
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An amino acid based system for CO2 capture and catalytic utilization to produce formates†
Duo
Wei
 ,
Henrik
Junge
* and
Matthias
Beller
*
,
Henrik
Junge
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, Rostock, 18059, Germany. E-mail: henrik.junge@catalysis.de; matthias.beller@catalysis.de
First published on 2nd March 2021
Abstract
Herein, we report a novel amino acid based reaction system for CO2 capture and utilization (CCU) to produce formates in the presence of the naturally occurring amino acid L-lysine. Utilizing a specific ruthenium-based catalyst system, hydrogenation of absorbed carbon dioxide occurs with high activity and excellent productivity. Noteworthy, following the CCU concept, CO2 can be captured from ambient air in the form of carbamates and converted directly to formates in one-pot (TON > 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). This protocol opens new potential for transforming captured CO2 from ambient air to C1-related products.
000). This protocol opens new potential for transforming captured CO2 from ambient air to C1-related products.
Introduction
Carbon dioxide concentration in the atmosphere and global warming is ever-increasing with the enormous global energy demand supplied by consuming fossil fuels (mainly coal, oil, and natural gas).1,2 CO2 capture and storage (CCS) enable the use of fossil fuels with significantly lower CO2 emissions than usual.3 CCS is based on the separation of CO2 from energy conversion or other industrial processes, followed by compression, transport, and storage. However, CCS processes are meanwhile energy intensive as the electricity burden with amine scrubbing (113 kW h per mt CO2 removed) constitutes the minimum work to separate and compress CO2 (150 bar). Indeed, in two demonstration units, Boundary Dam and Thompsons, 210–220 kW h per mt were required for this purpose.4 Developing novel CO2 capture and utilization (CCU) methods for converting CO2 from air or flue gas not only saves energy from CCS (mainly CO2 desorption and compression steps) but also provides C1-related products (Scheme 1a).5–12 It's thus an important opportunity for developing a sustainable economy.13–15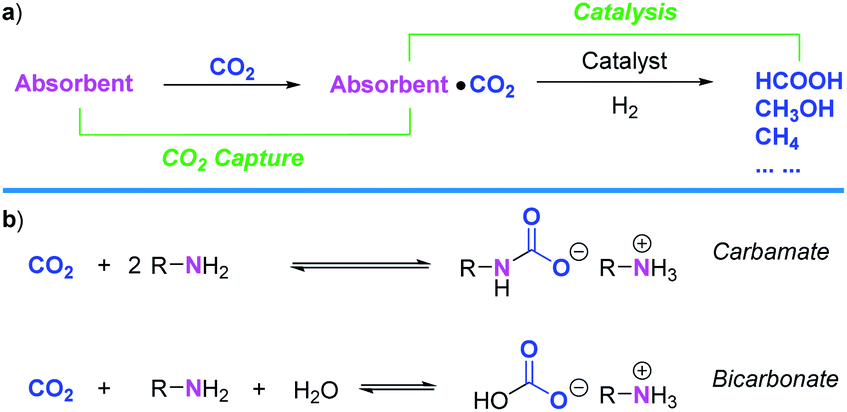 | ||
| Scheme 1 (a) Schematic CCU concept for CO2 hydrogenation to C1 products. (b) Reaction pathways for CO2 absorption with amines under aqueous conditions. | ||
In nature, inorganic carbon (particularly CO2) is converted to organic compounds by living organisms, which is known as carbon fixation, with photosynthesis as the most prominent example.16 It is estimated that approximately 258 billion tons of CO2 are converted into biomass by photosynthesis annually.17 As the most abundant protein on the Earth, ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO) is involved in the first major step of carbon fixation by plants and other photosynthetic organisms.18,19L-Lysine (Lys) is one of the six crucial amino acids (AAs) that are part of the active site of RuBisCO and it stabilizes CO2 in the form of carbamate for subsequent enzyme catalysis.20
By contrast, in industry e.g., power plants, the most common process for capturing CO2 relies on the use of aqueous amine solutions (Scheme 1b).3,21,22 However, the maximum CO2 absorption capacity for an amine system varies based on which products are formed. When carbamates are the preferred products, this capacity is 50 mol% per amines at most. If bicarbonates are mainly formed, this capacity could reach up to 100 mol% per amines. Alkanolamines have been extensively investigated as chemical absorbents;23 however, their large-scale use also created some environmental concerns. Substituting such conventional amine absorbents with high boiling and innocuous natural AAs in combining CO2 capture and catalysis is therefore highly relevant. Noteworthy, CO2 capture with aqueous AAs,24–27 including Lys25 was already reported, but not its direct valorization. Based on the infusive phenomenon of carbon fixation by RuBisCO and our long-term interest in CO2 reduction, we report herein a CCU process which enables CO2 capture from ambient air and its conversion to formate in the presence of L-lysine. Moreover, to the best of our knowledge, there exists no example of catalytic hydrogenation of CO2 assisted by AAs.
Several Rh- and Ru-based homogeneous catalysts have been previously reported for CO2 capture and in situ hydrogenation to C1 products (Fig. 1).8,28 In 2013, pioneering work was performed by the group of He utilizing RhCl3·3H2O and phosphine ligands, for instance CyPPh2, DPEphos, and PPh3, as catalysts where gaseous CO2 was absorbed by polyethyleneimine (PEI),29 amidines,30 and potassium phthalimide31 as well as hydrogenated in situ to formates or formic acid.
 | ||
| Fig. 1 Representative catalysts and absorbents for CO2 hydrogenation to C1 products applied in CCU processes. The highest TON (turnover number) of formates or methanol are shown in parentheses, respectively. | ||
In addition, ruthenium complexes have also been proven to be suitable catalysts for the hydrogenation of captured CO2 to formate or methanol. In 2014, Heldebrant and co-workers captured CO2 by DBU in methanol to form the methyl carbonate, which then was hydrogenated to formates catalyzed by [RuCl2(PPh3)3].32 One year later, Milstein and co-workers reported a CCU approach, where CO2 reacted with aminoethanols yielding oxazolidinones which were hydrogenated to CH3OH in 78–92% yield with a Ru-PNN pincer catalyst.33 In the same year, the Sanford group reported the CO2 capture with NHMe2 to form carbamate and subsequent hydrogenation to a mixture of DMF and CH3OH catalyzed by Ru-MACHO-BH complex.34 Employing the same catalyst and tetramethylguanidine (TMG),35 metal hydroxides,36 pentaethylenehexamine (PEHA),37–39 a mixture of metal hydroxides,40 or a tertiary amine41 with ethylene glycol as CO2 absorbent systems, Prakash and his colleagues combined CO2 capture from air with subsequent hydrogenation to produce formates or methanol. Recently, the group of Heldebrant reported a method where epoxides reacted with CO2 leading to cyclic carbonates. Then, in situ hydrogenation took place into methanol and glycol, with Ru-MACHO as catalyst.42
Compared to methanol, no hydrogen is lost in the form of water when formic acid or formate salts are produced by CO2 hydrogenation. Currently, formic acid is industrially produced by carbonylation of methanol to methyl formate and subsequent hydrolysis.43 It is mainly used as a preservative and antibacterial agent in livestock feed, e.g. silage and winter feed for cattle. In addition, formic acid is utilized in the production of leather and in dyeing and finishing textiles. More recently, it also gained interest as hydrogen storage medium as it contains 4.4 wt% of hydrogen with 53 g H2 per L of volumetric storage density.7
Results and discussion
CO2 capture with amino acids
For the development of a CCU concept to produce formic acid or formates, suitable CO2 absorbents must be used. Inspired by the carbon fixation pattern in nature, specifically RuBisCO, we considered applying AAs for this purpose.24–27 Thus, at the start of our investigations, we evaluated the ability of 12 different AAs, including the 6 ones involved in the active site of RuBisCo and some analogues to capture CO2. For this purpose, CO2 (2 bar) was charged into an aqueous solution of the respective AAs (5 M) and stirred at r.t. for 2–18 h.As shown in Table S1,† most of the tested systems such as L-proline, L-glutamine, and L-histidine achieved only small to moderate amounts of CO2 capture, around 0.1 mol of CO2 per mol of AA (CO2/AA), (Table S1, entries 1–11†). Interestingly, in the presence of L-lysine (Lys), a significantly improved performance (3.63 mmol of captured CO2, corresponding to 0.73 CO2/Lys) was obtained in 18 h (Table S1, entry 12†). Such high CO2 capture efficiency could be attributed to the basic side chain of Lys, as its pKa value is 10.7.
Thus, we investigated the effect of Lys for CO2 absorption under various conditions (Table 1 and Fig. S2 to S11†). As mentioned vide supra, carbon dioxide can be captured in form of Lys carbamates26 or Lys ammonium bicarbonate.44 Applying 20 bar of CO2, 0.83 CO2/Lys were obtained within only 0.5 h leading to carbamates and bicarbonate (ratio of 1:4, 98% total yield; Table 1, entry 1). A similar result was observed after 3 h (Table 1, entry 2). Also, at lower CO2 pressure (2 bar), significant absorption was achieved with 69–98% total yield of carbamates and bicarbonate within 0.5–18 h (Table 1, entries 3–5). Interestingly, in these cases (0.5 h and 3 h), mainly Lys carbamates were obtained. This shows that initially the corresponding carbamates are formed, which subsequently form bicarbonate.
| Entry | CO2 source | Time | Carbamatesb [mmol] | Bicarbonateb [mmol] | Yieldc [%] | CO2/Lysd |
|---|---|---|---|---|---|---|
| a Conditions: Lys (5.0 mmol), H2O (1.0 mL), stirred at r.t. Air bubbling: 1 L min−1. b Determined by 13C NMR-quant with THF (406.2 μL, 5.0 mmol) as internal standard. c Total yield of carbamates and bicarbonate based on Lys. d Mols of CO2 captured per mol of Lys. e THF (1 mL) as solvent. f Neat condition (without solvent). g Lys 20.0 mmol. n.d. = not detectable. Experiments were performed at least twice; average values are used (st. dev. < 10%). | ||||||
| 1 | CO2 (20 bar) | 0.5 h | 0.75 | 3.40 | 98% | 0.83 |
| 2 | CO2 (20 bar) | 3 h | 0.45 | 3.80 | 94% | 0.85 |
| 3 | CO2 (2 bar) | 0.5 h | 1.53 | 0.37 | 69% | 0.38 |
| 4 | CO2 (2 bar) | 3 h | 1.83 | 1.22 | 98% | 0.61 |
| 5 | CO2 (2 bar) | 18 h | 1.25 | 2.38 | 98% | 0.73 |
| 6e | CO2 (2 bar) | 3 h | 1.18 | n.d. | 47% | 0.24 |
| 7f | CO2 (2 bar) | 3 h | 0.30 | n.d. | 12% | 0.06 |
| 8 | Air | 1 d | 1.40 | n.d. | 56% | 0.28 |
| 9 | Air | 2 d | 1.95 | n.d. | 78% | 0.39 |
| 10 | Air | 4 d | 2.42 | n.d. | 97% | 0.48 |
| 11 | Air | 8 d | 2.45 | n.d. | 98% | 0.49 |
| 12g | Air | 4 d | 8.20 | n.d. | 82% | 0.41 |
Besides water, the aprotic solvent THF was applied. After 3 h exclusively the carbamate was formed (1.18 mmol corresponding to 0.24 CO2/Lys, Table 1, entry 6). A much lower CO2/Lys ratio (0.06) was observed under neat conditions (without solvent, Table 1, entry 7). Next, to demonstrate the viability of our general CCU methodology, ambient air, containing ca. 415 ppm (parts per million) CO2, was bubbled through Lys solution for 1–8 days (Fig. S1†). Indeed, up to 0.49 mol CO2 per mol Lys were absorbed representing a yield of 98% with carbamates as sole products. Performing the reaction on multi-g scale (20 mmol Lys), 8.20 mmol CO2 were captured corresponding to 0.41 CO2/Lys and 82% carbamate yield (Table 1, entry 12).
Catalytic hydrogenation of CO2 to formate
Next, to identify a suitable reduction system, various metal pincer complexes were tested for the hydrogenation of gaseous CO2 in the presence of different amino acids (Tables 2 and S2, Fig. S12 and S13†). To our delight, testing the Ru-MACHO-BH complex (Ru-1, 0.2 mol%) in H2O/THF (1:1 mixture) revealed significant activity in the presence of Lys for the hydrogenation of gaseous CO2 to formate (71% yield based on Lys) at 145 °C (Table S2, entry 1†).
|
|
|||
|---|---|---|---|
| Entry | Cat. [μmol, ppm] | Formateb [mmol] | % Yieldc (TON)d |
| a Conditions: catalyst, Lys (5.0 mmol), H2O (5.0 mL), THF (5.0 mL), CO2 (20 bar), H2 (60 bar), 145 °C, 12 h. b Determined by 1H NMR with DMF (250 μL, 3.24 mmol) as internal standard. c Calculated by formate [mmol]/Lys [mmol]. d Calculated by formate [mmol]/catalyst [mmol]. e 3 h. n.d. = not detectable. Experiments were performed at least twice; average values are used (st. dev. < 10%). | |||
| 1 | Ru-1 [2.0, 400] | 4.37 | 87 (2187) |
| 2 | Ru-1 [0.2, 40] | 3.89 | 78 (19440) |
| 3 | Ru-1 [0.02, 4] | 3.95 | 79 (197559) |
| 4 | Ru-2 [0.02, 4] | 4.24 | 85 (212139) |
| 5 | Ru-2 [0.01, 2] | 1.48 | 30 (147906) |
| 6e | Ru-1 [0.02, 4] | 2.77 | 55 (138510) |
| 7e | Ru-2 [0.02, 4] | 2.90 | 58 (144990) |
| 8e | Ru-3 [0.02, 4] | 0.29 | 6 (14580) |
| 9e | Ru-4 [0.02, 4] | 2.35 | 47 (117450) |
| 10e | Fe-1 [0.02, 4] | n.d. | — |
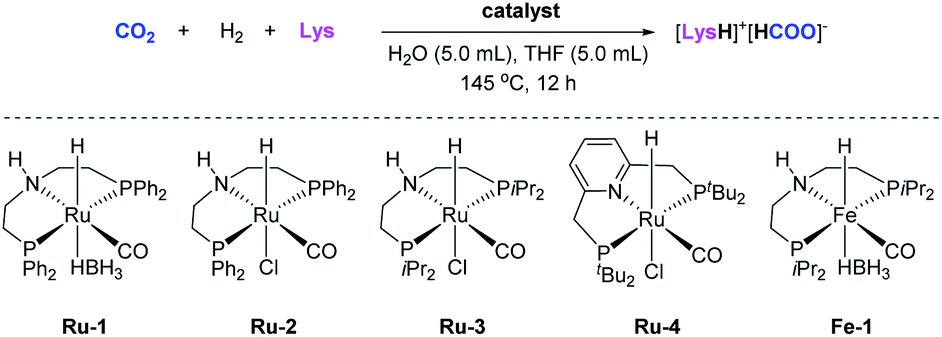
On the other hand, L-cysteine, L-histidine, L-serine, and L-threonine led to formates in much lower yields (up to 13%), while other AAs, such as glycine, L-proline, and L-glutamine showed no activity at all in the presence of catalyst Ru-1, (Table S2, entries 2–12†). Taking Lys as a benchmark CO2 absorbent, the TON of formate can be considerably increased from 2187 to 197559 when decreasing the loading of Ru-1 from 400 ppm (based on Lys) to 4 ppm (Table 2, entries 1–3). With 4 ppm of Ru-MACHO (Ru-2) as catalyst, the highest TON 212139 was achieved (Table 2, entry 4). Interestingly, in these reactions, CO2 was selectively converted to formate in up to 87% yield with less than 1% of formamide. Next, several ruthenium pincer complexes were tested at 4 ppm loading for the hydrogenation of gaseous CO2 in the presence of Lys within 3 h (Table 2, entries 6–10). Ru-1 and Ru-2 gave formate in 55% and 58% yields, respectively, whereas Ru-MACHOiPr (Ru-3) was less active leading to formate in only 6% yield. With Milstein's Ru-PNP complex (Ru-4) as catalyst, formate was obtained in 47% yield. However, no formate can be detected in the reaction catalyzed by Fe-MACHOiPr-BH complex (Fe-1).
Several blank reactions were also carried out (Table S3†): in the absence of either Lys, Ru-1, or CO2, no formate was detectable. These results clearly demonstrate that Lys and Ru-1 are both crucial to promote the hydrogenation of CO2 from air to formate. Reactions with other solvents, for example, triglyme, methanol, ethylene glycol or their 1:1 mixture with water could not improve the reaction efficiency (Table S4†). When replacing THF with the more eco-friendly green solvent 2-methyltetrahydrofuran (2-MTHF),45 a comparable yield of formate (86%) was observed. Lowering the temperature from 145 to 105 °C, the yield of formate decreased only slightly from 79% to 64% (Table S5†).
Development of a general CCU concept
After having studied the individual processes of (a) CO2 absorption and (b) CO2 reduction in the presence of Lys, the overall CCU concept was demonstrated by combining CO2 capture and in situ hydrogenation to formate (Table 3 and Fig. S14–S16†).|
|
||||
|---|---|---|---|---|
| Entry | Captured CO2 [mmol] | Cat. [μmol] | Formateb [mmol] | % Yieldc (TON)d |
| a Conditions: CO2 captured from air within 4 d applying 5 mmol Lys, given amount of catalyst dosed from stock solution, H2O (5.0 mL), THF (5.0 mL), H2 (80 bar), 145 °C, 12 h. b Determined by 1H NMR with DMF (250 μL, 3.24 mmol) as internal standard. c Calculated by formate [mmol]/captured CO2 [mmol]. d Calculated by formate [mmol]/catalyst [mmol]. e CO2 captured with 20 mmol Lys. Experiments were performed at least twice; average values are used (st. dev. < 10%). | ||||
| 1 | 2.42 | Ru-1 [2.0] | 1.10 | 46 (551) |
| 2 | 2.42 | Ru-1 [0.85] | 1.15 | 48 (1353) |
| 3 | 2.42 | Ru-1 [0.17] | 1.02 | 42 (6004) |
| 4 | 2.42 | Ru-1 [0.02] | 1.10 | 45 (54998) |
| 5 | 2.42 | Ru-2 [0.02] | 1.04 | 43 (52245) |
| 6e | 8.20 | Ru-1 [0.08] | 2.40 | 29 (29993) |
| 7e | 8.20 | Ru-2 [0.08] | 3.31 | 40 (41330) |
| 8e | 8.20 | Ru-2 [0.04] | 1.00 | 12 (25110) |

Using captured CO2 (2.42 mmol) as substrate in the presence of Ru-1 (2.0 μmol) as catalyst, 46% formate yield (based on captured CO2) was obtained (TON 551; Table 3, entry 1). The highest TON reached 54998 with 0.02 μmol Ru-1, while the yield was maintained at 45% (Table 3, entries 2–4). Ru-2 showed comparable activity for the hydrogenation of captured CO2 yielding 43% of formate (Table 3, entry 5). With 8.20 mmol captured CO2, 29% of formate were obtained with Ru-1 at 0.08 μmol loading (Table 3, entry 6). 3.31 mmol formate (40% yield) were obtained with the same amount of Ru-2 (Table 3, entry 7).
Finally, some Lys analogues and derivatives as well as selected benchmark amines35,37,39 were applied according to our overall protocol (Fig. 2). In the presence of 6-aminohexanoic acid and 1,5-diaminopentane, 0.12 and 0.82 CO2/amine were achieved and formates were obtained in yields of 25% and 34%, respectively. Noticeably, 2,3-diaminopropanoic acid and the simplest amino acid glycine did not show any activity in both CO2 absorption and hydrogenation processes. In the case of TMG and PEHA, CO2 was captured with 0.86 and 0.83 CO2/TMG or PEHA, respectively. However, the presence of TMG inhibited the hydrogenation of CO2, whereas PEHA led to formate and formamides in 38% and 8% yield, respectively. Applying the inorganic base NaOH36 resulted in a CO2/base ratio of 1.08 and 23% formate yield. All these experiments demonstrate the superiority of using Lys for carbon dioxide capture and direct valorizations. It also indicates the crucial presence of an α-amino acid moiety and an additional amine function in the side chain of AA.
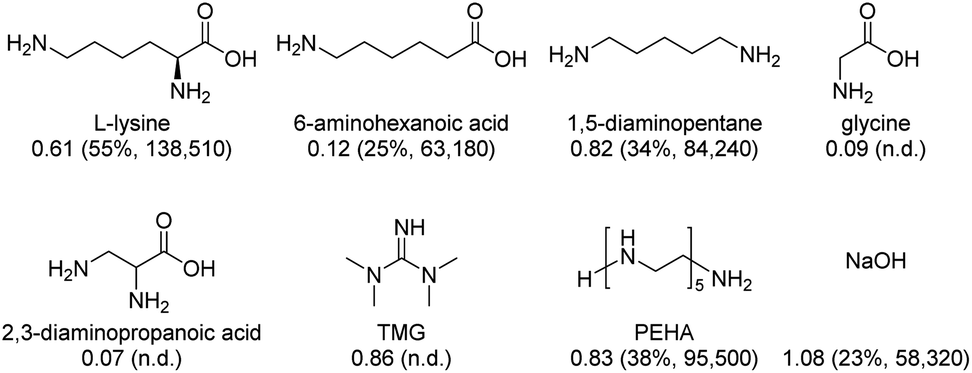 | ||
| Fig. 2 Various Lys analogues and benchmark amines applied in the CO2 absorption and hydrogenation processes performed under conditions in Table 1, entry 4 and Table 2 entry 6, respectively. CO2/amine (mols of CO2 captured per mol of amine) are shown with yield and TON of formates in parentheses; n.d. = not detectable. | ||
To rationalize the perfect selectivity towards formates in the current study, we conducted further experiments by heating up the mixture of formic acid and Lys or PEHA in H2O at 145 °C (Table S6†). Indeed, Lys led to formate in quantitative yield without any formamide detectable after 12 h, whereas PEHA gave 28% yield of formamide along with 71% formate. Obviously, the less basic conditions applying Lys (pH 10.2 for a 5 M aqueous solution) prevented the formation of formamides taking place in the presence of PEHA (pH 13.4).
Conclusions
In conclusion, we described an amino acid based catalyst system for the highly relevant CO2 capture and utilization (CCU) process to produce formates in one-pot. The naturally occurring amino acid L-lysine affords formate generation with a high efficiency. Among the investigated catalysts, the most active ones are identified with Ru-MACHO complexes (Ru-1 and Ru-2) for the hydrogenation of gaseous CO2 (TON > 210000) and the in situ hydrogenation of captured CO2 (TON > 50000). Noteworthy, in the present CCU concept, CO2 can be captured from ambient air in the form of carbamates and hydrogenated to formate directly.
Author contributions
D. W. conducted all the experimental work, collected and analyzed the data. D. W., H. J. and M. B. wrote the paper. H. J. and M. B. proposed and supervised the project. All the authors discussed the results and commented on the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the State of Mecklenburg-Vorpommern, the Danish government for funding the CADIAC excellence cluster, and the Leibniz-Program Cooperative Excellence K308/2020. We thank the analytical team of LIKAT for their kind support. This work is in memory of Prof. Dr Paul Kamer.References
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- G. T. Rochelle, in Absorption-Based Post-combustion Capture of Carbon Dioxide, ed. P. H. M. Feron, Woodhead Publishing, 2016, pp. 35–67, DOI:10.1016/B978-0-08-100514-9.00003-2.
- M. Aresta, Carbon dioxide as chemical feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- W.-H. Wang, X. Feng and M. Bao, Transformation of Carbon Dioxide to Formic Acid and Methanol, Springer, Singapore, 2018 Search PubMed.
- K. Dong, R. Razzaq, Y. Hu and K. Ding, Top. Curr. Chem., 2017, 375, 203–228 CrossRef PubMed.
- Y. Li, Z. Wang and Q. Liu, Chin. J. Org. Chem., 2017, 37, 1978–1990 CrossRef CAS.
- W. H. Bernskoetter and N. Hazari, Acc. Chem. Res., 2017, 50, 1049–1058 CrossRef CAS PubMed.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 RSC.
- A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- A. Bar-Even, E. Noor, N. E. Lewis and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8889–8894 CrossRef CAS PubMed.
- R. J. Geider, E. H. Delucia, P. G. Falkowski, A. C. Finzi, J. P. Grime, J. Grace, T. M. Kana, J. La Roche, S. P. Long and B. A. Osborne, Glob. Change Biol., 2001, 7, 849–882 CrossRef.
- T. D. Sharkey, Photosynth. Res., 2019, 140, 235–252 CrossRef CAS PubMed.
- U. Feller, I. Anders and T. Mae, J. Exp. Bot., 2008, 59, 1615–1624 CrossRef CAS PubMed.
- B. Stec, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18785–18790 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS.
- F. Barzagli, F. Mani and M. Peruzzini, Energy Environ. Sci., 2009, 2, 322–330 RSC.
- P. D. Vaidya and E. Y. Kenig, Chem. Eng. Technol., 2007, 30, 1467–1474 CrossRef CAS.
- A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu and Q.-W. Song, Angew. Chem., Int. Ed., 2012, 51, 11306–11310 CrossRef CAS PubMed.
- A.-A. Al-Terkawi, F. Lamaty and T.-X. Métro, ACS Sustainable Chem. Eng., 2020, 8, 13159–13166 CrossRef CAS.
- Y. Yamamoto, J. y. Hasegawa and Y. Ito, J. Phys. Org. Chem., 2012, 25, 239–247 CrossRef CAS.
- S. Shen, Y.-n. Yang, Y. Bian and Y. Zhao, Environ. Sci. Technol., 2016, 50, 2054–2063 CrossRef CAS PubMed.
- H.-C. Fu, F. You, H.-R. Li and L.-N. He, Front. Chem., 2019, 7, 1–15 CrossRef PubMed.
- Y.-N. Li, L.-N. He, A.-H. Liu, X.-D. Lang, Z.-Z. Yang, B. Yu and C.-R. Luan, Green Chem., 2013, 15, 2825–2829 RSC.
- Y.-N. Li, L.-N. He, X.-D. Lang, X.-F. Liu and S. Zhang, RSC Adv., 2014, 4, 49995–50002 RSC.
- S. Zhang, Y. N. Li, Y. W. Zhang, L. N. He, B. Yu, Q. W. Song and X. D. Lang, ChemSusChem, 2014, 7, 1484–1489 CrossRef CAS PubMed.
- M. Yadav, J. C. Linehan, A. J. Karkamkar, E. Van Der Eide and D. J. Heldebrant, Inorg. Chem., 2014, 53, 9849–9854 CrossRef CAS PubMed.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 16873–16876 CrossRef CAS PubMed.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS PubMed.
- S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS PubMed.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- R. Sen, C. J. Koch, A. Goeppert and G. K. S. Prakash, ChemSusChem, 2020, 13, 6318–6322 CAS.
- J. Kothandaraman and D. J. Heldebrant, RSC Adv., 2020, 10, 42557–42563 RSC.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–22, DOI:10.1002/14356007.a12_013.pub3.
- C. Perinu, B. Arstad and K.-J. Jens, Energy Procedia, 2013, 37, 7310–7317 CrossRef CAS.
- A.-G. Sicaire, M. A. Vian, A. Filly, Y. Li, A. Bily and F. Chemat, in Alternative Solvents for Natural Products Extraction, Springer, 2014, pp. 253–268 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00467k |
| This journal is © The Royal Society of Chemistry 2021 |