
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed cascade reactions of triazoles with organoselenium compounds – a combined experimental and mechanistic study†
Fang
Li
 ,
Chao
Pei
and
Rene M.
Koenigs
*
,
Chao
Pei
and
Rene M.
Koenigs
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 24th March 2021
Abstract
Herein, we report on our studies on the reaction of organoselenium compounds with triazoles under thermal conditions using simple Rh(II) catalysts. These reactions do not provide the product of classic rearrangement reactions. Instead two different cascade reactions were uncovered. While allyl selenides react in a cascade of sigmatropic rearrangement and selenium-mediated radical cyclization reaction to give dihydropyrroles, cinnamyl selenides undergo a double rearrangement reaction cascade involving a final aza-Cope reaction to give the product of 1,3-difunctionalization. Theoretical and experimental studies were conducted to provide an understanding of the reaction mechanism of these cascade reactions. The former provide an important insight into fundamental question on the nature of the ylide intermediate in rearrangement reactions and reveal that organoselenium compounds take up multiple roles in rearrangement reactions and mediate a free ylide reaction mechanism.
Introduction
Sigmatropic rearrangement reactions constitute an important transformation for the construction of new C–C or C–heteroatom bonds via ylide intermediates.1,2 In this context, the formation of the pivotal ylide intermediate via the reaction of an electrophilic metal carbene complex with nucleophilic species, such as sulfides, ethers, amines, and others, has witnessed significant interest.2 Recently, important developments in the direction of asymmetric, metal-catalyzed [2,3]-sigmatropic rearrangements have been achieved using rhodium,3a copper,3b nickel,3c or enzyme catalysts.3d Similarly, latest developments in photochemical carbene transfer reactions have further stimulated this research area and emphasize the viability of sigmatropic rearrangement reactions from ylide intermediates under metal-free conditions.4 Despite these advances the reaction mechanism of such sigmatropic rearrangement reactions remains unclear and it is an ongoing riddle, if these reactions proceed via an ylide intermediate with the catalyst still bound, or if the catalyst dissociates to form a free ylide intermediate (Scheme 1a).5,6 Only recently, the Tantillo group reported on computational investigations on the reaction mechanism of [2,3]-sigmatropic rearrangement reactions of chalcogenonium ylides, and concluded that the crucial intermediate in the rearrangement step was strongly dependent on the nature of the chalcogenonium ylide.7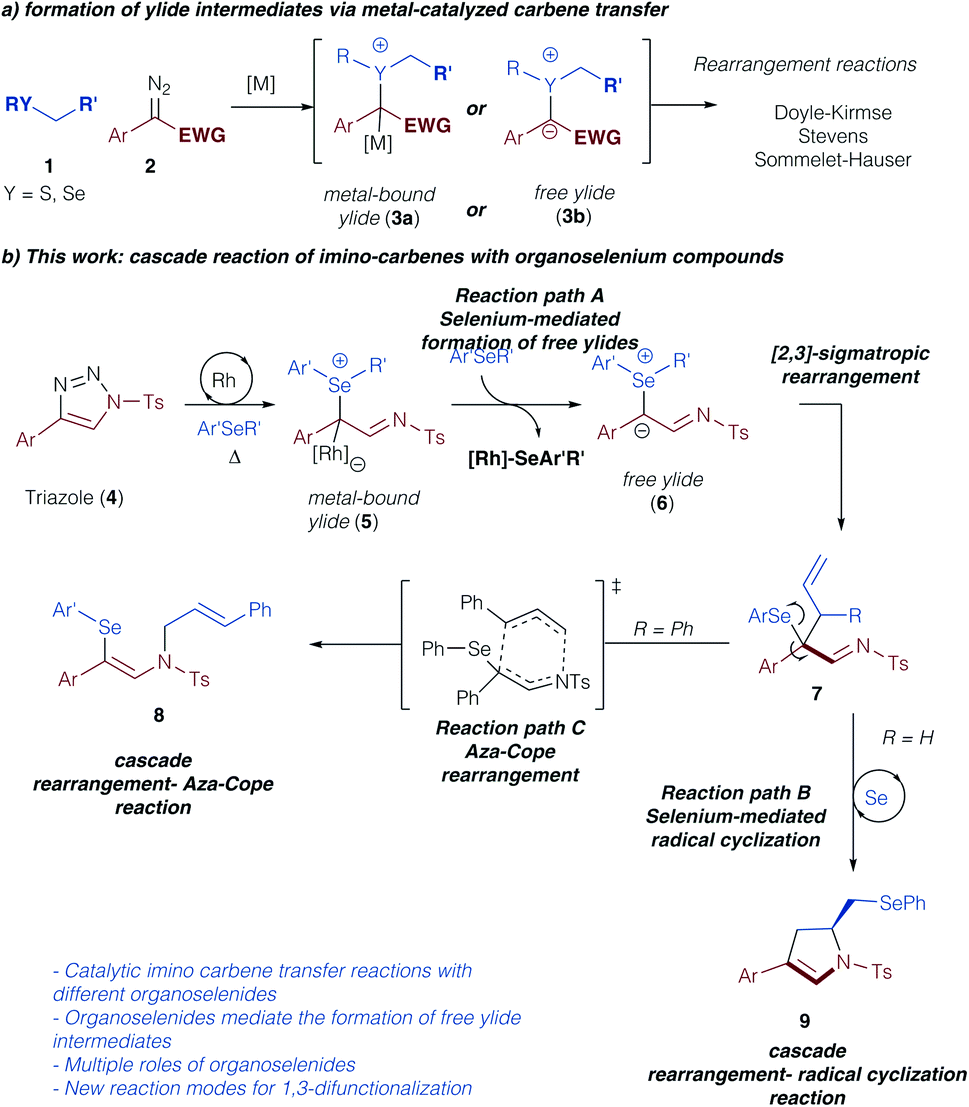 | ||
| Scheme 1 Ylide intermediates in sigmatropic rearrangement reactions. | ||
Triazoles represent a safe, non-diazo containing precursor to access the carbene reactivity following Dimroth rearrangement under thermal conditions as pioneered by Fokin and Murakami.8–10 Owing to our interest in rearrangement reactions of group VI elements and the nature of the respective ylide intermediates,5,6,11 we became intrigued in studying the reaction of triazoles with organoselenium compounds.11–13 Specifically, we assumed that the thermally forcing conditions that are required for the metal-catalyzed carbene transfer reactions from triazole precursors should influence the formation of metal-bound or free ylide intermediates. Moreover, the increased nucleophilicity and basicity of selenium vs. sulfur might lead to unexpected interactions of the selenium compounds with the catalyst and consequently impact the formation of a free ylide intermediate (7, Scheme 1b, path A).11 Last, the thermal reaction conditions should be ideally suited to access the unique reactivity of organoselenium compounds that can behave as versatile Lewis acidic catalysts or radical initiators.14 In this context, we hypothesized that a cascade reaction sequence of sigmatropic rearrangement reactions followed by selenium-mediated radical cyclization reaction could be achieved to rapidly construct a dihydropyrrole reaction product in a one-step procedure (9, Scheme 1b, path B). Alternatively, a rearrangement – aza-Cope reaction cascade could lead to the product 1,3-difunctionalization (8, Scheme 1b, path C). Accompanied by computational studies on the reaction mechanism, this combined experimental and computational study should therefore provide insight into the most demanding questions on the reaction mechanism of rearrangement reactions and open up new reaction pathways of ylide intermediates via cascade reactions.
Results and discussion
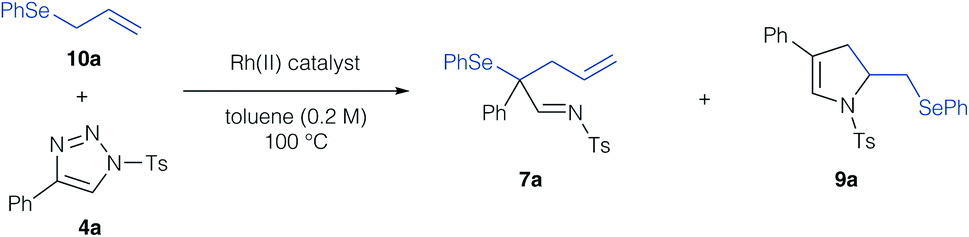
In a first step, we investigated the reaction of allylic selenides (10a, 11) and triazole 4a in the presence of Rh2(OAc)4 as catalyst to examine the viability of the [2,3]-sigmatropic rearrangement – cyclization reaction sequence. Indeed, allyl selenide 10a gave a mixture of the product of [2,3]-sigmatropic rearrangement (7a) and the dihydropyrrole 9a in good overall yield (Scheme 2a, 71% combined yield). The formation of the dihydropyrrole reaction product 9a raised our interest in further studying this reaction, and if the reaction conditions could be further improved to increase the yield of 9a. We initially examined, if 9a might be formed under thermal conditions from the rearrangement product 7a. Control experiments revealed the initial formation of the rearrangement product 7a as the sole product after 1 hour reaction time in 29%. When then treating isolated 7a under thermal conditions (100 °C) a near quantitative yield of 9a was observed, which underlines the relevance of a rearrangement step prior to the formation of the dihydropyrrole. In the case of cinnamyl selenide 11 however, 8a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8a′ was obtained as a 1:1 mixture as the sole reaction product, which corresponds to a formal 1,3-difunctionalization reaction of the imino carbene (Scheme 2b). We reason this unusual reactivity by the steric influence of the substituent of the allyl group. While an unsubstituted allyl group preferentially reacts in a [2,3]-sigmatropic rearrangement, steric hindrance in case of a cinnamyl-group results in preferential 1,3-difunctionalization reaction.
:8a′ was obtained as a 1:1 mixture as the sole reaction product, which corresponds to a formal 1,3-difunctionalization reaction of the imino carbene (Scheme 2b). We reason this unusual reactivity by the steric influence of the substituent of the allyl group. While an unsubstituted allyl group preferentially reacts in a [2,3]-sigmatropic rearrangement, steric hindrance in case of a cinnamyl-group results in preferential 1,3-difunctionalization reaction.
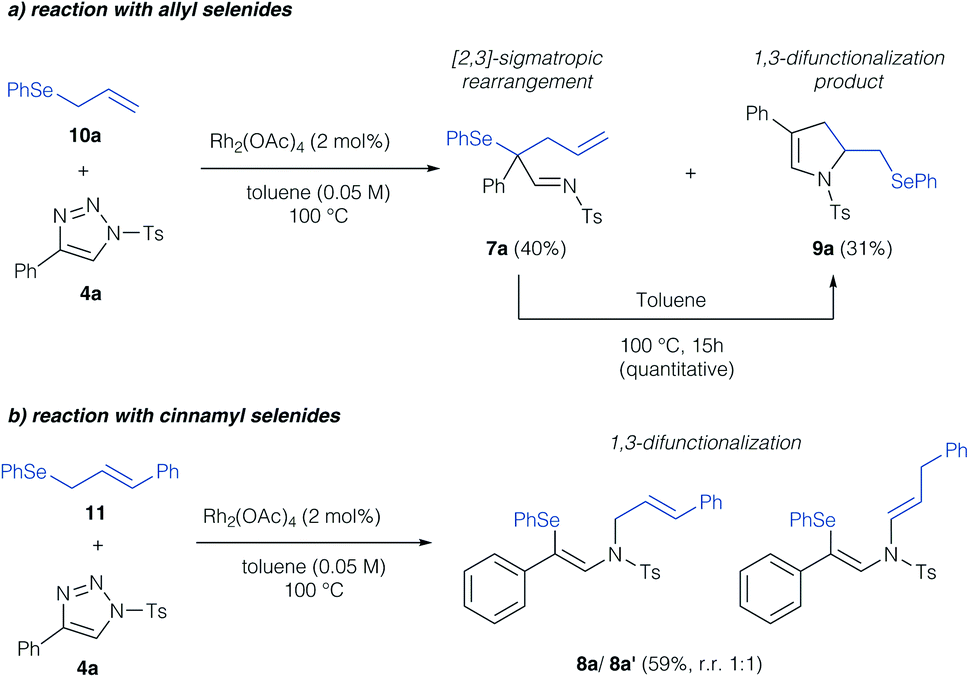 | ||
| Scheme 2 Initial studies on the reaction of organoselenides with triazoles. | ||
We next embarked on the reaction optimization (Table 1) and could identify that the reaction concentration had a significant effect on the product yield. When increasing the reaction concentration to 0.2 M, the dihydropyrrole reaction product was obtained in 86% yield and not even trace amounts of the rearrangement product were observed by NMR analysis of the crude reaction mixture. Further attempts to increase the yield concerned different reaction stoichiometry, solvents, reaction temperature or catalysts (Table 1, entries 4–10 and Table S1 in ESI†),15 yet no further improvement could be achieved. Importantly, high-boiling aromatic solvents gave high yields, chlorinated solvents resulted in slightly diminished yields. Importantly, Rh2(cap)4 resulted only in the decomposition of the triazole reaction partner 4a and no product formation was observed (Table 1, entry 11). To probe the potential of asymmetric carbene transfer reactions, we briefly evaluated the use of chiral Rh(II) catalysts, yet only a racemic mixture of product 9a was obtained (Table 1, entries 12 and 13).
|
|
|||
|---|---|---|---|
| Entrya | Catalyst | Further changes | Yieldb (%) (7a/9a) |
| a Reaction conditions: in an oven dried test tube 10a (1.0 equiv., 0.2 mmol) 4a (1.0 equiv.) and catalyst (2 mol%) were dissolved in 1 mL toluene and heated to 100 °C for 15 hours. b Isolated yields. | |||
| 1 | Rh2(OAc)4 | 0.05 M | 40/31 |
| 2 | Rh2(OAc)4 | 0.1 M | —/47 |
| 3 | Rh2(OAc)4 | — | —/86 |
| 4 | Rh2(OAc)4 | 3 equiv. Se 10a | —/63 |
| 5 | Rh2(OAc)4 | 2 equiv. triazole 4a | —/32 |
| 6 | Rh2(OAc)4 | 80 °C | —/22 |
| 7 | Rh2(OAc)4 | 120 °C | —/58 |
| 8 | Rh2(Oct)4 | — | —/21 |
| 9 | Rh2(Piv)4 | — | —/72 |
| 10 | Rh2(esp)2 | — | —/66 |
| 11 | Rh2(cap)4 | Dec. of 4a | |
| 12 | Rh2(S-DOSP)4 | —/62 (rac) | |
| 13 | Rh2(S-BTPCP)4 | —/36 (rac) | |
Polar coordinating solvents, such as 1,4-dioxane, were incompatible, which we assume to result from competing ylide formation with 1,4-dioxane solvent molecules. Increasing the amount of either reactant, reducing the reaction temperature or changes to the reaction concentration did not further improve the reaction yield (for details, please see Table S1 in ESI†).15
With the optimized conditions for this unusual 1,3-difunctionalization in hand, we next studied the substrate scope of this reaction (Scheme 3). We first studied the influence of the N-protecting group of the triazole reaction partner. Alkyl groups, electron-donating, and electron-withdrawing substituent at the aryl sulfonyl group were tolerated and in all cases the dihydro pyrrole was obtained as the only reaction product. Notably, the sterically demanding 2,4,6-tri-isopropyl phenyl protecting group resulted only a low yield of the cyclization product 9f. Alkyl sulfonyl protecting groups were poorly tolerated and the dihydro pyrrole was obtained in low yield (9g). In case of the low-yielding reactions (9f, 9g) the product of [2,3]-sigmatropic rearrangement was not observed and the dihydropyrrole was the only reaction product.
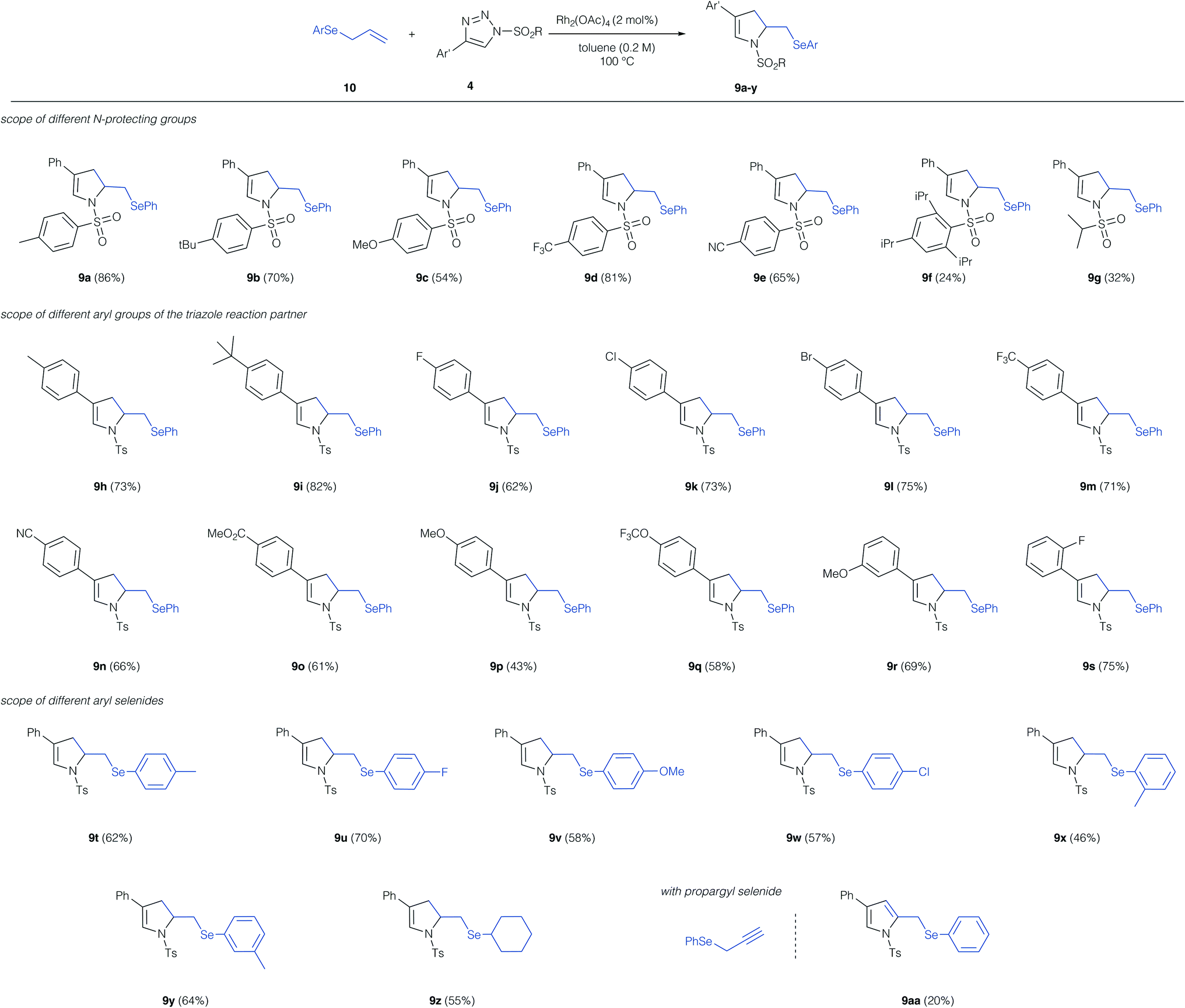 | ||
| Scheme 3 Investigations on the substrate scope of the cascade reaction of allyl selenides with triazoles. Reaction conditions: 10 (0.2 mmol, 1.0 equiv.), 4 (0.2 mmol, 1.0 equiv.) and Rh2(OAc)4 (2 mol%) are dissolved in toluene (1.0 mL) and heated to 100 °C for 15 h. Isolated yields are reported. | ||
We next studied the influence of the triazole reaction partner. Alkyl groups and halogens on the aromatic ring of triazole were well tolerated and the dihydropyrrole products were obtained in good yield. Similarly, electron-withdrawing groups, such as trifluoromethyl, nitrile or an ester proved compatible and the dihydropyrroles 9m, 9n and 9o were obtained in good yield. A strongly electron-donating methoxy-group in the para-position resulted in a significant reduction of the yield of product 9p, while no such effect was observed for an electron-donating group in the meta-position (9r). Finally, we studied different aryl and alkyl allyl selenides in this reaction; here alkyl groups, halogens and ethers were tolerated in ortho-, meta-, and para-position of the aromatic ring and the dihydropyrroles 9t–z were obtained in good yield. Finally, we employed a propargyl selenide, which reacted in a similar fashion to the cyclization product, yet, in this case the pyrrole product 9aa was obtained, albeit in low yield.
Studies on the reaction mechanism
To rationalize for the observed reaction of allyl selenides with triazoles and to gain an understanding on the relevance of metal-bound ylide vs. free ylide intermediates, we studied this reaction by DFT calculations (Scheme 4).15 These studies revealed an important interaction of the Lewis-basic selenide 10a with Rh2(OAc)4: one molecule of the selenide preferentially coordinates to the catalyst and both the selenium-bound rhodium catalyst and the free rhodium catalyst can undergo the formation the rhodium imino-carbene complex INT1 with similar absolute activation free energy. It is important to note that the formation of INT1via high-lysing TS1 is the rate-determining step of this reaction and that the Dimroth rearrangement of triazole readily proceeds with a rather low activation barrier.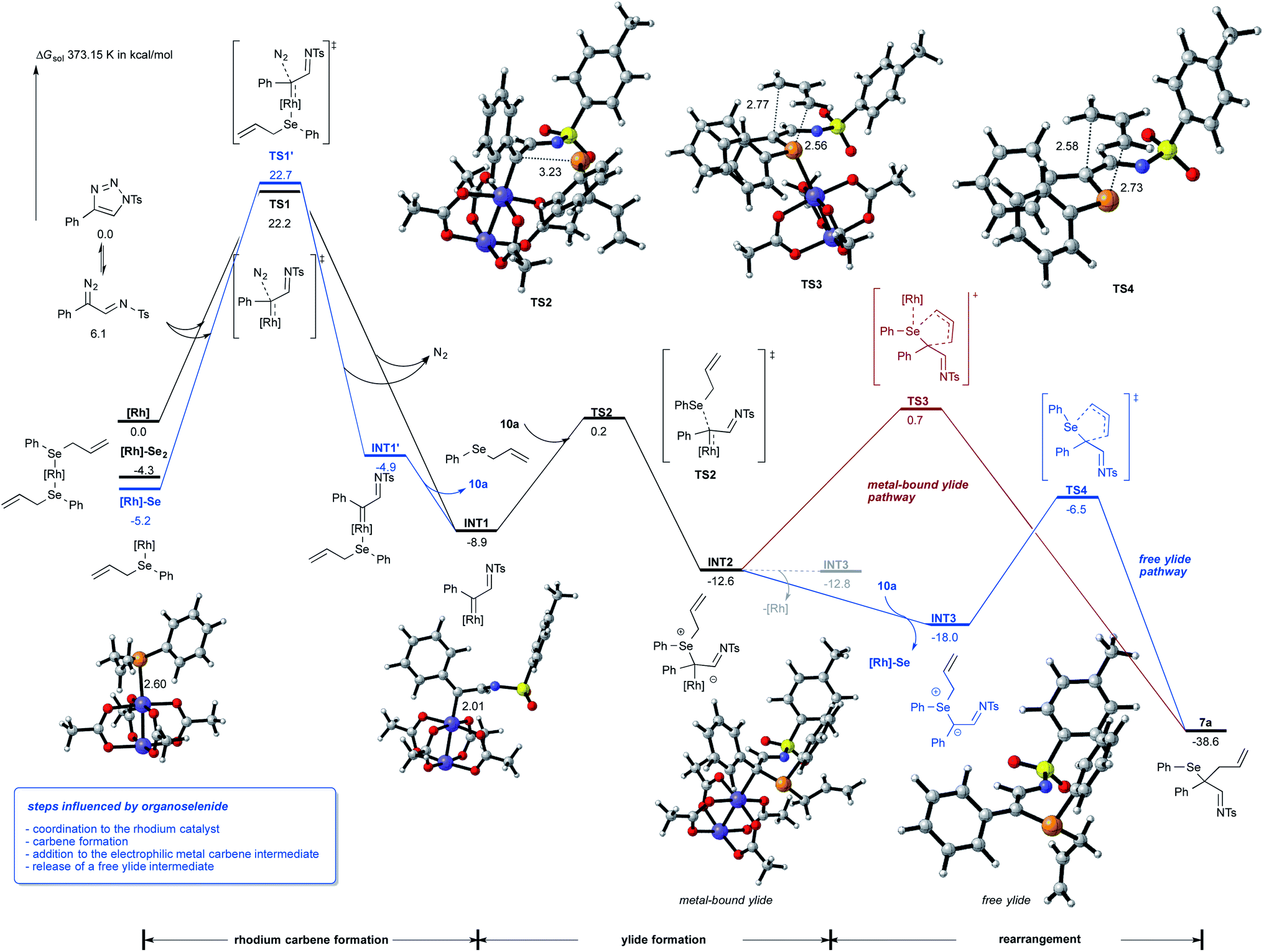 | ||
| Scheme 4 Influence of organoselenides on the catalyst, ylide intermediate and the rearrangement reaction. Calculations were performed at 373.15 K at the B3LYP-D3/def2-tzvp/SMD(toluene)//B3LYP-D3/def2-SVP/SMD(toluene) level of theory. | ||
We next assessed different reaction pathways involving one electron or two electron pathways for the rhodium imino-carbene intermediate INT1. A first pathway suggests the participation of a radical mechanism that involves the initial homolysis of the C–Se bond to form an allyl and selenyl radical. Analysis of the reaction of imino-carbene intermediate INT1 with the selenyl radical however gave no productive transition states. Contrarily, the addition of the allyl radical to the imino-carbene intermediate INT1 is a viable, low-energy process that could lead to the rearrangement product 7a. However, this one electron pathway is not favorable from both experiment and calculations, as no by-product formation from homocoupling of allyl or selenyl radical were observed and the bond cleavage of the C–Se bond of 10a is a high-energy process that requires an activation free energy of 32.4 kcal mol−1 (for details, please see Schemes S6 and S10 in ESI†).
We next assessed two electron pathways to account for the rearrangement reaction. A first pathway suggests a concerted reaction mechanism, that proceeds via 1,3 functionalization of INT1 and would directly lead to the dihydropyrrole reaction product viaTS5 with an activation free energy of 17.8 kcal mol−1 (please see Scheme S4 in ESI† for details).15 The formation of a metal-bound selenium ylide INT2 proceeds TS2 with an activation free energy of only 9.1 kcal mol−1 and is thus the favored reaction pathway.
The reaction of this metal-bound ylide intermediate can now proceed via different pathways: (a) transfer of the allyl group to the pendant nitrogen atom of the imino carbene proceeds viaTS6 and a very high activation free energy of 30.1 kcal mol−1 (please see Scheme S4 in ESI† for details);15 (b) sigmatropic rearrangement viaTS3 and a reasonable activation free energy of 13.3 kcal mol−1, which proceeds with concomitant transfer of the rhodium complex to the more Lewis-basic selenium atom (Scheme 4, red pathway) or (c) dissociation of the rhodium complex and formation of the free ylide intermediate INT3, which is 5.4 kcal mol−1 energetically more favored over the metal-bound ylide INT2. Importantly, one molecule of allyl selenide 10a promotes the formation of the free ylide via coordination to the backside of rhodium dimer and formation of [Rh]-Se catalyst as by-product. In the absence of selenide 10a the free ylide is very similar in energy as the metal-bound ylide and thus the back reaction to the metal-bound ylide can readily occur (see formation of INT3via the dotted line). In the last step, the free ylide can then undergo a very facile rearrangement reaction viaTS4 to give the product of classic [2,3]-sigmatropic rearrangement 7a (Scheme 4, blue pathway).
Next, we examined the reaction of the rearrangement product 7a and how the formation of the dihydropyrrole can be rationalized. We initially hypothesized that this step can be mediated either by a reaction mechanism that involves a selenyl cation or a selenium radical, which can be formed by hetero- or homolytic cleavage of the C–Se bond of 7a. Analysis of the bond dissociation energies revealed that homolysis of the C–Se bond is significantly favored (18.2 vs. 93.9 kcal mol−1 for homo vs. heterolytic bond cleavage; please also see Scheme S6 in ESI†),15 which now rationalizes a radical pathway for the cyclization reaction to give the dihydropyrrole product.
For the cyclization reaction, we could identify a reaction pathway that proceeds via radical addition of the selenyl radical to the terminal carbon of the double bond to give a secondary alkyl radical INT4. Subsequent cyclization and cleavage of a new selenyl radical viaTS10 furnishes the dihydropyrrole product with a total energy barrier of 19.5 kcal mol−1. An alternative pathway suggests the direct cyclization of an enamine radical that is formed via the homolytic cleavage of the C–Se bond. However, this pathway proceeds via relatively high-lying transition state (ΔGact = 25.6 kcal mol−1, for details, please see Scheme S8 in ESI†) and is thus not feasible.15 Studies aiming at the identification of radical pathways from the rhodium carbene complex INT1, did not lead to the desired reaction product (please see Scheme S10 in ESI†). This data is now suggesting that the cyclization reaction proceeds via a radical chain reaction (Scheme 5).
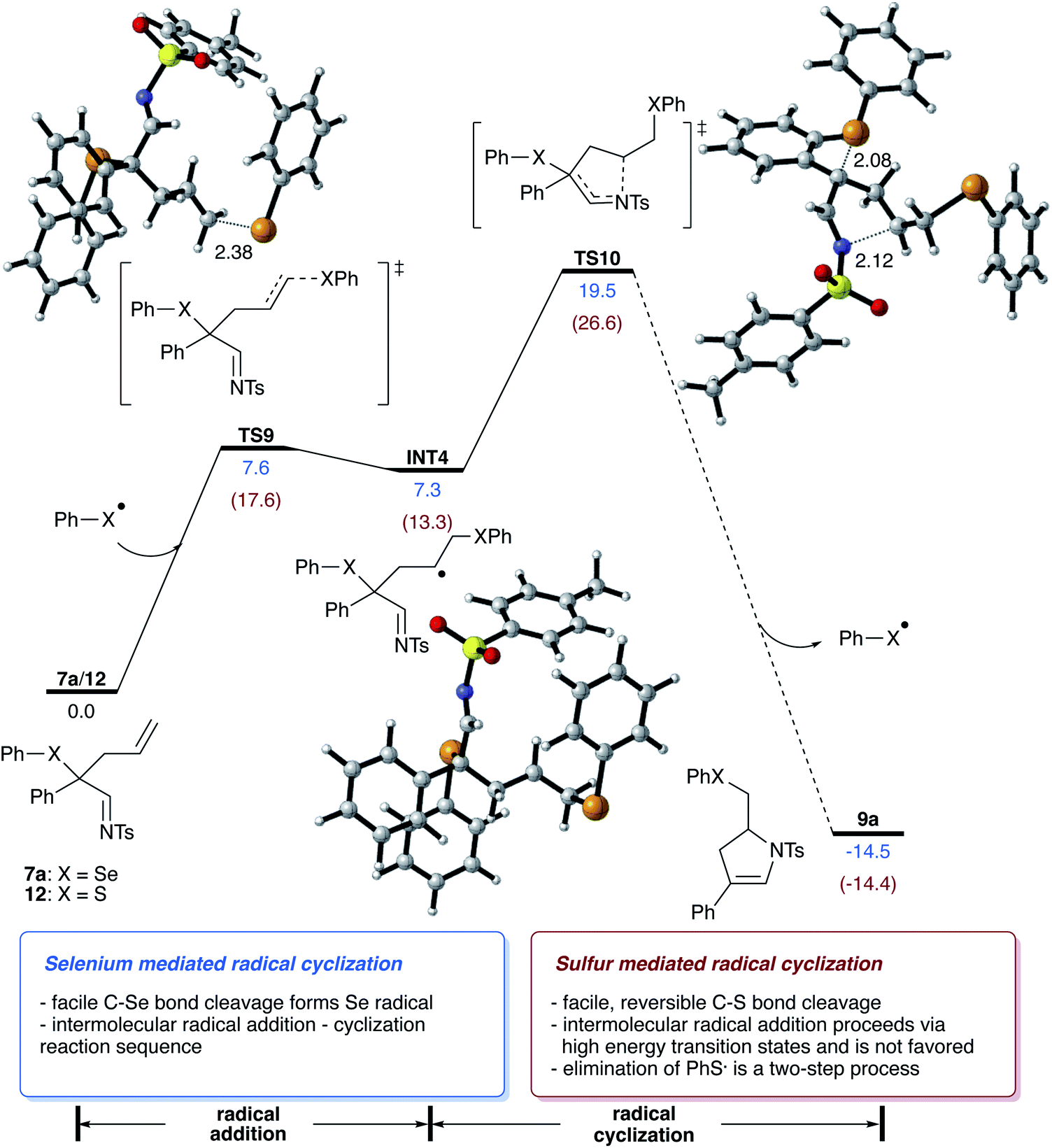 | ||
| Scheme 5 Studies on the cyclization reaction of homoallyl selenides and the formation of the dihydropyrrole. Calculations were performed at 373.15 K at the (U)B3LYP-D3/def2-tzvp/SMD(toluene)//(U)B3LYP-D3/def2-SVP/SMD(toluene) level of theory. Values in blue for 7a. Values in red for 12. | ||
In this context, we also performed theoretical calculations on the reaction of sulfide homologue of 12. In contrast to selenide 7a, the homolytic C–S bond cleavage of 12 proceeds with lower activation free energy and is thus a reversible process (ΔG = 14.1 kcal mol−1). In the case of sulfide 12, the radical cyclization process is a viable reaction pathway, yet radical addition to homoallylic sulfide 12 is significantly disfavored energetically and requires an activation free energy of 17.6 kcal mol−1, which is 10.0 kcal mol−1 higher compared to the radical addition of selenide 7a, which can now reason for the missing reactivity of sulfide 12 in the radical cyclization reaction.
To support these theoretical findings, we studied control experiments to identify, if the cyclization reaction indeed proceeds via a radical, intermolecular pathway. We thus examined a crossover experiment involving two different homoallyl selenides 7b and 7a (Scheme 6a). All four potential isomers (9ab, i, a, t) were formed, which is supportive of a reaction mechanism, in which the arylselenium group is transferred in an intermolecular fashion. To probe the radical character of this reaction, we examined the reaction in the presence of TEMPO as a radical trapping agent (Scheme 6b). No formation of the cyclization product 9a was observed, instead, the TEMPO adduct of the selenyl radical 13 could be determined by GCMS analysis and further substantiates the radical reaction mechanism.
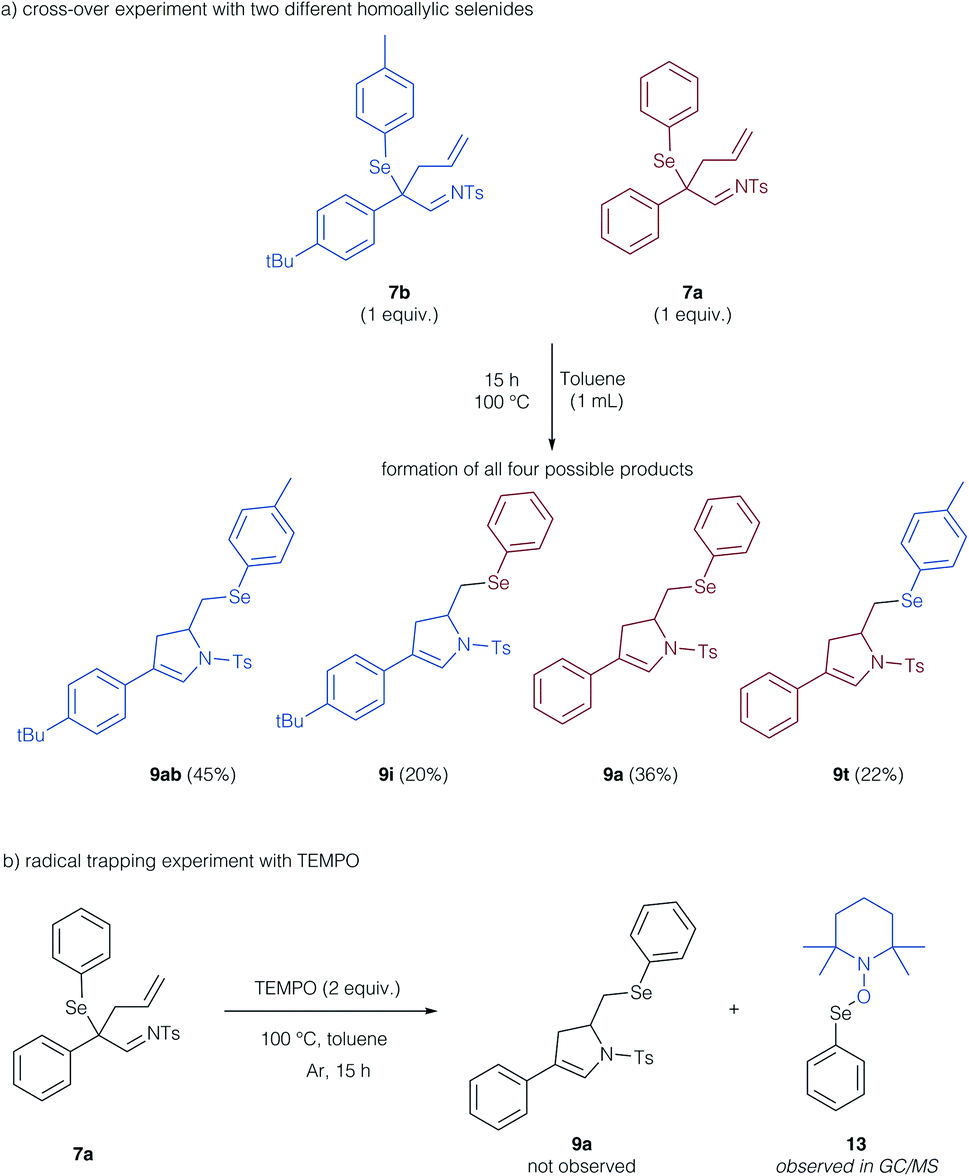 | ||
| Scheme 6 Control experiments on the formation of the dihydro pyrrole product. | ||
Next, we re-visited the reaction of cinnamyl selenide 11 that did not give the product of [2,3]-sigmatropic rearrangement, instead the product of 1,3-difunctionalization was observed (cf.Scheme 2b). The computational analysis suggests that a mechanism leading towards direct 1,3-difunctionalization proceeds via the unfavorable transition state TS3Ph with an activation free energy of 17.3 kcal mol−1 and thus does not represent a low-energy reaction pathway. Instead, the [2,3]-sigmatropic rearrangement process is favored by 9.7 kcal mol−1, to give the classic [2,3]-sigmatropic rearrangement product viaTS1Ph and an activation free energy of only 7.6 kcal mol−1 (for details, please see Scheme S11 in ESI†).15 This [2,3]-sigmatropic rearrangement product can subsequently undergo an aza-Cope rearrangement via low-lying TS2Ph to give the observed reaction product, which can be reasoned by formation of the higher-substituted double bond in the reaction product 8a. The comparison of this aza-Cope reaction pathway for cinnamyl- vs. allyl-substituted selenides shows a marked difference in the activation free energy of this step (Scheme 7). In the case of cinnamyl-substituted selenide 11, this step is significantly favored due to the stabilization of the transition state by π-interaction of the migrating cinnamyl group with the aromatic ring adjacent to the former carbene. This interaction is not possible in the case of the simple allyl-substituted selenide 10 and results in a higher activation free energy and thus preferential homolytic cleavage of the C–Se bond. Moreover, while the aza-Cope rearrangement is an exergonic reaction for cinnamyl selenide 11, it is an endergonic for allyl selenide and thus the aza-Cope rearrangement is disfavored for the latter for kinetic and thermodynamic reasons (Scheme 7a). A control experiment with a crotyl-substituted selenide 14 further underlines the relevance of this π-interaction (Scheme 7b). As in the case of the allyl selenide 10a, selective formation of the dihydropyrrole 15 is observed due to the absence of this π-interaction.
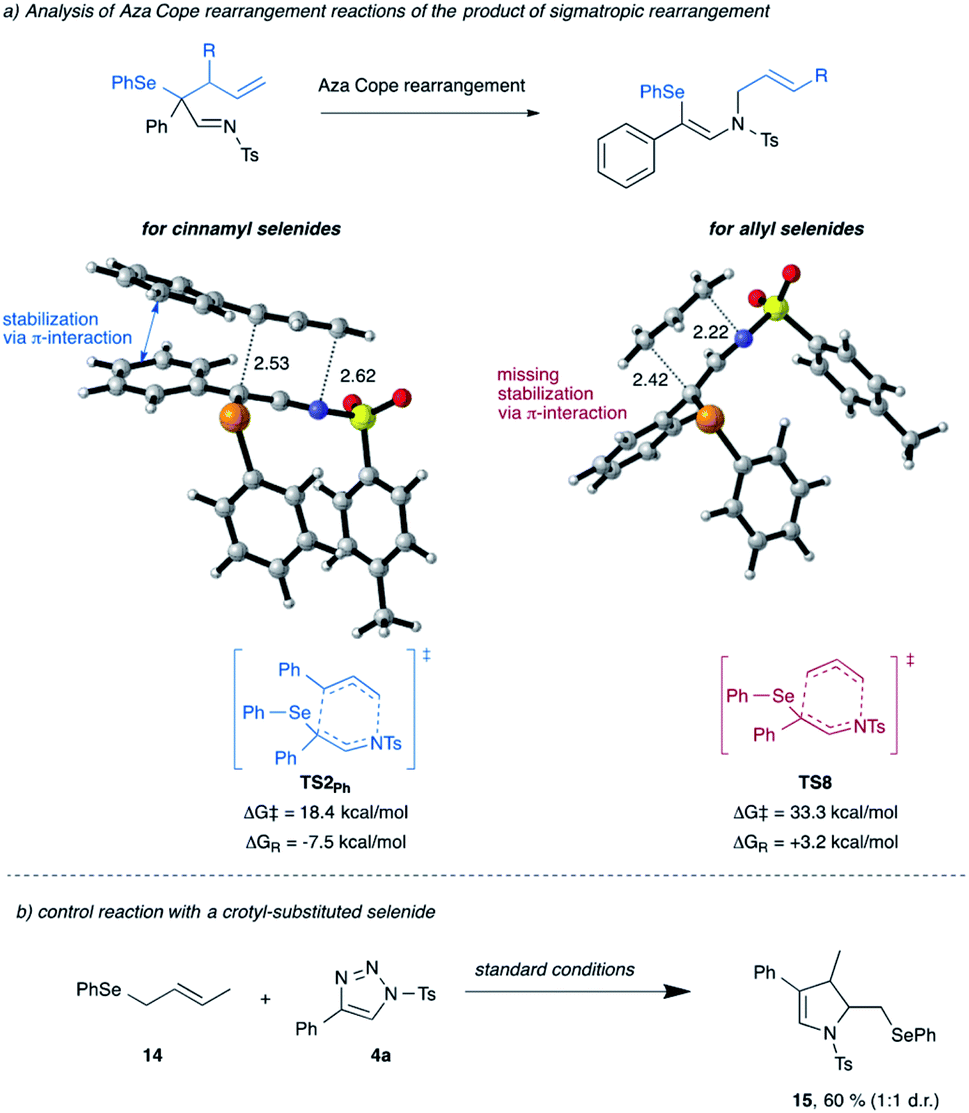 | ||
| Scheme 7 Relevance of the aza-Cope rearrangement on 1,3-difunctionalization or dihydropyrrole formation. Calculations were performed at 373.15 K at the B3LYP-D3/def2-tzvp/SMD(toluene)//B3LYP-D3/def2-SVP/SMD(toluene) level of theory. | ||
Conclusions
In summary, we herein report on the thermal reaction of triazoles with organoselenium compounds in the presence of Rh(II) catalysts. Depending on the organoselenium compound, two distinct cascade reactions of imino carbene intermediates were observed. DFT analysis of the reaction mechanism has demonstrated that these reactions proceed via a free ylide intermediate, the formation of which is facilitated by displacement of the ylide-ligand with a selenide ligand at the rhodium catalysts. The free ylide intermediate can then undergo [2,3]-sigmatropic rearrangement. Under the present reaction conditions the products of [2,3]-sigmatropic rearrangement can undergo, depending on the substitution pattern, either a radical cyclization reaction mediated by a selenyl radical or an aza-Cope rearrangement to give the final 1,3-difunctionalization products. The organoselenium compound thus plays an important role in the development of cascade reactions of imino carbenes and acts as substrate, mediator for the formation of the free ylide intermediate and initiator for the radical cyclization in case of allyl selenides.Author contributions
FL performed the experiments. CP performed theoretical calculations. RMK conceived this project and wrote the manuscript. All authors contributed to the editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RMK thanks the German Science Foundation and the Fonds of the Chemical Industry for financial support. Funded by the Federal Ministry of Education and Research (BMBF) and the Ministry of Culture and Science of the German State of North Rhine-Westphalia (MKW) under the Excellence Strategy of the Federal Government and the Länder. FL and CP gratefully acknowledge the China Scholarship Council for generous support.Notes and references
- (a) Z. Sheng, Z. Zhang, C. Chu, Y. Zhang and J. Wang, Tetrahedron, 2017, 73, 4011–4022 CrossRef CAS; (b) J. D. Neuhaus, R. Oost, J. Merad and N. Maulide, Top. Curr. Chem., 2018, 376, 15 CrossRef PubMed; (c) Y. Nishibayashi and S. Uemura, in Organoselenium Chemistry: Synthesis and Reactions, ed. T. Wirth, Wiley-VCH, 2012, pp. 287–320 Search PubMed.
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (b) H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (c) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (d) Y. Zhang and J. Wang, Coord. Chem. Rev., 2010, 254, 941–953 CrossRef CAS.
- (a) Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970 CrossRef CAS PubMed; (b) B. Xu and U. K. Tambar, Angew. Chem., Int. Ed., 2017, 56, 9868–9871 CrossRef CAS PubMed; (c) X. Li, Y. Tang, W. Yang, F. Tan, L. Lin, X. Liu and X. Feng, J. Am. Chem. Soc., 2018, 140, 3299–3305 CrossRef PubMed; (d) V. Tyagi, G. Sreenilayam, P. Bajaj, A. Tinoco and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 13562–13566 CrossRef CAS PubMed.
- (a) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203 CrossRef CAS PubMed; (b) S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC; (c) J. Yang, J. Wang, H. Huang, G. Qin, Y. Jiang and T. Xiao, Org. Lett., 2019, 21, 2654–2657 CrossRef CAS PubMed; (d) K. Orlowska, K. Rybicka-Jasinska, P. Krajewski and D. Gryko, Org. Lett., 2020, 22, 1018–1021 CrossRef CAS PubMed; (e) Z. Yang, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2019, 25, 6703–6706 CrossRef CAS PubMed.
- K. J. Hock and R. M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566–13568 CrossRef CAS PubMed.
- S. Jana, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2021, 27, 1270–1281 CrossRef CAS PubMed.
- C. J. Laconsay and D. Tantillo, ACS Catal., 2021, 11, 829–839 CrossRef CAS.
- (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed; (b) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS PubMed.
- (a) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (b) P. Anbarasa, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 46, 3004–3023 CrossRef.
- (a) T. Miura, Y. Fujimoto, Y. Funakoshi and M. Murakami, Angew. Chem., Int. Ed., 2015, 54, 9967–9970 CrossRef CAS PubMed; (b) T. Miura, T. Nakamuro, K. Hiraga and M. Murakami, Chem. Commun., 2014, 50, 10474–10477 RSC; (c) T. Miura, T. Tanaka, T. Biyajima, A. Yada and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 3883–3886 CrossRef CAS PubMed; (d) S. Chuprakov, B. T. Worrell, N. Selander, R. K. Sit and V. V. Fokin, J. Am. Chem. Soc., 2014, 136, 195–202 CrossRef CAS PubMed; (e) M. Zibinsky and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 1507–1510 CrossRef CAS PubMed; (f) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802–6805 CrossRef CAS PubMed; (g) T. Miura, T. Nakamura, S. G. Stewart, Y. Nagata and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 3334–3338 CrossRef CAS PubMed; (h) K. Pal, G. S. Sontakke and C. M. R. Volla, Org. Lett., 2019, 21, 3716–3720 CrossRef CAS PubMed; (i) K. Pal, A. Hoque and C. M. R. Volla, Chem.–Eur. J., 2018, 24, 2558–2564 CrossRef CAS PubMed; (j) A. C. S. Reddy, K. Ramachandran, P. M. Reddy and P. Anbarasan, Chem. Commun., 2020, 56, 5649–5652 RSC; (k) F. Li, C. Pei and R. M. Koenigs, Org. Lett., 2020, 22, 6816–6821 CrossRef CAS PubMed.
- (a) S. Jana and R. M. Koenigs, Org. Lett., 2019, 21, 3653–3657 CrossRef CAS PubMed; (b) S. Jana, P. Aseeva and R. M. Koenigs, Chem. Commun., 2019, 55, 12825–12828 RSC.
- Selected references on organoselenium compounds: (a) T. Wirth, Organoselenium Chemistry, ed. T. Wirth, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) V. K. Jain, Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments, Ed. V. K. Jain and K. I. Priyadarsini, Royal Society of Chemistry, 2018, pp. 1–33 Search PubMed; (c) T. Wirth, Angew. Chem., Int. Ed., 2000, 39, 3740–3749 CrossRef CAS.
- Y. Nishibayashi, K. Ohe and S. Uemura, J. Chem. Soc., Chem. Commun., 1995, 1245–1246 RSC.
- (a) A. Breder and C. Depken, Angew. Chem., Int. Ed., 2019, 58, 17130–17147 CrossRef CAS PubMed; (b) W. R. Bowman, Organoselenium Chemistry: Synthesis and Reactions, ed. T. Wirth, Wiley-VCH, Weinheim, 2012, p. 111 Search PubMed; for selected references: (c) X. Zhao, Z. Yu, T. Xu, P. Wu and H. Yu, Org. Lett., 2007, 9, 5263–5266 CrossRef CAS PubMed; (d) J. Trenner, C. Depken, T. J. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS; (e) E. Tang, Y. Zhao, W. Li, W. Wang, M. Zhang and X. Dai, Org. Lett., 2016, 18, 912–915 CrossRef CAS PubMed.
- For details, please see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00495f |
| This journal is © The Royal Society of Chemistry 2021 |