
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning energy landscapes and metal–metal interactions in supramolecular polymers regulated by coordination geometry†
Nils
Bäumer
a,
Kalathil K.
Kartha
a,
Stefan
Buss
 b,
Iván
Maisuls
b,
Jasnamol P.
Palakkal
c,
Cristian A.
Strassert
b and
Gustavo
Fernández
*a
b,
Iván
Maisuls
b,
Jasnamol P.
Palakkal
c,
Cristian A.
Strassert
b and
Gustavo
Fernández
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 36, 48149 Münster, Germany. E-mail: fernandg@uni-muenster.de
bCeNTech, CiMIC, SoN, Institut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Heisenbergstraße 11, 48149, Germany
cTechnische Universität Darmstadt, Department of Materials and Earth Sciences, Alarich-Weiss-Straße 2, 64287 Darmstadt, Germany
First published on 23rd February 2021
Abstract
Herein, we exploit coordination geometry as a new tool to regulate the non-covalent interactions, photophysical properties and energy landscape of supramolecular polymers. To this end, we have designed two self-assembled Pt(II) complexes 1 and 2 that feature an identical aromatic surface, but differ in the coordination and molecular geometry (linear vs. V-shaped) as a result of judicious ligand choice (monodentate pyridine vs. bidentate bipyridine). Even though both complexes form cooperative supramolecular polymers in methylcyclohexane, their supramolecular and photophysical behaviour differ significantly: while the high preorganization of the bipyridine-based complex 1 enables an H-type 1D stacking with short Pt⋯Pt contacts via a two-step consecutive process, the existence of increased steric effects for the pyridyl-based derivative 2 hinders the formation of metal–metal contacts and induces a single aggregation process into large bundles of fibers. Ultimately, this fine control of Pt⋯Pt distances leads to tuneable luminescence—red for 1vs. blue for 2, which highlights the relevance of coordination geometry for the development of functional supramolecular materials.
Introduction
Self-assembled nanomaterials have emerged in the past decades as potential candidates for various applications.1–5 Fueled by the desire to control the pathways6–9 and the resulting functional properties10–12 of such supramolecular assemblies, detailed structure–property relationships have been successfully established,13,14 with particular focus on organic compounds.15–17 In recent years, molecular design strategies18,19 have been successfully exploited to drive and control molecular self-assembly processes.20–22 The intriguing possibilities that arise from adding metal ions into the mix23–26 have kickstarted the relatively young field of metal-based supramolecular polymers.26 Due to the wide variety of available ligands and metal ions capable of forming supramolecular polymers, the structural diversity of such compounds is immense.27–31 Consequently, metal complexes have been investigated as supramolecular building blocks in a highly diverse manner for manifold applications.32–34 However, these miscellaneous studies render a structure-based optimization of these systems challenging. For example, geometrical modifications are often accompanied by additional structural differences, such as inclusion or variation of linkers, solubilizing chains or coordinating atoms.35–38 Further, electronic effects are rarely discussed and can be difficult to predict.39,40 Particularly, in contrast to discrete supramolecular systems such as helicates,41,42 catenanes43,44 or cages,45,46 the coordination geometry is often underappreciated in the context of metallosupramolecular polymerization. While there are countless studies utilizing mono-,47–49 bi-,50–52 and tridentate53–55 ligands, the effect of changing the ligand system is typically excluded from discussion and often consequential differences in molecular design render such comparisons unfeasible. Moreover, a simple coordination geometry change (cis-/trans-isomerism) is difficult to investigate due to the rapid interconversion between both species in solution.56,57Mono- and polydentate ligands in complexes of d8 transition metals based on N-heterocyclic aromatics have attracted considerable attention in supramolecular chemistry.58 Owing to their preorganized square-planar geometry favoring stacking,59,60 they are prone to undergo supramolecular polymerization, gaining further stabilization by metal–metal and/or hydrogen bonding interactions.61,62 In addition, the intriguing photophysical properties of these molecules provide access to various applications, such as in optoelectronics or sensing.63–65 However, while many studies have focused on modifying ligands to create diverse binding motifs or differences in sterics, there is a distinct lack of structure–property relationships in terms of coordination geometry changes.
To bridge this knowledge gap, we have herein designed and synthesized Pt(II) complexes 1 and 2 (Scheme 1), which feature a similar ligand design but differ in the coordination geometry (bidentate V-shaped “cis” 1vs. monodentate linear “trans” 2). This structural modification greatly influences the molecular preorganization of the system, enabling the establishment of distinct non-covalent interactions, which in turn dictate the photophysical properties and the energy landscapes of the systems. While the molecular geometry for 1 enforces short Pt⋯Pt contacts driven by an efficient overlap of the aromatic oligophenyleneethynylene (OPE) ligands, the out-of-plane arrangement of the Cl ligands at the Pt(II) center for 2 hinders the formation of short Pt⋯Pt contacts by precluding a parallel molecular packing, leading to a minor translational offset. Ultimately, the distinct interplay of interactions for 1 and 2 leads to significant differences in luminescence and energy landscapes – two-step vs. single-step self-assembly for 1 and 2, respectively. Detailed investigation of these complexes allowed us to unravel for the first time how changes in the coordination geometry affect supramolecular polymerization.
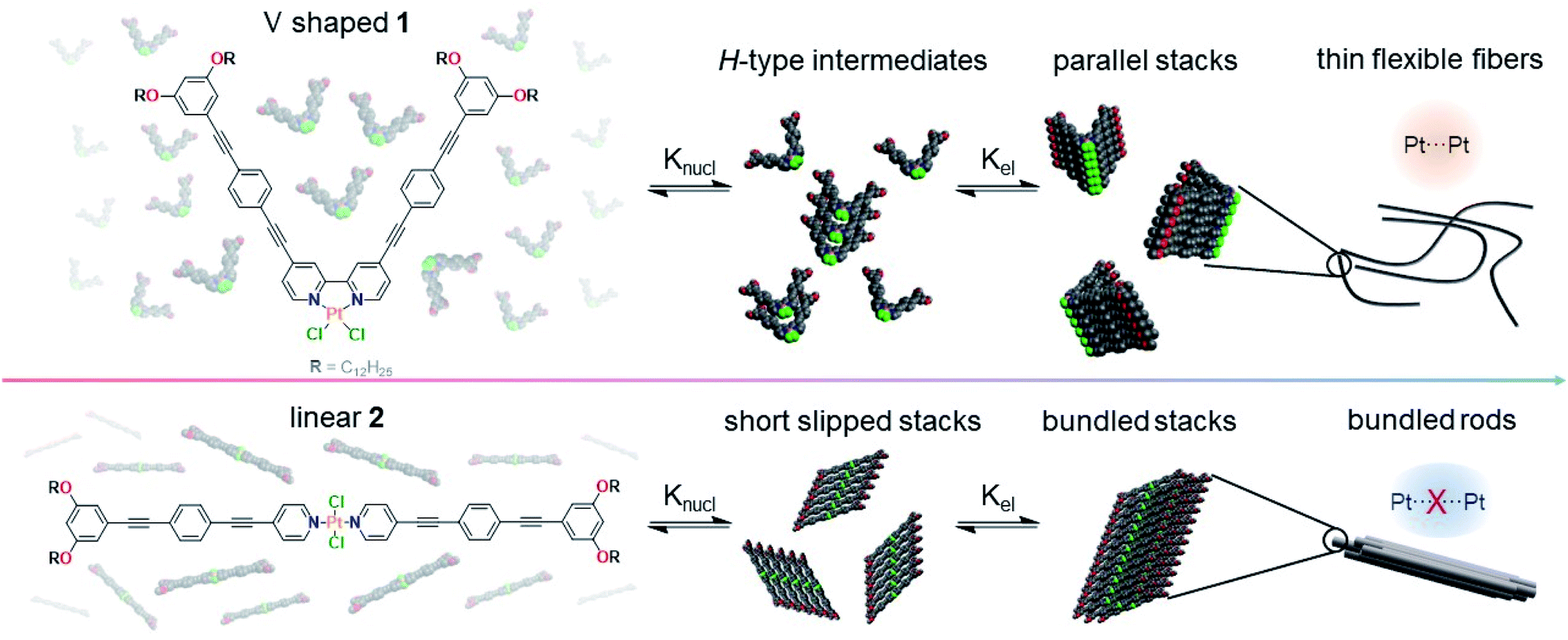 | ||
| Scheme 1 Chemical structures of Pt(II) complexes 1 and 2 and cartoon representation of their supramolecular polymerization in MCH leading to different emission properties induced by close (top, orange) or weak Pt⋯Pt contacts (bottom, blue). Alkyl chains have been reduced to methyl groups for better visibility. | ||
Results and discussion
Photophysical analysis
Initially, we probed the self-assembly of 1 and 2 by solvent-dependent UV/Vis studies (Fig. 1a and d). Complex 1 exhibits two high-energy absorption bands in all investigated solvents (around 310 and 330 nm in MCH, n-hexane, chloroform and DCM; and at 310 and 349 nm in THF). These bands can be readily assigned to intraligand (IL) transitions into excited states with π–π* as well as n–π* character.66 Furthermore, two additional high-energy absorption bands (around 349 and 373 nm) can also be observed in solvents of slightly higher polarity (DCM and chloroform). In low-polarity solvents (MCH and n-hexane), these bands only appear as shoulders. Additionally, one low-energy absorption band (423 and 429 nm in DCM and chloroform, respectively) can be observed in higher polarity solvents and two low-energy absorption bands (404 and 428 nm) in apolar solvents (MCH and n-hexane). Based on the strong solvatochromic effect in the low energy region of the UV/Vis spectrum, we assign these bands to transitions into metal-to-ligand charge-transfer (MLCT) states.67 Moreover, a minor contribution of ligand-to-ligand charge-transfer (LLCT) character involving the halogen and bipyridine ligands is also expected, as previously reported for this spectral region.68 On this basis, we conclude that non-polar solvents (such as MCH or n-hexane) induce aggregation, whereas more polar solvents (such as chloroform) favor the molecularly dissolved state.69 On the other hand, solvent-dependent UV/Vis studies of 2 reveal a similar aggregation propensity in non-polar media, although differences are also appreciable (Fig. 1d). In particular, the spectra in most solvents at low concentration (10−5 M) are almost identical, showing an absorption maximum at ∼350 nm and a shoulder around 370 nm. These spectral signatures can be assigned to transitions into metal perturbed IL (main peak) as well as MLCT states (shoulder).70,71 The only exception is the spectrum in n-hexane, where a double band at 353 and 365 nm with an additional absorption maximum at 401 nm are noticed. Additionally, a significant hypochromism compared to all other solvents can be observed, which is concomitant with the increase in optical density in the low energy region (>400 nm). An identical trend is observed for solutions in MCH at slightly higher concentrations (3 × 10−5 M). Consequently, we identified solvents of high polarity (such as chloroform) as good solvents, while low polarity solvents (such as MCH and n-hexane) can be considered as poor solvents suitable for aggregation studies. | ||
| Fig. 1 Solvent-dependent UV/Vis spectra of 1 (a) and 2 (d) at 10−5 M and 298 K; VT-photoluminescence spectra of 1 (b) and 2 (e) at 3 × 10−6 M (b) and 30 × 10−6 M (e); cooling rate: 1 K min−1. The photoluminescence spectrum of 1 in chloroform at 298 K has been added for reference; time-resolved photoluminescence decay of Agg1 (c) and Agg2 (f) in deaerated MCH at 298 K and 10−5 M, including the residuals (λexc = 376.7 nm, λem = 690 nm for Agg1; λexc = 376.7 nm, λem = 550 nm for Agg2). Inset: photographs of the used solutions at ambient conditions and under UV light (λexc = 405 nm) at 298 K and 77 K, respectively. | ||
Subsequently, we performed variable temperature (VT)-photoluminescence studies of 1 in MCH between 368 K and 273 K (Fig. 1b and S13†). During initial cooling, only minor changes in the emission were observed; however, below 350 K, a broad emission between 600 and 850 nm showed up, a behavior that has been typically attributed to a 3MMLCT state for structurally related compounds.72 In addition, 1 showed a featureless emission band at 560 nm at lower temperature (<308 K), which can be assigned to 1(d,π)–π* excited states.73
To further probe the existence of metal–metal interactions, we performed additional photoluminescence measurements. Initially, the emission spectra of 1 in a molecularly dissolved state (in chloroform) and in an aggregated form (in MCH) were compared to confirm whether the observed emission is caused by aggregation, or by a subsequent decrease in molecular rotation during cooling (Fig. 1b). To our satisfaction, no emission at low energies was observed in chloroform, confirming that aggregation-induced emission takes place. In order to assign these emission bands, we turned to phosphorescence lifetime measurements. To this end, we prepared Agg1 by cooling a hot monomer solution (1 × 10−5 M) of 1 in MCH from 368 K to 298 K at a rate of 1 K min−1. Afterwards, the solution was deaerated by purging with argon (Fig. 1c and S14–S20†). The excited state of Agg1 shows a relatively long average lifetime of 0.67 μs (amplitude weighted). Cooling the aggregate solution to 77 K extends the long lifetimes to 20.6 μs. Interestingly, Gray and co-workers were able to correlate a linear relationship between the emission maximum from the 3MMLCT state to the inverse cube of the Pt⋯Pt distance for a structurally related compound Pt(bpy)Cl2.74 Based on this linear relationship, the Pt⋯Pt distance in Agg1 was estimated to be ∼3.30 Å. Additionally, the emission at 560 nm observed at temperatures below 308 K can be assigned to an IL state, based on the relatively short average lifetime of 1.2 ns.
The self-assembly of 2 was also probed using VT-fluorescence spectroscopy revealing minor emission above 600 nm (Fig. 1e). Instead, a relatively sharp emission with a maximum at 380 nm typical for an IL 1π–π* state accompanied by a vibronic structure can be observed (Fig. S21†). These findings can be attributed to stretching of the phenyl rings as well as the ethynylene units.75–77
Based on the results obtained from VT-luminescence, we envisage that the supramolecular packing of 2 can be described as a nearly parallel, short slipped aggregate with Pt⋯Pt distances (>4.0 Å) larger than the sum of van der Waals radii (3.5 Å),78 as previously observed for related OPE-based bispyridyldichlorido Pt(II) complexes.47 To validate this hypothesis, we conducted photoluminescence lifetime measurements (Fig. 1f and S21–S25†) using the free pyridyl ligand as reference compound (Fig. S26 and S27†). The average lifetime of the excited state of Agg2 was only 9.7 ns, while for the free ligand a lifetime of 0.8 ns was obtained. Due to the comparable order of magnitude as well as the generally similar emission profile, the luminescence of the free ligand likely originates from the OPE system, while the emission of Agg2 can be best described as originated by metal perturbed IL states, without any MMLCT contributions; here, the short lifetime is related to a fast radiationless deactivation promoted by thermally accessible low-lying metal centered (MC) states (d–d*) with dissociative character caused by the weak ligand field of the Cl ligands and the non-coplanar arrangement of the pyridine ligands.
From these results, we understand that in the case of Agg1, the MMLCT states act as energy traps from which the thermal population of such MC states is slowed down, which hampers non-radiative deactivation and leads to longer lifetimes. The out of plane torsional distortion of the pyridine ligands in Agg2 probably hinders efficient Pt⋯Pt interactions, as previously observed in Pt(II) complexes with C^N^N luminophores and diverse ancillary ligands.79
Energy landscape elucidation
VT-UV/Vis studies in MCH (3 × 10−6 M) were initially conducted to gain a better understanding of the self-assembly behaviour of 1 (Fig. 2a and S28†). At 368 K, 1 shows two absorption bands at 450 nm and 475 nm, which can be attributed to the MLCT state of the molecularly dissolved state.69 Cooling down to 278 K leads to a decrease of the maxima at 450 nm and 475 nm and a simultaneous increase of an absorption band at 430 nm, with an appreciable second absorption maximum at 404 nm. A similar hypsochromic shift of 4 and 35 nm is observed for the high energy absorption bands at 313 and 367 nm, respectively. This spectral pattern is in agreement with an H-type aggregation process.80 In contrast to 1, 2 requires significantly higher concentrations to aggregate in MCH solution (Fig. 2b, c and S30†). At high temperatures (>300 K), a minor hyperchromism with no isosbestic points can be observed, a phenomenon that has been previously assigned to the planarization of the aromatic surface.81,82 Further cooling below a critical temperature (300 K) leads to a sudden drop in absorbance and a concurrent aggregate band formation at 400 nm that spreads up to ca. 700 nm (Fig. 2b). Additionally, the absorption maximum shows a bathochromic shift from 350 to 367 nm, indicating that the monomers of 2 are packed with a slight translational offset.83 The increase in optical density in the low energy region above 450 nm indicates the formation of poorly solvated aggregates. Plotting the degree of aggregation (αagg) of 1vs. temperature (T) at 475 nm shows a steep, non-sigmoidal curve (Fig. 2c), indicating a cooperative supramolecular polymerization (Fig. 2c).84 Similarly, a cooperative aggregation mechanism could be identified for 2 by plotting αaggvs. T at 400 nm (Fig. 2c). These observations could be confirmed by denaturation studies, revealing poorly solvated aggregates that disassemble in a clearly non-sigmoidal fashion (Fig. S31†). | ||
| Fig. 2 VT-UV/Vis spectra of 1 (a) and 2 (b) between 368 K and 278 K or 273 K at 3 × 10−6 M (a) and 30 × 10−6 M (b); cooling rate: 1 K min−1. (c) Plot of αaggvs. T for 1 (λ = 475 nm) and 2 (λ = 400 nm) derived from cooling experiments and fitting to the nucleation–elongation model.84 | ||
In comparison to 1, the self-assembly of 2 shows a significantly more pronounced all-or-nothing behavior, which is reflected in the larger nucleation penalty of 2 (22.5 vs. 17.3 kJ mol−1) in combination with a much larger elongation enthalpy (189 vs. 49 kJ mol−1, respectively, Table 1). The smaller nucleation penalty of 1 can be rationalized by the molecular preorganization induced by the bipyridine binding site, which forces a planar coordination environment. In contrast, trans-oriented monodentate pyridine complexes are arranged out-of-plane, typically allowing both inter- and intramolecular hydrogen bonds between the halogens and the aromatic C–H groups from the pyridine moieties.37,49 This potential steric hindrance during the initial nucleus formation is also reflected in the nucleation constant (Kn), which is one order of magnitude lower for 2 than it is for 1. On the other hand, the elongation constant (Ke) remains within the same order of magnitude leading to a noticeably smaller cooperativity factor (1.2 × 10−4vs. 27.1 × 10−4) for 2. This is a direct consequence of the change in coordination geometry, as the linear complex 2 can balance the steric demand and attractive intermolecular interactions, while a translational offset for 1 would not be feasible. Notably, careful investigation of the VT spectral changes of 1 reveals a transition between two species during cooling from 368 K to 313 K (Fig. 3a and b). Most likely, this transition corresponds to the aggregation of the monomer Mon1 into an intermediate species Int1. In a second step, between 313 K and 283 K, a second transformation occurs, where Int1 converts to the thermodynamic aggregate Agg1 (Fig. 3a). Monitoring the absorption changes at 370 nm as well as the depletion of the monomer at 475 nm allows us to qualitatively estimate the relative concentration of all active species during the aggregation process (Fig. 3c and S32; see ESI† for methodology).75 Time-dependent UV/Vis studies at variable concentrations (Fig. 3d, for full spectra see Fig. S33†) reveal that the transformation of Int1 to Agg1 is accelerated upon increasing concentration, indicating that Int1 is an on-pathway intermediate towards Agg1.85 The self-assembly of 1 follows a two-step dual cooperative process, as shown in the qualitative energy diagram depicted in Fig. 3e. Based on the small changes in UV/Vis during the second step, both Int1 and Agg1 can be identified as H-type aggregates with similar packing, where Agg1 represents a more ordered structure. Additionally, given the rapid Int1 → Agg1 transformation and the fact that the monomer depletion does not seem to be influenced by the onset of elongation of Agg1, we assign Int1 as an on-pathway transient species, possibly a dimer or short oligomer. In contrast, the self-assembly of 2 follows a one-step cooperative process with a higher activation energy compared to 1, which is evident from the decrease in Kn as well as a sharp decrease in Te.86 The observation of pathway complexity only for compound 1 clearly reflects the noteworthy influence of coordination geometry on self-assembly. In fact, our results reveal that on-pathway intermediates can be stabilized by decreasing steric demands (in-plane arrangement of Cl ligands for 1vs. out-of-plane arrangement for 2). This behavior strongly differs from the majority of systems exhibiting off-pathway kinetic species,85,87 where increased steric interactions stabilize the kinetic product.
| 1 | 2 | |
|---|---|---|
| c [10−6 M] | 3.0 | 30 |
| ΔHe [kJ mol−1] | −48.9 ± 0.3 | −189.1 ± 1.3 |
| ΔHn [kJ mol−1] | −17.3 ± 0.2 | −22.5 ± 1.9 |
| ΔS° [kJ mol−1] | −0.034 ± 0.001 | −0.535 ± 0.004 |
| ΔG° [kJ mol−1] | −38.9 ± 0.4 | −29.7 ± 1.8 |
| T e [K] | 351.6 ± 0.1 | 299.9 ± 0.1 |
| K e [104 M−1] | 32.3 | 10.0 |
| K n [M−1] | 876 | 12 |
| σ [10−4] | 27.1 | 1.2 |
 | ||
| Fig. 3 VT-UV/Vis spectra of 1 between 363 K and 313 K (a) and 313 K and 283 K (b) at 3 × 10−6 M; cooling rate: 1 K min−1. (c) Relative concentration of the different species involved in the aggregation process plotted vs. T estimated based on the spectral changes at λ = 475 nm and λ = 370 nm during VT-UV/Vis studies. (d) Time-dependent transformation of Int1 to Agg1 at different concentrations at 303 K monitored at λ = 370 nm over time after rapid temperature drop from the molecularly dissolved state at 368 K. (e) Qualitative energy landscape depicting pathway complexity for 1 and a single-step assembly behavior for 2. | ||
Structural and morphological elucidation
Solvent- and temperature-dependent 1H NMR studies were used to examine the supramolecular packing of 1. Initially, we recorded a 1H NMR spectrum of monomeric 1 in CDCl3 at 1 mM and subsequently added increasing volume fractions of MCH-d14 to this solution. The stepwise addition of MCH-d14 leads to marked signal shifts accompanied by progressive broadening, suggesting a self-assembly process (Fig. 4a). Throughout the entire polarity spectrum, most aromatic protons undergo a significant upfield shift indicating aromatic interactions.88,89 The two proton signals corresponding to the peripheral phenyl ring (He and Hf) represent the only exception, as these two signals show an initial minor downfield shift followed by a more significant upfield shift accompanied by broadening with MCH contents larger than 40%. This observation indicates the proximity of an electron rich group, such as O,90 a behaviour that could be confirmed by VT-1H NMR studies (Fig. S34 and S35†). These results match well with a parallel arrangement of the molecules within the stack, as most protons would thus exhibit interactions with the aromatic moieties. | ||
| Fig. 4 (a and c) Partial solvent dependent (CDCl3/MCH-d14, 298 K) 1H NMR spectra of 1 (a) and 2 (c) at 1 × 10−3 M. (b and d) Schematic representation of the packing modes of Agg1 (b) and Agg2 (d) with the most relevant intermolecular distances highlighted with arrows. The alkyl chains have been reduced to methyl groups in this representation to enhance visibility. | ||
Additional 2D ROESY studies of 1 (Fig. S36†) also support this hypothesis. In such packing, the outer aromatic protons would come in close contact with the alkoxy oxygens of neighboring moieties, thus explaining the initial minor downfield shift observed for those signals. Such parallel stacks would enable short Pt⋯Pt contacts, as previously suggested by spectroscopic studies. Further, we infer from these studies that the intermediate species Int1 represents a less ordered parallel arrangement of the aromatic moieties due to steric effects of the peripheral alkyl chains. At low temperatures, the additional planarization and decreased flexibility of the OPE backbone can facilitate a more beneficial stacking leading to better defined UV/Vis absorption bands. Unfortunately, attempts to further inspect the molecular packing of Agg1 using XRD analysis proved unsuccessful (Fig. S37†) despite the seemingly high order within the stack. In particular, between 2θ = 12° and 30°, a very broad featureless reflex is obtained, which can be rationalized by a potential flexibility of the 1D stack (vide infra). Based on the relatively long phosphorescence lifetimes and the emission profile of Agg1, we propose that the experimentally observed parallel stacking possibly results in close Pt⋯Pt contacts with distances shorter than <3.5 Å.
Subsequently, we also unraveled the packing mode of Agg2. Solvent-dependent 1H NMR (Fig. 4c) in CDCl3/MCH-d14 shows spectral changes typical for the planarization of pyridyl-based Pt(II) complexes, namely a pronounced downfield shift of the alpha proton of the pyridine moiety (8.87 ppm to 9.03 ppm) due to the increased intramolecular hydrogen bonding between the proton Ha and the Cl ligand. Additionally, the remaining protons show only negligible shifts without any signal broadening. Another interesting finding is the downfield shift of the peripheral protons He and Hf (Fig. 4c), which might be explained by intermolecular close contacts with electron rich groups, for example the O atoms of the neighboring molecule within the 1D stack.90 Thus, the downfield shift suggests a relatively short-slipped J-type packing, as also indicated by UV/Vis studies and supported by XRD (Fig. S38†). Given that the proton signals remain relatively sharp during the whole experiment, the observed spectral changes should be attributed to the planarization and pre-nucleation of 2 only, which fits with the all-or-nothing aggregation behavior observed for 2 in VT-UV/Vis. In fact, if the MCH content is further increased above 60%, rapid precipitation of the large aggregates occurs, as evident from the disappearance of the NMR signals.
Combining all results for 2, we assume the formation of initial 1D stacks with a short-slipped molecular packing, which can then agglomerate laterally via alkyl chain interdigitation to give larger, poorly solvated bundles of fibers.
In order to correlate the packing mode of 1 and 2 with the aggregate morphology, we employed atomic force microscopy (AFM) and scanning electron microscopy (SEM). AFM studies of Agg1 reveal the formation of flexible fibers with a height between 1.5 and 1.8 nm (Fig. 5a, b and S40–S43†), which agrees with the molecular dimensions of 2 (length of the aromatic moiety = 1.8 nm). The values obtained from height analysis also support the interpretation of the alkoxy chains wrapping themselves around the aromatic stack. The fibers show a relatively high polydispersity with lengths as small as 50 nm up to 1 μm. This observation can be rationalized by the spin-coating technique used during sample preparation which can lead to fractured aggregates, especially if thin morphologies with high aspect ratio are investigated.48,75,91 Notably, a dense network of fibers could be observed at higher concentrations (5 × 10−5 M, Fig. 5a and S43†). The high flexibility of these structures along with the absence of high crystallinity and long-range order may be one possible explanation for the observation of a broad reflex during XRD measurements as opposed to the sharp reflexes obtained for Agg2. For this sample, AFM discloses the formation of large, poorly solvated bundles of stiff fibers with heights of more than 100 nm and lengths of multiple microns (Fig. 5c, S44 and S45†), as also supported by SEM (Fig. 5d and S46, S47†). The resulting structures indicate a higher long-range order compared to Agg1, which explains the different results obtained during XRD measurements.
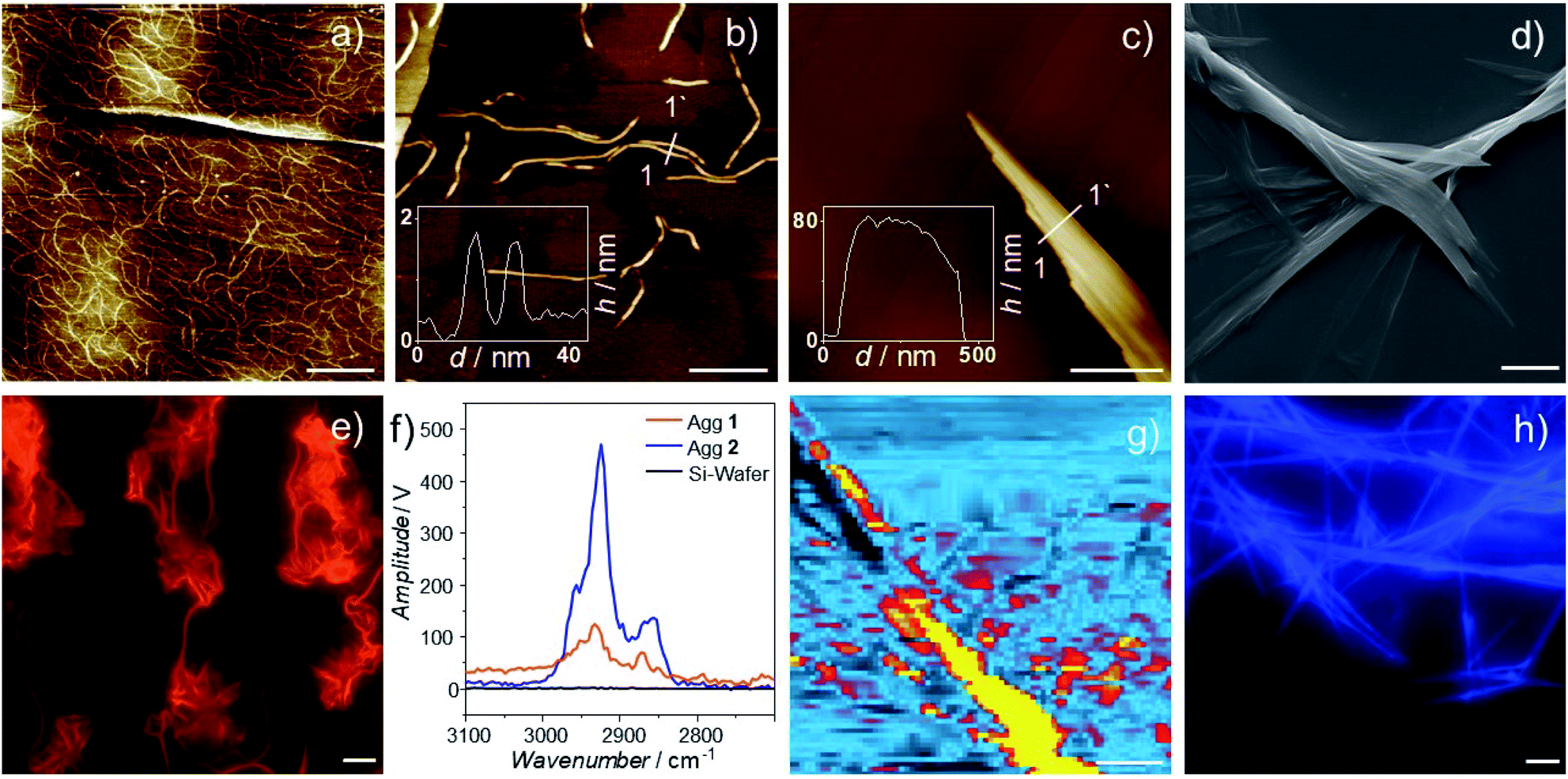 | ||
| Fig. 5 AFM height images of Agg1 (a and b) and Agg2 (c) at 273 K on HOPG after cooling from 368 K with a cooling rate of 1 K min−1. (a) c = 5 × 10−5 M; (b and c) c = 2 × 10−5 M. (d) SEM image of Agg2. (e and h) Luminescence micrographs of Agg1 and Agg2 (λexc = 375 nm). (f) AFM-IR spectra of Agg1 and Agg2. (g) IR image of Agg2 measured with λ = 2925 cm−1. The scale bars correspond to 400 nm (a), 50 nm (b), 800 nm (c), 4000 nm (d), 10 μm (e), 2000 nm (g) and 10 μm (h) respectively. | ||
Further, we attempted to microscopically correlate the morphologies observed during AFM with the luminescence properties identified in solution. To this end, we used photo-luminescence microscopy and could indeed observe emissive supramolecular structures, whose emission properties matched well with previous observations (Fig. 5e and h). For solutions containing Agg1 (5 × 10−5 M), highly flexible fibers with intense dark orange emission could be observed (Fig. 5e and S48†). In contrast, for Agg2, the stiff bundled fibers observed in AFM and SEM showed the typical blue fluorescence expected for metal-perturbed OPE ligands (Fig. S49†).86,87
Ultimately, we examined the fibers of Agg1 and Agg2 by AFM-IR to gain a more detailed understanding of their chemical environment (for details, see ESI†). To the best of our knowledge, this is the first time that AFM-IR is used to investigate a supramolecular polymer. Interestingly, for both polymers, two sharp IR bands that are typical for alkyl C–H stretching are observed at 2925 (νanti) and 2856 cm−1 (νsym) (Fig, 5f and S50, S51†).92 These values are characteristic of interdigitation of the alkyl chains, which further supports that both polymers form an effective alkyl shell around their respective aromatic core.92,93 This observation enabled us to create an IR image of the larger morphologies of Agg2, by scanning the thermal expansion attributed to the IR absorption at 2925 cm−1. The resulting chemical image of Agg2 discloses a clear match between alkyl stretching vibrations and aggregate morphology, supporting an effective shielding of the aromatic backbone by the aliphatic chains (Fig. 5g and S53†). These results further allow us to explain why both polymers exhibit identical behavior during PeakForce Tunneling AFM (Fig. S54–S57†), as the insulating alkyl shell effectively shields the central polymer backbone.94,95
Conclusions
In conclusion, we have unraveled the subtle interplay between coordination geometry, energy landscapes and metal–metal interactions in the context of supramolecular polymerization. For this purpose, we have designed two Pt(II) complexes that primarily differ in the molecular shape (V-shaped 1vs. linear 2) as a result of different coordination geometry (cis or trans) determined by the choice of ligand (bipyridyl vs. bispyridyl). Preorganized V-shaped Pt(II) complex 1 self assembles in a two-step cooperative process into 1D fibers with short metal–metal contacts and red luminescence. In contrast, the monomer units of linear Pt(II) complex 2 are unable to establish short Pt⋯Pt contacts as a result of more significant steric effects than 1, leading to a single cooperative step into microscopic bundles of fibers with short-slipped molecular arrangement and blue luminescence. Interestingly, the increase in molecular preorganization for 1 not only enables short Pt⋯Pt contacts, but also promotes the emergence of pathway complexity via the controlled formation of an on-pathway kinetic species. These results pave the way towards functional materials based on self-assembled supramolecular polymers with potentially relevant optoelectronic applications. Work in our laboratory is underway to optimize the molecular design in order to access metallosupramolecular polymers with potential semiconductive properties arising from Pt⋯Pt interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. B., K. K. K. and G. F. acknowledge the European Commission (ERC-StG-2016 SUPRACOP-715923) for funding. CAS acknowledges the DFG (EXC 1003 Cluster of Excellence “Cells in Motion”; SPP “Light Controlled Reactivity of Metal Complexes”) for funding. JPP acknowledges the DFG 429646908 for funding. We thank Prof. Lambert Alff for access to XRD measurements. IM thanks the Alexander von Humboldt foundation for a postdoctoral fellowship.Notes and references
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Nat. Chem., 2015, 7, 281–294 CrossRef CAS PubMed.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400–405 CrossRef CAS PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S.-I. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363–14367 CrossRef CAS PubMed.
- M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed.
- L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed.
- Y. Kim, W. Li, S. Shin and M. Lee, Acc. Chem. Res., 2013, 46, 2888–2897 CrossRef CAS PubMed.
- X. Feng, B. Shen, B. Sun, J. Kim, X. Liu and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 11355–11359 CrossRef CAS PubMed.
- E. E. Greciano, J. Calbo, J. Buendía, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2019, 141, 7463–7472 CrossRef CAS PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- S. Ogi, N. Fukaya, D. Arifin, B. B. Skjestad, Y. Hijikata and S. Yamaguchi, Chem.–Eur. J., 2019, 25, 7303–7307 CrossRef CAS PubMed.
- F. Tantakitti, J. Boekhoven, X. Wang, R. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. Olvera de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- A. M. Oliver, J. Gwyther, C. E. Boott, S. Davis, S. Pearce and I. Manners, J. Am. Chem. Soc., 2018, 140, 18104–18114 CrossRef CAS PubMed.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339–2343 CrossRef CAS PubMed.
- N. Fukaya, S. Ogi, M. Kawashiro and S. Yamaguchi, Chem. Commun., 2020, 56, 12901–12904 RSC.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- H. Ouchi, T. Kizaki, M. Yamato, X. Lin, N. Hoshi, F. Silly, T. Kajitani, T. Fukushima, K.-I. Nakayama and S. Yagai, Chem. Sci., 2018, 9, 3638–3643 RSC.
- Y. Wang, Y. Kim and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 13122–13126 CrossRef CAS PubMed.
- M. Y. Leung, S. Y. L. Leung, K. C. Yim, A. K. W. Chan, M. Ng and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 19466–19478 CrossRef CAS PubMed.
- A. S. Y. Law, L. C. C. Lee, M. C. L. Yeung, K. K. W. Lo and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 18570–18577 CrossRef CAS PubMed.
- K. K. Zhang, M. C. L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 9594–9605 CrossRef CAS PubMed.
- Z. Gao, Y. Han, Z. Gao and F. Wang, Acc. Chem. Res., 2018, 51, 2719–2729 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- Q. Wan, X.-S. Xiao, W.-P. To, W. Lu, Y. Chen, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS PubMed.
- K. K. Kartha, N. K. Allampally, A. T. Politi, D. D. Prabhu, H. Ouchi, R. Q. Albuquerque, S. Yagai and G. Fernández, Chem. Sci., 2019, 10, 752–760 RSC.
- J. Chen, L. Ao, C. Wei, C. Wang and F. Wang, Chem. Commun., 2019, 55, 229–232 RSC.
- N. Zhou, R. Hailes, Y. Zhang, Z. Chen, I. Manners and X. He, Polym. Chem., 2020, 11, 2700–2707 RSC.
- S.-C. Chan, M. C. W. Chan, Y. Wang, C.-M. Che, K.-K. Cheung and N. Zhu, Chem.–Eur. J., 2001, 7, 4180–4190 CrossRef CAS.
- J. Ni, L.-Y. Zhang, H.-M. Wen and Z.-N. Chen, Chem. Commun., 2009, 3801–3803 RSC.
- T. J. Wadas, Q.-M. Wang, Y.-J. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS PubMed.
- N. K. Allampally, C.-G. Daniliuc, C. A. Strassert and L. de Cola, Inorg. Chem., 2015, 54, 1588–1596 CrossRef CAS PubMed.
- C. Wang, Z. Chen, M. Liu, H. Zhong and F. Wang, Polym. Chem., 2019, 10, 3210–3216 RSC.
- L. Herkert, A. Sampedro and G. Fernández, CrystEngComm, 2016, 18, 8813–8822 RSC.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1977, 99, 1316–1332 CrossRef CAS.
- C. B. Aakeröy and K. Epa, in Electronic Effects in Organic Chemistry, ed. B. Kirchner, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 125–147 Search PubMed.
- B. Hasenkopf, J.-M. Lehn, N. Boumediene, A. Dupont-Gervais, A. Van Dorsselaer, B. O. Kneisel and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956–10962 CrossRef.
- B. Hasenknopf, J.-M. Lehn, B. O. Kneisel, G. Baum and D. Fenske, Angew. Chem., Int. Ed., 1996, 35, 1838–1840 CrossRef CAS.
- J.-P. Sauvage, Chem. Commun., 2005, 1507–1510 RSC.
- J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC.
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS.
- Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889–3919 RSC.
- N. K. Allampally, M. J. Mayoral, S. Chansai, M. C. Lagunas, C. Hardacre, V. Stepanenko, R. Q. Albuquerque and G. Fernández, Chem.–Eur. J., 2016, 22, 7810–7816 CrossRef CAS PubMed.
- N. Bäumer, K. K. Kartha, N. K. Allampally, S. Yagai, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 15626–15630 CrossRef PubMed.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192–5200 CrossRef CAS PubMed.
- Y. Ai, Y. Li, H. L.-K. Fu, A. K.-W. Chan and V. W.-W. Yam, Chem.–Eur. J., 2019, 25, 5251–5258 CrossRef CAS PubMed.
- A. Kobayashi, Y. Fukuzawa, H.-C. Chang and M. Kato, Inorg. Chem., 2012, 51, 7508–7519 CrossRef CAS PubMed.
- I. Stengel, C. A. Strassert, L. de Cola and P. Bäuerle, Organometallics, 2014, 33, 1345–1355 CrossRef CAS.
- Y. Atoini, E. A. Prasetyanto, P. Chen, S. Silvestrini, J. Harrowfield and L. de Cola, Chem.–Eur. J., 2018, 24, 12054–12060 CrossRef CAS PubMed.
- Y. Chen, C.-M. Che and W. Lu, Chem. Commun., 2015, 51, 5371–5374 RSC.
- W. Wu, J. Zhao, H. Guo, J. Sun, S. Ji and Z. Wang, Chem.–Eur. J., 2012, 18, 1961–1968 CrossRef CAS PubMed.
- H. Ube, Y. Yasuda, H. Sato and M. Shionoya, Nat. Commun., 2017, 8, 14296 CrossRef CAS PubMed.
- D. Cornacchia, L. Cerasino, C. Pacifico and G. Natile, Eur. J. Inorg. Chem., 2008, 1822–1829 CrossRef CAS.
- Y. Han, Z. Gao, C. Wang, R. Zhong and F. Wang, Coord. Chem. Rev., 2020, 414, 213300 CrossRef CAS.
- G. V. Janjić, P. V. Petrović, D. B. Ninković and S. D. Zarić, J. Mol. Model., 2011, 17, 2083–2092 CrossRef PubMed.
- P. V. Petrović, G. V. Janjić and S. D. Zarić, Cryst. Growth Des., 2014, 14, 3880–3889 CrossRef.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS PubMed.
- C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- K. M.-C. Wong, W.-S. Tang, X.-X. Lu, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2005, 44, 1492–1498 CrossRef CAS PubMed.
- W. Yang and J. Zhao, Eur. J. Inorg. Chem., 2016, 5283–5299 CrossRef CAS.
- M. Milkevitch, E. Brauns and K. J. Brewer, Inorg. Chem., 1996, 35, 1737–1739 CrossRef CAS.
- J. Burgess, Spectrochim. Acta, Part A, 1970, 26, 1369–1374 CrossRef CAS.
- A. Vogler and H. Kunkley, Inorg. Chem., 1990, 9, 201–220 CAS.
- P. M. Gidney, R. D. Gillard and B. T. Heaton, J. Chem. Soc., Dalton Trans., 1973, 132–134 RSC.
- M. A. M. Meester, D. J. Stufkens and K. Vrieze, Inorg. Chim. Acta, 1975, 14, 33–36 CrossRef CAS.
- M. Textor and W. Ludwig, Helv. Chim. Acta, 1972, 55, 184–198 CrossRef CAS.
- V. M. Miskowski, V. H. Houlding, C.-M. Che and Y. Wang, Inorg. Chem., 1993, 32, 2518–2524 CrossRef CAS.
- R. Büchner, C. T. Cunningham, J. S. Field, R. J. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711–718 RSC.
- W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261–6265 CrossRef CAS.
- J. Matern, K. K. Kartha, L. Sánchez and G. Fernández, Chem. Sci., 2020, 11, 6780–6788 RSC.
- W. Lu, W.-M. Kwok, C. Ma, C. T.-L. Chan, M.-X. Zhu and C.-M. Che, J. Am. Chem. Soc., 2011, 133, 14120–14135 CrossRef CAS PubMed.
- H.-Y. Chao, W. Lu, Y. Li, M. C. W. Chan, C.-M. Che, K.-K. Cheung and N. Zhu, J. Am. Chem. Soc., 2002, 124, 14696–14706 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. Sanning, L. Stegemann, P. R. Ewen, C. Schwermann, C. G. Daniliuc, D. Zhang, N. Lin, L. Duan, D. Wegner, N. L. Doltsinis and C. A. Strassert, J. Mater. Chem. C, 2016, 4, 2560–2565 RSC.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259–4265 CrossRef CAS PubMed.
- K. Seehafer, M. Bender and U. H. F. Bunz, Macromolecules, 2014, 47, 922–927 CrossRef CAS.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291–5301 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. de Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- G. Das, S. Cherumukkil, A. Padmakumar, V. B. Banakar, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202015390.
- E. E. Greciano, B. Matarranz and L. Sánchez, Angew. Chem., Int. Ed., 2018, 57, 4697–4701 CrossRef CAS PubMed.
- S. Lahiri, J. L. Thompson and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 11315–11319 CrossRef CAS.
- C. Giessner-Prettre, B. Pullman, P. N. Borer, L. S. Kan and P. O. Ts'o, Biopolymers, 1976, 15, 2277–2286 CrossRef CAS PubMed.
- X. Lu, Z. Guo, C. Sun, H. Tian and W. Zhu, J. Phys. Chem. B, 2011, 115, 10871–10876 CrossRef CAS PubMed.
- A. Suzuki, K. Aratsu, S. Datta, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi, M. Hollamby, F. Silly and S. Yagai, J. Am. Chem. Soc., 2019, 141, 13196–13202 CrossRef CAS PubMed.
- B. H. Stuart, Infrared spectroscopy: Fundamentals and Applications, Wiley, Chichester, 2008, p. 72 Search PubMed.
- G. Fernández, F. García, F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2009, 7155–7157 RSC.
- K. Sugiyasu, Y. Honsho, R. M. Harrison, A. Sato, T. Yasuda, S. Seki and M. Takeuchi, J. Am. Chem. Soc., 2010, 132, 14754–14756 CrossRef CAS PubMed.
- M. Hecht, T. Schlossarek, M. Stolte, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 12979–12983 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, synthetic details, complete spectroscopic characterization, variable temperature/solvent UV-Vis studies, 1H NMR studies, photoluminescence studies, AFM imaging, phosphorescence lifetime measurements, XRD diffraction. See DOI: 10.1039/d1sc00416f |
| This journal is © The Royal Society of Chemistry 2021 |