrons and holes as demonstrated by its stable photocurrent of 1.74 μA cm−2 under visible light irradiation, which was much higher than that of WO3 (1.23 μA cm−2) (Fig. 2c). This agreed well with the result of linear sweep voltammetry (Fig. 2d), namely, enhanced current density was acquired on Au/WO3 compared with that of WO3. Therefore, the aforementioned three requirements for designing a high-performance photocatalyst are met, highlighting the great promise of Au/WO3 as a highly efficient photocatalyst for ethane oxidation under visible light irradiation.
Visible-light driven photocatalytic ethane oxidation over Au/WO3
As shown in Fig. S6,† photocatalytic ethane oxidation was carried out in a sealed reactor under visible light irradiation (λ >420 nm, 100 mW cm−2). Besides ethane and oxygen, water was also introduced into the reactor to dissolve oxygen to enable a thermodynamically favorable photocatalytic process. As expected, the two-hour room-temperature photocatalytic reaction over Au/WO3 catalyst (35 °C as measured by the thermocouple) resulted in an impressive yield of CH3CH2OOH (1887 μmol gcat−1), together with some CH3CHO (621 μmol gcat−1) and CH3COOH (585 μmol gcat−1), surpassing current reports (at room temperature) even though their yields were obtained under UV light or simulated sunlight irradiation (Table 1).
Table 1 Reported photocatalytic activities of ethane oxidation
| Catalyst |
Oxidant |
Light |
Pressure |
Temp. |
Yield (μmol gcat−1 h−1) |
Ref. |
| ZnO |
O2 |
UV |
Gas flow with C2H6:O2:He of 3:1:10 |
220 °C |
336 (CH3CHO) |
1
|
| 280 (CO2) |
| 28 (CO) |
| 24 (HCHO) |
| 11 (CH3CH2OH) |
| MoO3/SiO2 |
O2 |
UV |
Gas flow with C2H6:O2:He of 3:1:10 |
220 °C |
2400 (CH3CHO) |
19
|
| 880 (HCHO) |
| 200 (CO2) |
| 92 (CH3CH2OH) |
| BiOX-POM |
O2 |
UV + visible (cold white LED lamp) |
Gas flow with C2H6:O2 of 1:1 |
RT |
72 (CH3CHO) |
20
|
| V2O5/SiO2 |
O2 |
UV |
Gas flow with C2H6:O2:He of 3:1:10 |
220 °C |
2480 (CH3CHO) |
21
|
| 280 (HCHO) |
| 80 (CO2) |
| 40 (CO) |
| Au/TiO2 |
H2O |
UV, 59 mW cm−2 |
Gas flow with C2H6:H2O:Ar of 1:1:8 |
RT |
0.07 (CO2) |
22
|
| Pd–MoO3/SiO2 |
CO2 |
UV, 40 mW cm−2 |
Gas flow with C2H6:CO2 of 1:1 |
150 °C |
28 (CH3CHO) |
23
|
| 5 (C2H5CHO) |
| 3 (CH3CH2OH) |
| 4 (CH3CHO) |
| 3 (CO) |
| ZnO–TiO2/SiO2 |
CO2 |
UV, 40 mW cm−2 |
Gas flow with C2H6 and CO2 |
200 °C |
25 (C2H5CHO) |
24
|
| 8 (C4H10) |
| 4 (CH3CHO) |
| 3 (CO) |
| Pd/TiO2 |
CO2 |
UV |
0.1 MPa C2H6 and 0.1 MPa CO2/Ar |
RT |
231 (C2H4) |
25
|
| 118 (CO) |
| 22 (CH4) |
| Au/WO3 |
O2 |
Visible (λ >420 nm), 100 mW cm−2 |
1.0 MPa 4:1 C2H6/N2 and 1.5 MPa O2 |
RT |
944 (CH3CH2OOH) |
This work |
| 311 (CH3CHO) |
| 293 (CH3COOH) |
| 100 °C |
5617 (CH3CH2OOH) |
This work |
| 1799 (CH3CHO) |
| 665 (CH3COOH) |
| 449 (CH3CH2OH) |
Here, considering the intrinsic issue of visible-light photocatalysis, namely, the contradiction between visible light absorption and sufficient driving force, we propose that the introduction of thermal energy, which can help to break the kinetic limitation of the reaction, will greatly promote ethane oxidation under visible light irradiation. By analysing the band structure of Au/WO3, the driving forces for O2 reduction (i.e., energy difference between the redox potential of dissolved O2/˙O2− and the conduction band minimum) and H2O oxidation (i.e., energy difference between the redox potential of ˙OH/H2O and the valence band maximum) were found as small as 0.05 and 0.25 V, respectively. The introduction of thermal energy by simply increasing the reaction temperature will enhance the kinetic energy of the reactants, thus providing additional kinetic driving force for the semi-reactions. The compensation effect of the kinetic driving force with regards to the potential driving force is highly expected to lead to highly efficient ethane oxidation under visible light irradiation.
To demonstrate above hypothesis, photocatalytic ethane oxidation was carried out at 50, 100, 150, 180, and 200 °C. The results are shown in Fig. 3a, where the yield of CH3CH2OOH reached 11233 μmol gcat−1 at 100 °C, which was six times of that obtained at room temperature. The production of CH3CHO and CH3COOH was also enhanced, with a new high-value product, CH3CH2OH, being obtained. These products were further evidenced by 1H NMR (Fig. S7†) and GC-MS (Fig. S8–S11†). The significant production efficiencies outperform not only all the reported values (Table 1), but also those obtained with other catalysts under the same test condition (Fig. S12†), indicating the superiority of the Au/WO3 catalyst. Herein, Au contributed to both charge separation and light harvest due to SPR as demonstrated by the fact that loading other metals without evident SPR effect showed much poorer efficiencies. However, although the increase of the Au loading amount from 0.08 to 0.33 wt% promoted ethane oxidation, excessive Au loading of 0.69 wt% resulted in the decreased yield (Fig. S13†) because Au nanoparticles could also act as the recombination centers of electrons and holes.53,54 Furthermore, different from other reports with CH3CHO as the major product, the selectivity of CH3CH2OOH we obtained was as high as 63.6%, marking an effective way to produce such an easy-to-decompose substance which is promising for cancer therapy. As-produced liquid could be further purified by column chromatography, etc., for further application. The apparent quantum efficiency over Au/WO3 at 100 °C was measured as high as 17.9% at 450 nm, again demonstrating the excellent ethane oxidation performance achieved by thermo-photo catalysis. The decrease of the yield in the temperature range from 150 to 200 °C was due to the overoxidation of ethane into CO2 and the decreased solubility of feed gases, which were supported by the enhanced CO2 yield and lowered total yield, respectively.
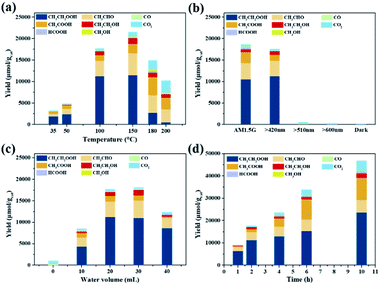 |
| | Fig. 3 Photocatalytic ethane oxidation over Au/WO3. (a) Visible-light driven ethane oxidation at various temperatures (reacting for 2 h in 20 mL water). (b) Ethane oxidation under various lights (reacting for 2 h in 20 mL water at 100 °C). (c) Visible-light driven ethane oxidation with various water volumes (reacting for 2 h at 100 °C). (d) Visible-light driven ethane oxidation with various reaction times (in 20 mL water at 100 °C). | |
In contrast, as shown in Fig. 3b, in the process at 100 °C without illumination, negligible oxidation products were detected, evidencing the little contribution of thermal catalysis to ethane oxidation at this temperature. In addition, the activity under simulated sunlight irradiation (AM1.5G) was just slightly higher than that obtained under visible light irradiation, indicating that the activity of ethane oxidation was directly correlated to photo energy (as distributed by light wavelengths). Furthermore, light irradiation with wavelengths above 510 nm resulted in very low yields of ethane oxidation products because these lights cannot be absorbed by Au/WO3 (with a band gap of 2.43 eV) to generate electrons and holes. This also implied the very limited contribution of hot electrons of Au derived from the plasmonic effect to ethane oxidation. These observations demonstrated that ethane oxidation under illumination at 100 °C still followed a photocatalytic pathway. Therefore, our hypothesis (that the introduction of thermal energy can accelerate the photocatalytic process of ethane oxidation under visible light irradiation by enhancing the driving force) was exactly confirmed.
Water plays a key role in photocatalytic ethane oxidation since negligible product was detected in the reaction without water (Fig. 3c). Besides enabling a process with dissolved O2 as the reduced species to meet the thermodynamic requirement of photocatalysis, water also accelerates ethane oxidation via the following two aspects. On the one hand, water is the source of some ROS such as ˙OH and H2O2. On the other hand, water could serve as an effective solvent for the oxidation products, cleaning the catalyst surface and regenerating the active sites. Both aspects could be promoted at an appropriately elevated temperature due to the enhanced water dissociation (to form OH− which can be oxidized into ˙OH more easily) and the accelerated desorption of the products. However, excessive amount of water led to the decrease of the yield, because in such a three-phase system, the contact between the feed gases and catalyst could be suppressed by water. The prolongation of the reaction from 2 to 10 hours showed a nearly linear enhancement in ethane oxidation (Fig. 3d), indicating there was no evident photo-corrosion. However, the time extension mainly promoted the overoxidation into CH3COOH and CO2 while lowered the selectivity of CH3CH2OOH. Thus, considering the time effectiveness, activity, and selectivity, two-hour reaction was the best.
Reaction pathway
Since multiple oxidation products with various oxidation degrees were detected, visible-light driven ethane oxidation over Au/WO3 is highly possible to undergo a stepwise process. When using CH3CH2OH as the reactant, its conversion efficiency was as high as 96.9%, generating the same types of products as those obtained from ethane oxidation (Fig. 4a), indicating that CH3CH2OH was an important intermediate species. The major product was CH3COOH, while the generation of CH3CH2OOH, as the most abundant and desirable product, showed a poor selectivity of 13.2% (much below that of 63.6% in ethane oxidation), suggesting that CH3CH2OH might not be the main intermediate to CH3CH2OOH. Besides, the conversion efficiency of CH3CHO was just 60.0%, with CH3COOH as the dominant product with a high selectivity of 91.3%, revealing that CH3CHO was a key intermediate to CH3COOH. Only minor amount of CH3COOH could be further converted. Above results implied CH3CH2OOH was not mainly produced from CH3CH2OH, CH3CHO, and CH3COOH, but, as a primary product directly generated from ethane.3,44
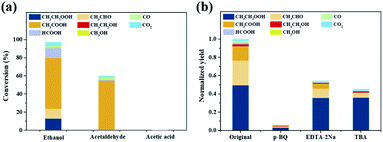 |
| | Fig. 4 Identification of the reaction pathway and active species in visible-light driven photocatalytic ethane oxidation over Au/WO3. (a) Conversion efficiency using ethanol, acetaldehyde, or acetic acid as the reactant. (b) Normalized yield obtained with the employment of trapping agents. | |
Moreover, to reveal the role of active species in photocatalytic ethane oxidation, trapping agents, including p-benzoquinone (p-BQ), tertiary butyl alcohol (TBA), and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA–2Na), were employed to trap ˙O2−, ˙OH, and holes, respectively. A small addition of p-BQ (10 mM) led to a 94.4% decrease in the total yield (Fig. 4b), indicating the vital importance of ˙O2−. In the photocatalytic reduction semi-reaction, ˙O2− was first produced, by which H2O2 and ˙OH could be formed successively. Therefore, trapping ˙O2− would result in an evident decrease of ROS and thereby a low yield. Moreover, ˙O2− played a key role for generating CH3CH2OOH, which would be revealed in the following part. In contrast, the effect induced by trapping holes was smaller, causing a 45.6% decrease of the total yield. As proposed in Fig. 2b, holes would react with H2O to form ˙OH, which could then contribute to ethane oxidation. Even though holes were trapped, ˙OH could still be produced from the reduction semi-reaction, thus the influence on the yield was not very much noticeable. Moreover, by trapping ˙OH, the production of CH3CH2OOH was slightly affected while the yields of CH3CHO and CH3COOH significantly decreased, indicating that ˙OH is directly correlated to the formation of CH3CHO and CH3COOH but not CH3CH2OOH.
Furthermore, isotope labeling experiments with the use of O182, H2O18, or D2O, were conducted to demonstrate the origin of each product. The products detected by GC-MS are shown in Table 2 (see Fig. S8–S11† for the GC-MS spectra). Employing O182 resulted in the formation of CH3CH2O18O18H, CH3CH2O18H, CH3CHO18, CH3CO18O18H, CH3CO18OH, and CH3COO18H, while the use of H2O18 generated CH3CHO18, CH3CO18O18H, CH3CO18OH, and CH3COO18H. Moreover, replacing general water by D2O made the hydroperoxyl group of CH3CH2OOH be fully deuterated and the carboxy group of CH3COOH be partially deuterated.
Table 2 Products obtained in isotope labeling experiments
| Reactant |
CH3CH2OOH |
CH3CH2OH |
CH3CHO |
CH3COOH |
| O182 |
CH3CH2O18O18H |
CH3CH2O18H |
CH3CHO18 |
CH3CO18O18H, CH3CO18OH, CH3COO18H and CH3COOH |
| H2O18 |
CH3CH2OOH |
CH3CH2OH |
CH3CHO18 |
CH3CO18O18H, CH3CO18OH, CH3COO18H and CH3COOH |
| D2O |
CH3CH2OOD |
CH3CH2OH |
CH3CHO |
CH3COOD and CH3COOH |
According to the above results, the reaction pathway was proposed as following. First of all, visible light was absorbed by Au/WO3 to generate active electrons and holes (eqn (1)). Considering the thermodynamic requirements of photocatalysis (Fig. 2b), photo-generated electrons would transfer from the conduction band of WO3 to Au nanoparticle to reduce dissolved O2 into ˙O2− (eqn (2)), which could be further converted into H2O2 (eqn (3)) and ˙OH (eqn (4)), while photo-generated holes remaining on WO3 would oxidize H2O into ˙OH (eqn (5)).
| | | ˙O2− + 2H+ + e− → H2O2 | (3) |
| | | H2O2 + H+ + e− → ˙OH + H2O | (4) |
CH3CH3 could react with either h+ (eqn (6)) or ˙OH (eqn (7)) to form an unstable intermediate radical CH3CH2˙.25 The participation of both h+ and ˙OH was supported by the result of trapping experiments, namely, trapping h+ or ˙OH did not completely inhibit the reaction. ˙O2− could be protonated by H+ in water to generate ˙OOH (eqn (8)),55,56 which then reacted with CH3CH2˙ to form CH3CH2OOH (eqn (9)). Since ˙OOH and CH3CH2˙ were generated on Au and WO3 respectively, the most favourable site for CH3CH2OOH production was the interface of Au and WO3. Such a pathway for CH3CH2OOH production agreed well with the result of isotope labeling experiments, namely, the O atoms in CH3CH2OOH were from O2 while the H atom in the hydroperoxyl group originated from water. The insertion of ˙O2− was the preliminary step for the generation of subsequent oxidation products, thus trapping ˙O2− led to a sharp drop of the yields (Fig. 4b).
| | | CH3CH3 + h+ → CH3CH2˙ + H+ | (6) |
| | | CH3CH3 + ˙OH → CH3CH2˙ + H2O | (7) |
| | | CH3CH2˙ + ˙OOH → CH3CH2OOH | (9) |
Since the O atom in CH3CH2OH came from O2 rather than H2O, it was most likely to be resulted with CH3CH2OOH as the intermediate as shown in eqn (10) and (11). Here, eqn (11) ensured that H2O was not the source of the H atom in the hydroxy group. Eqn (10) and (11) could proceed reversibly because CH3CH2OOH was detected as one of the products in photocatalytic conversion of CH3CH2OH (Fig. 4a).
| | | CH3CH2OOH ⇌ CH3CH2O˙ + ˙OH | (10) |
| | | CH3CH2O˙ + CH3CH3 ⇌ CH3CH2OH + CH3CH2˙ | (11) |
CH3CHO could be produced either from CH3CH2OH (eqn (12)) or directly from CH3CH2OOH (eqn (13)), by which the O atom in CH3CHO originated from O2. Moreover, the detected CH3CHO18 with H2O18 as the reactant was ascribed to the reversible reaction between CH3CHO and CH3CH(OH)2 (eqn (14)). The existence of CH3CH(OH)2 was evidenced by 1H NMR spectrum (Fig. S7†). In this case, both H atoms and at least one O atom(s) in the hydroxyl groups of CH3CH(OH)2 should be derived from H2O.
| | | CH3CH2OH + 2˙OH → CH3CHO + 2H2O | (12) |
| | | CH3CH2OOH → CH3CHO + H2O | (13) |
| | | CH3CHO + H2O ⇌ CH3CH(OH)2 | (14) |
CH3CH(OH)2 could be further oxidized into CH3COOH by ˙OH (eqn (15)), in which the H atom and at least one O atom(s) in the carboxy group originated from H2O, partially agreeing with the result of isotope labeling experiments (Fig. S11d†). Thus, there should be another pathway leading to the formation of CH3COOH in which both O atoms came from O2 and the H atom in the carboxy group was not derived from H2O. This might be achieved via eqn (16) and (17).
| | | CH3CH(OH)2 + 2˙OH → CH3COOH + 2H2O | (15) |
| | | CH3CH2OH + 2˙OOH → CH3CH(OOH)(OH) + H2O2 | (16) |
| | | CH3CH(OH)(OOH) → CH3COOH + H2O | (17) |
The cleavage of the C–C bond occurred in CH3CH2OH and CH3CHO because HCOOH and CH3OH were detected after photocatalytic oxidation of CH3CH2OH and CH3CHO (Fig. 4a). The production of HCOOH from CH3CH2OH was proposed via eqn (18) and (19), with the assistance of ˙OH. HCOOH could also be resulted by the reaction between CH3CHO and ˙OH (eqn (20)). ˙CH3 generated in eqn (18) and (20) could combine with ˙OH into CH3OH (eqn (21)).
| | | CH3CH2OH + ˙OH → ˙CH3 + CH2(OH)2 | (18) |
| | | CH2(OH)2 + 2˙OH → HCOOH + 2H2O | (19) |
| | | CH3CHO + ˙OH → ˙CH3 + HCOOH | (20) |
As-produced HCOOH could be decomposed into CO (eqn (22)) or further oxidized into CO2 by ˙OH (eqn (23)).
| | | HCOOH + 2˙OH → CO2 + 2H2O | (23) |
The reaction pathway was depicted in Fig. 5 based on above discussions. It should be noted that the reactions took place at the interface of Au and WO3 for relieving the limitation of mass transfer. Moreover, it could be easily observed that ˙OH was mainly responsible for further conversion of CH3CH2OOH. The high selectivity for CH3CH2OOH here was owed to its fast desorption from the catalyst surface due to the solvation effect of water and the elevated temperature to 100 °C, which supressed its further oxidation. This provides a guidance to develop more efficient photocatalytic processes for the production of CH3CH2OOH.
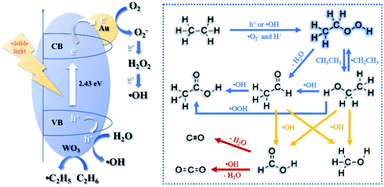 |
| | Fig. 5 Schematic reaction pathway for visible-light driven photocatalytic ethane oxidation over Au/WO3. | |
Conclusions
In summary, highly efficient visible-light driven photocatalytic ethane conversion into a radical reservoir (namely, CH3CH2OOH) was achieved over our created Au/WO3 catalyst. The yield of CH3CH2OOH obtained at room temperature in two hours was as high as 1887 μmol gcat−1, which was further enhanced by six times by simply elevating the temperature to 100 °C. Moreover, a significant apparent quantum efficiency of 17.9% was obtained at 450 nm. Such a promotion was owed to the compensation effect of the kinetic energy of the reactants with regards to the insufficient potential energy in photocatalytic ethane oxidation over the narrow-band-gap Au/WO3 catalyst. Furthermore, the reaction pathway was revealed by converting intermediate substances, trapping active species, and labeling reactants with isotopes, implying that ˙OH plays a key role to determine the selectivity and activity of CH3CH2OOH production. Here, the excellent catalytic performance and the thorough analysis of the reaction mechanism will pave the way for scalable ethane conversion into CH3CH2OOH, which shows a great promise for cancer therapy by regulating the oxidative stress.
Experimental
Materials
Monoclinic WO3 (99.8%, Macklin), P25 (99.5%, Sigma-Aldrich), ZnO (99.9%, Macklin), Bi2O3 (99.99%, Macklin), MoO3 (99.50%, Alfa Aesar), and CeO2 (99.99%, Macklin) were acquired to synthesize the catalysts. Freshly prepared 2 mg mL−1 HAuCl4 (Sigma-Aldrich) aqueous solution was used as Au precursor. Other metal precursors included H2PtCl6 (Sigma-Aldrich), PdCl2 (Sigma-Aldrich), RhCl3 (Alfa Aesar), AgNO3 (Sinopharm), Cu(NO3)2 (Sinopharm), and Fe(NO3)3 (Sinopharm). The free radical scavengers, p-benzoquinone (97%), EDTA–2Na (AR), and tertiary butyl alcohol (AR), were obtained from Macklin Co., Ltd. Feed gases, ethane (80% ethane + 20% nitrogen) and oxygen (99.99%), were supplied by Air Liquid Co., Ltd.
Catalyst preparation
Au was loaded on WO3via photo-deposition. Namely, 0.2000 g WO3 was added into 40 mL deionized water to form a 5 g L−1 suspension, followed by adding 100 μL freshly prepared 20 mg mL−1 HAuCl4 solution during vigorous stirring at 500 rpm (the theoretical Au loading amount: 1 wt%). The mixture was continuously stirred at 500 rpm for 20 min and then ultrasonicated for 20 min. Afterwards, the suspension was irradiated by an ultraviolet lamp (100 W, λ <400 nm) for 10 min while being stirred at 600 rpm. In this process, the color of the suspension was found to change from yellow to purple, demonstrating the reduction of Au3+. The as-obtained Au/WO3 suspension was washed and then collected via suction filtration with a 45 μm filter membrane. Lastly, the collected powder was dried at 60 °C for 12 h. Au/WO3 catalysts with 0.1, 0.5, and 1.5 wt% Au were also prepared, with 10, 50, and 150 μL HAuCl4 solutions, respectively. The real loading amount was confirmed by inductively coupled plasma mass spectrometry (ICP-MS).
In addition, Au was also loaded on commercial TiO2, ZnO, Bi2O3, MoO3, and CeO2 as control samples, using the same method as above-described. Moreover, to investigate the influence of different metal cocatalysts, Pt, Pd, Ag, and Rh were photo-deposited on WO3 with H2PtCl6, PdCl2, AgNO3, and RhCl3 as the precursors, while Fe and Cu were loaded on WO3via impregnation and subsequent calcination at 400 °C for 6 h.
Catalyst characterization
X-ray diffraction (XRD) patterns were obtained from Shimadzu X-ray diffractometer (XRD-6100). The structures and morphologies of the catalysts were characterized using a FEI Tecnai G2 F20 field-emission transmission electron microscope (TEM) at an accelerating voltage of 200 kV. Attached energy dispersive X-ray spectroscopy (EDS) was employed for elemental analysis of the as-synthesized samples. The surface areas of the samples were measured and calculated using the multipoint Brunauer–Emmett–Teller (BET) technique with a BET Sorptometer (ASAP 2020 PLUS HD88) based on the adsorption isotherms. The chemical state and composition of the catalyst surface were determined via an X-ray photoelectron spectroscopy (XPS) instrument (Thermo EscaLab 250Xi). The element contents were measured by inductively coupled plasma mass spectrometry (ICP-MS) (Thermo iCAP Q). Ultraviolet-visible (UV-vis) absorption spectra of the catalysts were obtained using an UV-vis spectrometer (Shimadzu UV-2600). 1H nuclear magnetic resonance (NMR) was conducted on a Bruker AVANCE III HD 400 NMR spectrometer using the liquid mixture obtained from ethane oxidation with deuterated water (D2O).
DFT calculations
DFT calculations were conducted using Gaussian 16 package.57 The geometric structure of the complex which contains one organic molecule (i.e., CH3CH2OOH or CH3CH2OH) and two water molecules was optimized using the B3LYP method58 combined with the 6-311G(d,p) basis set.59 The optimized structures were confirmed to be stable by frequency calculations. Afterwards, 1H NMR chemical shifts were computed by the gauge-independent atomic orbital (GIAO) model60 at the B3LYP/6-311++G(d,p) level,58,61 using the self-consistent reaction field (SCRF)62 technique in simulated water continuum. The geometric structure and NMR shifts of the reference tetramethylsilane (TMS) were calculated using the same method.
Photocatalytic activity test
As shown in Fig. S6,† photocatalytic ethane oxidation was carried out in a 100 mL sealed stainless steel reactor with a concentric quartz window on the top. A 300 W xenon lamp (CEL-HXF300, AULIGHT) equipped with an UVcut420 filter, as the light source (λ >420 nm), was placed right above the reactor with the light intensity kept at 100 mW cm−2. AM1.5G filter was employed to simulate the sunlight irradiation in some cases. In a typical test, 20 mg Au/WO3 and 20 mL deionized water were added into the Teflon vessel, after which the reactor was vacuumed and purged with the C2H6/N2 mixture gas for three times to remove any impurities in the system. Subsequently, 1.0 MPa C2H6/N2 mixture (4:1) and 1.5 MPa O2 were injected into the reactor. The photocatalytic reaction then started under visible light irradiation (λ >420 nm) at a certain temperature in the range of 35–200 °C during continuous stirring at 900 rpm. After reacting for 2 h, the reactor was placed in an ice bath for 30 min to ensure the termination of the reaction, and then as-produced CO and CO2 were measured by a gas chromatograph (GC) (Fuli-9800) equipped with a TDX-01 packed column and a thermal conductive detector (TCD) working at 80 and 100 °C, respectively. Afterwards, the suspension was filtered, and the liquid was collected for product detection. CH3CH2OH, CH3CHO, and CH3OH were analyzed by GC with a Porapak Q column and a flame ionization detector (FID) by injecting 1 μL liquid. The temperatures of the injector, column, and FID detector were 150, 120, and 150 °C, respectively. CH3CH2OOH, CH3COOH, and HCOOH were quantified by a high-performance liquid chromatograph (HPLC) (Shimadzu Essentia LC-16) with a C18 column (Φ 4.6 mm × 250 m) working at 35 °C.
The apparent quantum efficiency (AQE) was measured using a band-pass filter of monochromatic light of 450 nm (λ1/2 = ∼20 nm) at 100 °C following the same procedure as above described. All the products after two-hour reaction were quantified, and then the AQE was calculated via the following equations:
| | 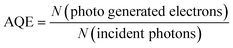 | (24) |
| | 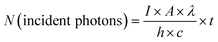 | (25) |
| | | N(photo generated electrons) = 2 × N(CH3CH2OOH) + 2 × N(C2H5OH) + 4 × N(CH3CHO) + 6 × N(CH3COOH) + N(CH3OH) + 5 × N(HCOOH) + 5 × N(CO) + 7 × N(CO2) | (26) |
where
I,
A,
λ, and
t are light intensity, irradiation area, light wavelength, and duration, respectively, while
h and
c represent Planck's constant and light speed, respectively.
Furthermore, to identify the role of free radicals in photocatalytic ethane oxidation, 10 mM free radical scavenger, i.e., p-benzoquinone (p-BQ), tertiary butyl alcohol (TBA), and ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA–2Na), was added into the catalyst suspension for photocatalytic activity test at 50 °C to avoid the decomposition of scavengers at high temperatures.
Isotope labeling experiment
Isotope labeling experiments were conducted simply by replacing general O2 and H2O with O182, H2O18, or D2O. The reaction temperature was controlled at 100 °C. The products were analyzed by gas chromatography-mass spectrometry (GC-MS) (Shimadzu QP 2020 NX) equipped with an RTX-WAX column and an electron ionization source working at 70 eV. The temperatures of the split injector, column, connector, and ionization source were set as 120, 40, 120, and 200 °C, respectively.
Electrochemical measurement
The photocurrent–time (i–t) curve and linear sweep voltammetry (LSV) curve were recorded for Au/WO3 and WO3 in a three-electrode cell with a 0.5 M Na2SO4 electrolyte under visible light irradiation. A fluorine-doped tin oxide glass coated with catalyst, Pt, and saturated Ag/AgCl performed as the working, counter, and reference electrodes, respectively. LSV measurement was preformed from 1.2 to 0.3 V (vs. SHE) at a sweep rate of 0.5 mV s−1. Mott–Schottky analysis was carried out at 500, 1000, 1500, and 2000 Hz in the same cell in dark.
Author contributions
Conceptualization, YHH; methodology, YHH; investigation, YZ, SF, SC, YT, CW and YHH; formal analysis, YZ, SF, and YHH; writing – original draft, SF; writing – review & editing, YHH; supervision, YHH.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- K. Wada, K. Yoshida, T. Takatani and Y. Watanabe, Appl. Catal., A, 1993, 99, 21–36 CrossRef CAS.
- Y. Zhu, S. Shi, C. Wang and Y. H. Hu, Int. J. Energy Res., 2020, 44, 708–717 CrossRef CAS.
- M. M. Forde, R. D. Armstrong, C. Hammond, Q. He, R. L. Jenkins, S. A. Kondrat, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. Willock, C. J. Kiely and G. J. Hutchings, J. Am. Chem. Soc., 2013, 135, 11087–11099 CrossRef CAS.
- R. Jin, M. Peng, A. Li, Y. Deng, Z. Jia, F. Huang, Y. Ling, F. Yang, H. Fu, J. Xie, X. Han, D. Xiao, Z. Jiang, H. Liu and D. Ma, J. Am. Chem. Soc., 2019, 141, 18921–18925 CrossRef CAS.
- F. Battin-Leclerc, O. Herbinet, P.-A. Glaude, R. Fournet, Z. Zhou, L. Deng, H. Guo, M. Xie and F. Qi, Angew. Chem., Int. Ed., 2010, 49, 3169–3172 CrossRef CAS PubMed.
- D. Chen, H. Jin, Z. Wang, L. Zhang and F. Qi, J. Phys. Chem. A, 2011, 115, 602–611 CrossRef CAS PubMed.
- W. Chamulitrat, N. Takahashi and R. P. Mason, J. Biol. Chem., 1989, 264, 7889–7899 CrossRef CAS.
- B. Kalyanaraman, C. Mottley and R. P. Mason, J. Biol. Chem., 1983, 258, 3855–3858 CrossRef CAS.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- Z. Zhou, J. Song, R. Tian, Z. Yang, G. Yu, L. Lin, G. Zhang, W. Fan, F. Zhang, G. Niu, L. Nie and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 6492–6496 CrossRef CAS PubMed.
- A. Caputo, M. Brunori and R. Giuliano, Cancer Res., 1961, 21, 1499 CAS.
- Y. Wan, G. Lu, J. Zhang, Z. Wang, X. Li, R. Chen, X. Cui, Z. Huang, Y. Xiao, J. Chelora, W. Zhang, Y. Liu, M. Li, H.-Y. Xie and C.-S. Lee, Adv. Funct. Mater., 2019, 29, 1903436 CrossRef.
- C. Wang, F. Cao, Y. Ruan, X. Jia, W. Zhen and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 9846–9850 CrossRef CAS PubMed.
- C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- C. T. Brigden, S. Poulston, M. V. Twigg, A. P. Walker and A. J. J. Wilkins, Appl. Catal., B, 2001, 32, 63–71 CrossRef CAS.
- K. Wada, K. Yoshida, Y. Watanabe and T. Suzuki, Appl. Catal., 1991, 74, L1–L4 CrossRef CAS.
- M. Somekh, A. M. Khenkin, A. Herman and R. Neumann, ACS Catal., 2019, 9, 8819–8824 CrossRef CAS.
- K. Wada, H. Yamada, Y. Watanabe and T.-a. Mitsudo, J. Chem. Soc., Faraday Trans., 1998, 94, 1771–1778 RSC.
- A. Tóth, T. Bánsági and F. Solymosi, Mol. Catal., 2017, 440, 19–24 CrossRef.
- X. Wang, Z. He, S. Zhong and X. Xiao, J. Nat. Gas Chem., 2007, 16, 173–178 CrossRef CAS.
- X.-T. Wang, S.-H. Zhong and X.-F. Xiao, J. Mol. Catal. A: Chem., 2005, 229, 87–93 CrossRef CAS.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280–9286 CrossRef CAS.
- N. Djeghri, M. Formenti, F. Juillet and S. Teichner, Faraday Discuss. Chem. Soc., 1974, 58, 185–193 RSC.
- M. P. Thekaekara, Sol. Energy, 1976, 18, 309–325 CrossRef.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS PubMed.
- D. P. DePuccio, P. Botella, B. O'Rourke and C. C. Landry, ACS Appl. Mater. Interfaces, 2015, 7, 1987–1996 CrossRef CAS PubMed.
- P. Y. Dong, G. H. Hou, X. U. Xi, R. Shao and F. Dong, Environ. Sci.: Nano, 2017, 4, 539–557 RSC.
- L. Chen and S. C. Tsang, Sens. Actuators, B, 2003, 89, 68–75 CrossRef CAS.
- K. Wenderich, A. Klaassen, I. Siretanu, F. Mugele and G. Mul, Angew. Chem., Int. Ed., 2014, 53, 12476–12479 CAS.
- C. A. Bignozzi, S. Caramori, V. Cristino, R. Argazzi, L. Meda and A. Tacca, Chem. Soc. Rev., 2013, 42, 2228–2246 RSC.
- T. Zhu, M. N. Chong and E. S. Chan, ChemSusChem, 2014, 7, 2974–2997 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- B. Han and Y. H. Hu, J. Phys. Chem. C, 2015, 119, 18927–18934 CrossRef CAS.
- S. Fang, Z. Sun and Y. H. Hu, ACS Catal., 2019, 9, 5047–5056 CrossRef CAS.
- S. Fang, Y. Liu, Z. Sun, J. Lang, C. Bao and Y. H. Hu, Appl. Catal., B, 2020, 278, 119316 CrossRef CAS.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494–497 CrossRef CAS.
- B. Han, W. Wei, M. Li, K. Sun and Y. H. Hu, Chem. Commun., 2019, 55, 7816–7819 RSC.
- X. Wu, J. Lang, Y. Jiang, Y. Lin and Y. H. Hu, ACS Sustainable Chem. Eng., 2019, 7, 19277–19285 CrossRef CAS.
- C. Wang, S. Fang, S. Xie, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 7390–7394 RSC.
- F. Sastre, A. Corma and H. García, Appl. Catal., B, 2012, 128, 84–90 CrossRef CAS.
- Q. Li, H. Yue, C. Liu, K. Ma, S. Zhong, B. Liang and S. Tang, Chem. Eng. J., 2020, 395, 125120 CrossRef CAS.
- A. Tanaka, K. Hashimoto and H. Kominami, J. Am. Chem. Soc., 2014, 136, 586–589 CrossRef CAS PubMed.
- N. Minh Vuong, D. Kim and H. Kim, Sci. Rep., 2015, 5, 11040 CrossRef PubMed.
- Y. Wang, B. Zhang, J. Liu, Q. Yang, X. Cui, Y. Gao, X. Chuai, F. Liu, P. Sun, X. Liang, Y. Sun and G. Lu, Sens. Actuators, B, 2016, 236, 67–76 CrossRef CAS.
- L. Wang, C.-S. Tsang, W. Liu, X. Zhang, K. Zhang, E. Ha, W.-M. Kwok, J. H. Park, L. Y. Suk Lee and K.-Y. Wong, J. Mater. Chem. A, 2019, 7, 221–227 RSC.
- W. He, H.-K. Kim, W. G. Wamer, D. Melka, J. H. Callahan and J.-J. Yin, J. Am. Chem. Soc., 2014, 136, 750–757 CrossRef CAS PubMed.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- H. Li, Z. Bian, J. Zhu, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 4538–4539 CrossRef CAS PubMed.
- V. Jovic, W.-T. Chen, D. Sun-Waterhouse, M. G. Blackford, H. Idriss and G. I. N. Waterhouse, J. Catal., 2013, 305, 307–317 CrossRef CAS.
- H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507–20515 CrossRef CAS PubMed.
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
-
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, 2016 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterizations of Au/WO3, schematic reactor, 1H-NMR and GC-MS spectra of the products. See DOI: 10.1039/d1sc00694k |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab






