
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
5-Hydroxy-pyrrolone based building blocks as maleimide alternatives for protein bioconjugation and single-site multi-functionalization†
Ewout
De Geyter‡
 a,
Eirini
Antonatou‡
a,
Dimitris
Kalaitzakis‡
b,
Sabina
Smolen
a,
Abhishek
Iyer
a,
Laure
Tack
a,
Emiel
Ongenae
a,
Georgios
Vassilikogiannakis
*b and
Annemieke
Madder
*a
a,
Eirini
Antonatou‡
a,
Dimitris
Kalaitzakis‡
b,
Sabina
Smolen
a,
Abhishek
Iyer
a,
Laure
Tack
a,
Emiel
Ongenae
a,
Georgios
Vassilikogiannakis
*b and
Annemieke
Madder
*a
aOrganic and Biomimetic Chemistry Research Group OBCR, Department of Organic and Macromolecular Chemistry, Faculty of Sciences, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: annemieke.madder@ugent.be
bDepartment of Chemistry, University of Crete, Vasilika Vouton, 71003, Iraklion, Crete, Greece. E-mail: vasil@uoc.gr
First published on 4th March 2021
Abstract
Recent dramatic expansion in potential uses of protein conjugates has fueled the development of a wide range of protein modification methods; however, the desirable single-site multi-functionalization of proteins has remained a particularly intransigent challenge. Herein, we present the application of 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones (5HP2Os) as advantageous alternatives to widely used maleimides for the chemo- and site-selective labeling of cysteine residues within proteins. A variety of 5HP2O building blocks have been synthesized using a one-pot photooxidation reaction starting from simple and readily accessible furans and using visible light and oxygen. These novel reagents display excellent cysteine selectivity and also yield thiol conjugates with superior stability. 5HP2O building blocks offer a unique opportunity to introduce multiple new functionalities into a protein at a single site and in a single step, thus, significantly enhancing the resultant conjugate's properties.
Introduction
Chemo- and site-selective protein modifications are cornerstones of chemical biology as they offer a powerful strategy to both alter and study the properties of proteins. In recent years, a rapid increase in the demand for modified proteins for use in a range of applications (from fluorescent labeling to new conjugate-based therapeutic technologies) has led to increased attention being focused on the development of novel methods for protein modification.1–4 Site-selective modifications can be achieved via different strategies, including by incorporation of unnatural amino acids possessing distinct biorthogonal reactivities or by using enzymatic methods to modify a target tag or recognition pattern.5 The modifications of proteinogenic amino acid side-chains and the C- and N-termini of a protein are other widely used methods.6Despite recent advances in modifying Lys,7 Met,8–10 His,11 Tyr12–14 and Trp15,16 residues, cysteine is by far the most attractive target for chemo- and site-selective protein conjugation. Cysteine is considered the most robustly nucleophilic amino acid of the 20 canonical amino acids since it possesses a sulfhydryl group, which, under physiological conditions, has a high propensity to form the thiolate anion (pKa of 8.3). In addition, cysteine residues are low in natural abundance and can be introduced easily in a specific position into proteins by mutagenesis, thus allowing for site-selective modification. These innate properties mean that cysteine has the ideal profile for participation in site-specific protein manipulations targeting clinically useful protein conjugates, such as antibody–drug conjugates (ADCs).17
Within the toolbox of selective conjugation methods, maleimides are the most widely used cysteine modifiers (acting via a Michael addition reaction). However, maleimides are susceptible to hydrolysis, as well as having a tendency to undergo retro-Michael reactions and/or thiol exchange reactions leading to protein conjugates with poor stability.18–20 In response to these issues, a number of research groups have developed next-generation cysteine modifiers,21 with an emphasis on improving reactivity, specificity, stability and biocompatibility. Recent alternatives are included in Scheme 1A: dibromomaleimides,22 carbonylacrylic reagents (A),23 exocyclic olefinic succinimides (B),24 2-cyclopentenones (C),25 phosphonamidates (D),26 (di-)bromopyridazinediones (E),27 dinitroimidazoles (F)28 and chlorotetrazines (G).29 However, not all of these compounds are readily accessible nor do they have all the requisite characteristics to act as single-site multi-functionalization mediators that form protein conjugates. Efforts have been made to address the multi-functionalization issue with a recent one-pot thiol-amine bioconjugation strategy using dibromomaleimides for dual functionalisation in two steps.30
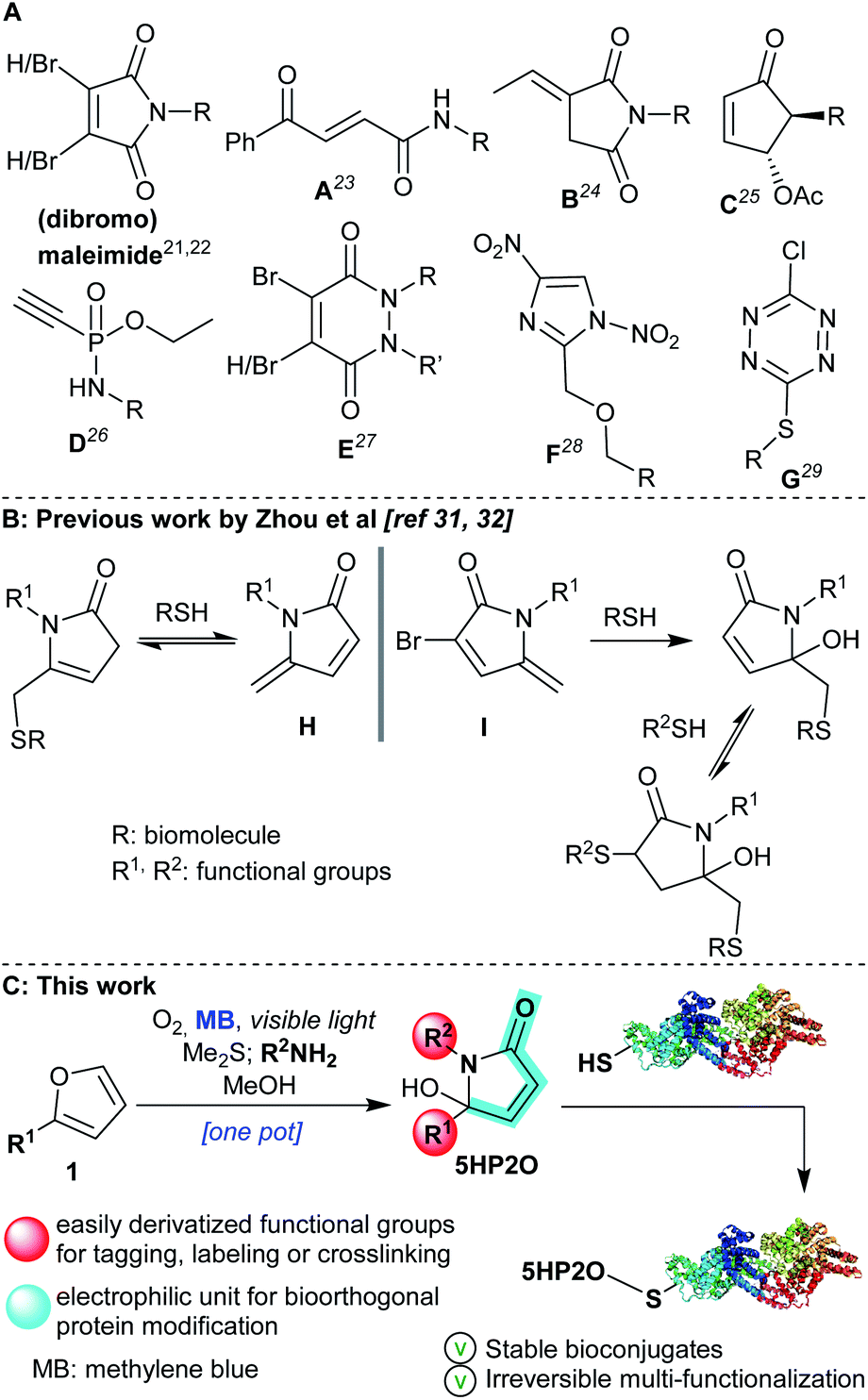 | ||
| Scheme 1 (A) Maleimide and next-generation reagents21–29 for the labeling of cysteine residues. (B) Previously studied thiol conjugations with pyrrolone H and I by Zhou et al.31,32 (C) Singlet oxygen-induced formation of 5HP2O as a candidate for cysteine multi-functionalization. | ||
Recently, two elegant studies were published by Zhou et al.,31,32 where methylene pyrrolone derivatives (H and I, Scheme 1B) were applied for cysteine modification. In the case of mono-substituted pyrrolone H, the reaction with thiols was characterised by its reversibility, a feature exploited in bioconjugation protocols where the removal of modifiers was desired.31 Insertion of a bromine atom at the 3-position of the pyrrolone backbone (I) proved crucial to achieving an irreversible thiol bioconjugation. It also provided the opportunity for a second thiol addition reaction, and, consequently, to access dual-functionalization. However, the second addition was reversible giving unstable double addition adducts.32
Over the last few years, our groups have established expertise in singlet oxygen-based synthetic chemistry33–36 and its application in DNA37,38 and peptide cross-linking.39,40 Driven by the facile nature of these protocols, we sought to develop a new and highly versatile maleimide alternative using the 5-hydroxy-1-(R2)-5-(R1)-1,5-dihydro-2H-pyrrol-2-one motif which we could gain ready access to (5HP2O, Scheme 1C). We anticipated that these easily synthesized new building blocks might offer not only excellent site selectivity but also previously unattainable irreversible multi-functionalization.
The facile preparation of 5HP2O building blocks relies on the use of simple and readily available starting materials (such as substituted furans and primary amines) and requires just one synthetic operation (Scheme 1C).41 This one-pot procedure provides the opportunity for the direct and controlled introduction of two functional groups of choice (R1 and R2 groups). In combination with the α,β-unsaturated moiety of the 5HP2O unit, a highly selective tag and modification procedure for cysteine residues should result. Moreover, the introduction of an alkyl group (R1) at the 5-position of the lactam's scaffold was considered crucial for attaining the desired enhanced stability, to hydrolysis and retro-Michael reaction, of the resultant thiol conjugates in comparison with their maleimide counterparts.
Results and discussion
Synthesis of 5HP2O building blocks and their reactivity towards thiols
We started our investigation with the γ-lactams 2 and 3 which were easily synthesized from furan 1a (Scheme 2).41 Specifically, 2-methylfuran (1a) was photooxidised in the presence of molecular oxygen and catalytic amounts of methylene blue (MB), followed by reduction with dimethylsulfide (Me2S). After the addition of the corresponding amine (benzylamine or ethanolamine), the desired 5HP2Os 2 and 3 were obtained in 62 and 64% isolated yields, respectively (Scheme 2). In order to study 5HP2O’s reactivity toward thiol conjugation, compound 2 was subjected to 1.1 equiv. of benzyl mercaptan in methanol in the presence of 0.3 equiv. of EDTA.Na4 (conditions A, Scheme 2). After 1 hour at room temperature, lactam 2 was fully converted to the conjugated product 4. The same results were obtained when either lactam 2 or 3 was subjected to 1-butanethiol in a mixture of water/acetonitrile (3/2) and at pH 8.0 (conditions B). Interestingly, in every case the conjugation proceeded exclusively at the 3 position of the lactam's backbone (products 4 and 5 were fully characterized by NMR experiments after separation of the diastereomers by flash-chromatography, see the ESI†). The regioselectivity of the reaction can be explained by the existence of an equilibrium between the 5HP2O and its open keto-form I, wherein the Michael addition proceeds at the position directed by the keto group (I → II, Scheme 2). Strong evidence for the proposed mechanism comes from the fact that the reaction of the methoxy-analogue of compound 2 (2-OMe, Scheme 2) with benzyl mercaptan, under conditions A, did not proceed even after 24 h.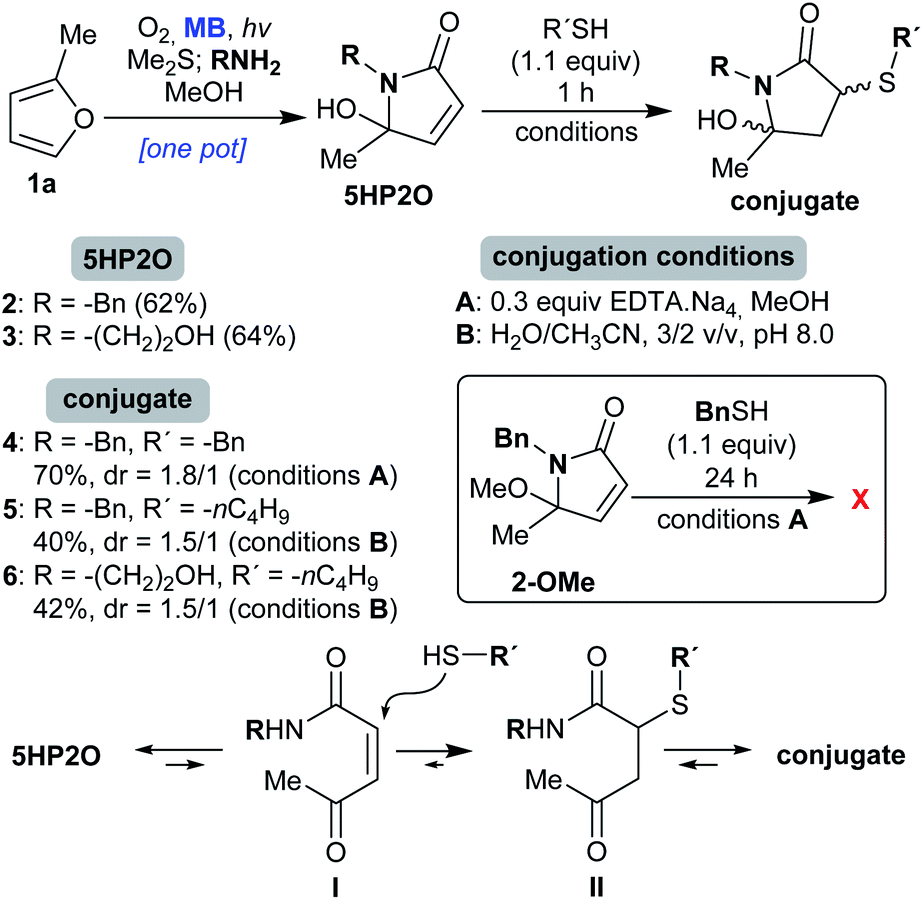 | ||
| Scheme 2 Conditions for the conjugation reaction of 5HP2O 2 and 3 with thiols and the proposed mechanistic explanation for the observed regioselectivity. | ||
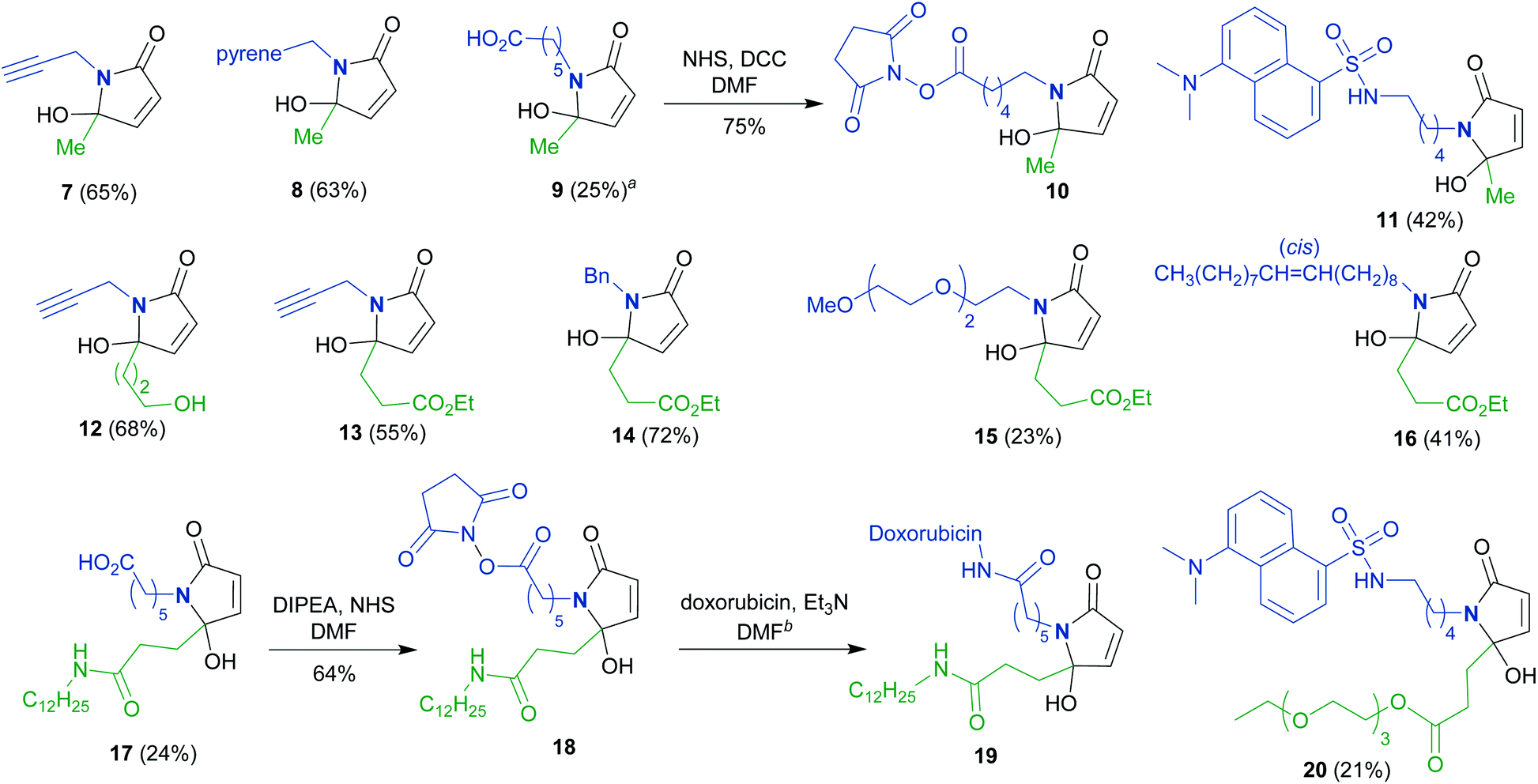
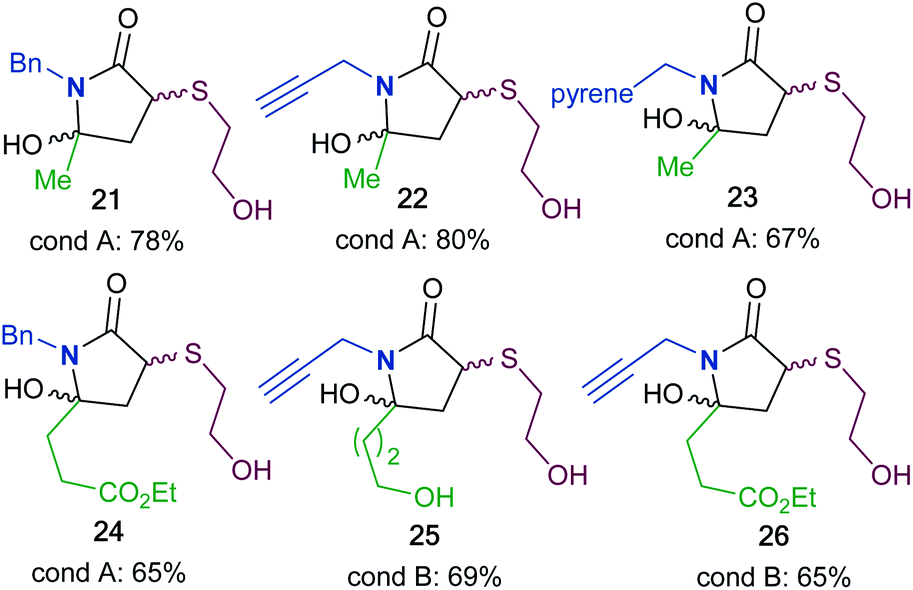
To synthesize a variety of 5HP2Os, different furan substrates (1a–1e, variability of the R1 group) were combined with various amines (variability of the R2 group) to give a series of functionalized derivatives (7–20, Table 1). Many of the R1 and R2 groups were carefully selected to allow for further derivatization: for instance, the use of propargyl amine (products 7, 12 and 13) results in a handle that can be subsequently modified via the alkyne–azide click reaction.42 The direct incorporation of a pyrene or a dansylcadaverine group results in products (8 and 11, respectively) with fluorescent properties. Additionally, compound 10 represents an alternative for the widely known commercially available SMCC linker. Ethyl-3-(furan-2-yl)propionate (1c) was combined with different amines to give bi-functionalized moieties consisting of an ester functionality at the R1 position and an alkyne (compound 13), PEG (compound 15) or a long chain unsaturated alkyl functionality (compound 16) at the R2 position. To further demonstrate the versatility of the 5HP2O synthesis, multi-functionalized building blocks were synthesized (17–20). Notably, building block 19 bears a long alkyl chain and the drug doxorubicin, while 20 contains a dansyl fluorophore and a short PEG moiety. Representative 5HP2O compounds were then tested in a simple thiol addition reaction with β-mercapto-ethanol under conditions A or B; in every case, the expected conjugates were obtained as a mixture of diastereomers (21–26, Table 2).
|
|
Building block and conjugate stability
The major drawback when using maleimides is their limited stability towards hydrolysis. To assess the hydrolytic stability of 5HP2Os, pyrrolone 3 was selected as the model compound and studied in different buffer solutions at pH 6, 7 and 8. After 24 hours of incubation at 37 °C in the appropriate buffer, no hydrolysed products were observed (see the ESI†). Unlike maleimides (we also tested the hydrolytic stability of N-benzyl maleimide – see the ESI†), 5HP2Os proved inert towards hydrolysis, giving them a considerable advantage with respect to shelf-life. We also investigated the hydrolytic stability of the resulting thiol conjugates. Specifically, a 2 mM solution of a selected conjugate (21) was incubated in different buffers at pH 7, 8 and 9 (37 °C, 24 h, see the ESI†). In any case 21 proved stable towards hydrolysis.Another drawback of maleimide-based protein conjugation is the fact that maleimide–thiol adducts can undergo thiol exchange reactions at physiological pH and temperature.43 Therefore, the stability of 21 towards thiol exchange was tested using 10 equiv. of glutathione at 37 °C (glutathione is found in high concentrations in cells). We found that trace amounts of the exchanged product were observed only after 4 days (see the ESI†). This observation contrasts with the behavior of the thiol conjugates coming from pyrrolones of type I (Scheme 1B),32 where thiol exchange was observed, most probably due to the presence of the sulfur group at the exocyclic position. As a consequence, 5HP2O building blocks fulfill all the criteria in terms of hydrolytic stability and Michael-addition irreversibility to be able to answer the previously unmet call for a selective method for dual-functionalization of cysteine residues.
Conjugations to peptides and proteins
Following the efficient conjugation of 5HP2Os with simple thiols, we embarked on extending the reactivity testing using N-acetyl-L-cysteine (NAC) (27) and glutathione (GSH) as model nucleophiles. Full conversion of 2 into the desired conjugate 28 was observed within 2.5 h under conditions B (Fig. 1A, see also the ESI†). Additionally, a direct comparison with benzyl-maleimide was made using, respectively, 5 and 20 equiv. of building block and following the kinetics of conjugation over time. Upon addition of the maleimide, immediate and full conversion was seen at a pH of 8 in accordance with the known fast reaction kinetics for maleimides. However, also with 20 equiv. of 5HP2O full conversion was reached after only 30 min. The expected GSH conjugates (29–33) with different 5HP2O building blocks (2, 3, 7, 15 and 16) were also prepared in phosphate buffered solution (Fig. 1A). The superior hydrolytic stability of our 5HP2O building blocks was illustrated via a direct comparison between NAC-conjugates with, respectively, benzyl-maleimide and our benzyl-5HP2O (2). NAC-conjugates were incubated for 24 hours in buffered aqueous media at pH of 7, 8 and 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 and at a concentration of 0.4 mM and analysed via HPLC. While for our 5HP2O conjugates no hydrolysis could be observed after 24 hours in all studied buffered media, maleimide conjugates hydrolysed up to 50% after 24 hours at a pH of 7 and more than 99% after 24 hours at a pH of 9 (see the ESI†). Following the hydrolytic stability studies, GSH exchange experiments were performed. NAC-conjugates were incubated in buffered media at a pH of 7.5 with 10 mM GSH at a concentration of 0.1 mM. Degradation of the NAC-conjugates was followed via HPLC (see the ESI†). Even with 100 equivalents of GSH, 70% of intact NAC–5HP2O conjugate is left after 5 days, while for maleimides only 20% of intact conjugate is left. The degradation of the NAC–maleimide conjugate is the result of the hydrolysis of the NAC-conjugate combined with GSH exchange as well as the hydrolysis of the exchanged GSH-conjugate.45 Detailed investigations on the influence of the nature of the N-substituent as well as the role of the exact structure and pKa of the thiol are currently ongoing and form the subject of a separate detailed study, which will be reported in due course.
44 and at a concentration of 0.4 mM and analysed via HPLC. While for our 5HP2O conjugates no hydrolysis could be observed after 24 hours in all studied buffered media, maleimide conjugates hydrolysed up to 50% after 24 hours at a pH of 7 and more than 99% after 24 hours at a pH of 9 (see the ESI†). Following the hydrolytic stability studies, GSH exchange experiments were performed. NAC-conjugates were incubated in buffered media at a pH of 7.5 with 10 mM GSH at a concentration of 0.1 mM. Degradation of the NAC-conjugates was followed via HPLC (see the ESI†). Even with 100 equivalents of GSH, 70% of intact NAC–5HP2O conjugate is left after 5 days, while for maleimides only 20% of intact conjugate is left. The degradation of the NAC–maleimide conjugate is the result of the hydrolysis of the NAC-conjugate combined with GSH exchange as well as the hydrolysis of the exchanged GSH-conjugate.45 Detailed investigations on the influence of the nature of the N-substituent as well as the role of the exact structure and pKa of the thiol are currently ongoing and form the subject of a separate detailed study, which will be reported in due course.
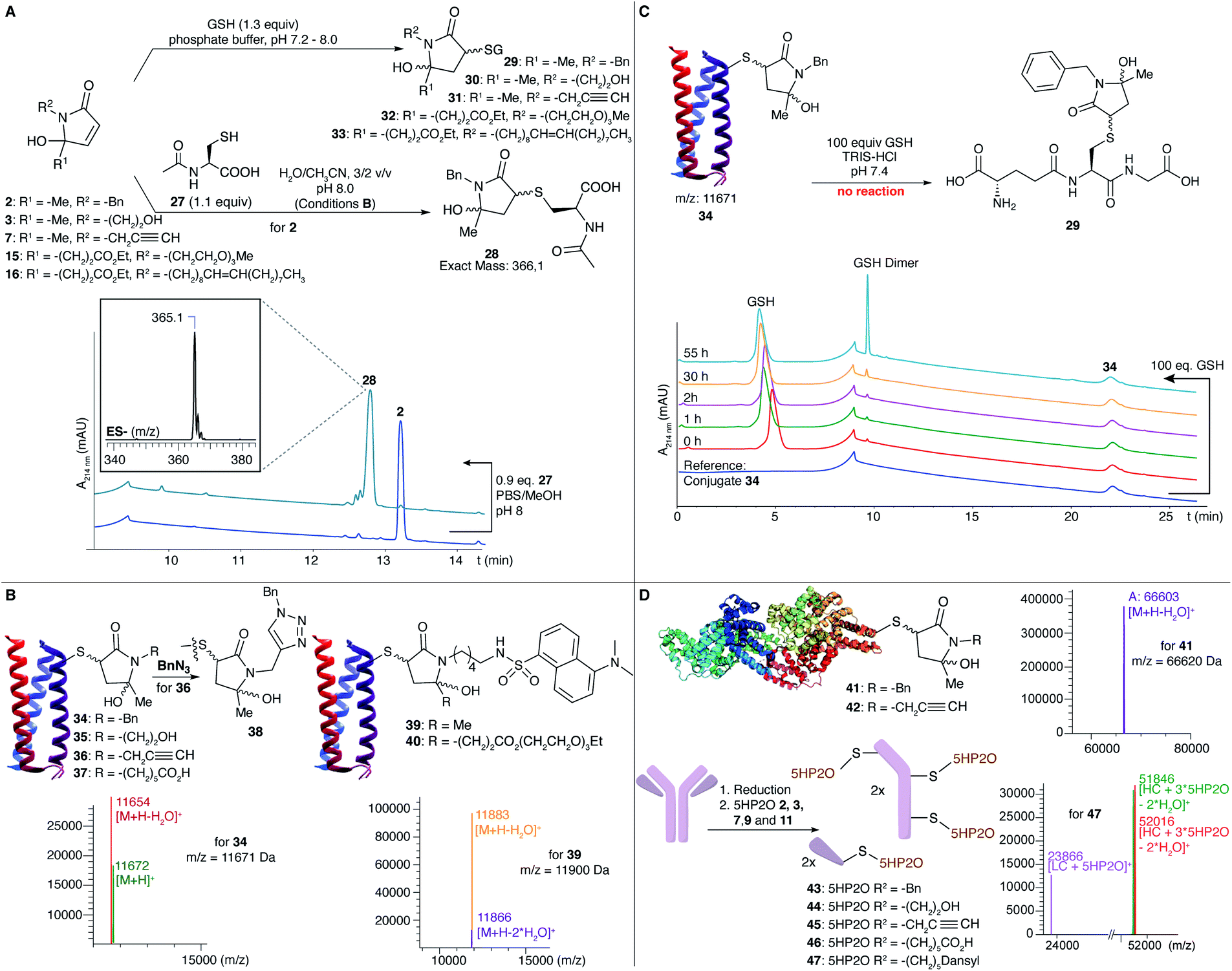 | ||
| Fig. 1 (A) N-Acetyl-L-cysteine and glutathione (GSH) conjugation to 5HP2O 2, 3, 7, 15 and 16. Conversion of 2 to 28 was observed by RP-HPLC and the product was analysed by LC-MS ([M − H]− = 365.1). (B) Conjugation of different 5HP2Os (using 10 equiv.) is achieved on alphabody (34–37, 39 and 40) at a pH of 8. (C) Conjugate 34 was incubated with 100 equiv. of GSH and the reaction was monitored by RP-HPLC. No thiol-exchanged product 29 was found. (D) Protein conjugates were efficiently prepared for BSA (41 and 42) and for the antibody trastuzumab (43–47) using 100 equiv. of 5HP2O at a pH of 8. | ||
To evaluate the applicability of 5HP2Os as genuine protein modifiers, bioconjugation experiments were performed using a model protein, MB23-Cys alphabody obtained from Complix N.V. (Fig. 1B). Alphabody proteins are of therapeutic interest because of their capacity to target protein–protein interfaces.46 The alphabody possesses one surface-exposed free cysteine residue and is an ideal model protein. The reaction was conducted using 10 equiv. of different 5HP2O building blocks (2, 3, 7 and 9) under Tris buffer conditions (10 mM, pH 8) at 25 °C. Efficient formation of the corresponding conjugates 34–37 was observed after 6–12 h. Additionally, protein conjugation conditions were screened in the pH range between 6 and 9 using 10 equiv. of 2. Limited conjugation was observed at a pH of 6, while full conjugation was seen in the pH range 7–9 (see the ESI†).
Notably, we could efficiently modify the alphabody with a fluorescent label using dansyl 5HP2O 11 (conjugate 39, Fig. 1B). For one-site bi-functionalization, an alphabody was treated with bi-functional 5HP2O 20 to yield an alphabody conjugate consisting of a dansyl fluorophore and a PEG moiety (40). Additionally, as a proof-of-concept, an initial experiment was performed to click benzyl-azide onto an alkyne-functionalised alphabody according to the well-known CuAAC reaction (product 38, Fig. 1B), illustrating the potential for versatile tandem 5HP2O–CuAAC modification protocols. Conjugate 34 was found to be hydrolytically stable in 10 mM Tris buffer at pH 7, 8 or 9 for up to 6 days. In contrast, when an alphabody was treated with 6-maleimidohexanoic acid under the same conditions, ring-opened hydrolysis products were observed (see the ESI†). Finally, the stability of the new alphabody conjugate towards thiol exchange was tested using 100 equivalents of glutathione. The RP-HPLC analysis showed no thiol exchange, with no peak for product 29 observed even after several days at 35 °C (Fig. 1C and see the ESI†).
To further illustrate the potential of 5HP2O, modification reactions were successfully performed on bovine serum albumin (BSA). When the protein was treated with 100 equiv. of building blocks 2 and 7, the mono-conjugates 41 and 42 were observed (Fig. 1D). Finally, exemplary application of the current conjugation protocol in a therapeutic context was obtained by conjugation of 2, 3, 7, 9 and 11 to the Her2-addressing IgG monoclonal antibody, trastuzumab. Trastuzumab (Fig. 1D) was first reduced using 20 equiv. of TCEP followed by the addition of 12.5 equiv. of 5HP2O per free cysteine. Labeling was confirmed by LC-MS analysis (conjugates 43–47), which showed a shift in the signal with a mass difference corresponding to one or more 5HP2O moieties in accordance with the number of solvent-exposed cysteine residues present in trastuzumab (Fig. 1D), illustrating the high degree of chemo selectivity.
Conclusions
To conclude, we have presented an efficient one-pot synthesis of a novel class of 5HP2O protein modifying agents. Functional groups at two different positions of these building blocks can be varied readily and at will (opening up further multi-functionalization possibilities, such as click reactions). The decorated 5HP2Os can be efficiently conjugated to thiols (cysteine modifiers). We have also established that the new 5HP2O modifiers have characteristics which offer significant performance enhancement when compared to the alternative maleimide-based bioconjugation methods. We have shown that the resulting conjugates are hydrolytically stable in buffered media and that the protein conjugate is stable towards thiol-exchange over prolonged periods of time which represent considerable advantages when compared to maleimide derivatives or the thiol conjugates of the 3-bromo-5-methylene pyrrolones32 which degrade through exchange. Furthermore, protein labeling with 5HP2Os resulted in conjugates with high stability. Therefore, we propose our 5HP2O building blocks as highly advantageous maleimide alternatives for protein modification and single-step multi-functionalization.Author contributions
E. D. G. was responsible for conceptualization, data curation, formal analysis, lead investigation (ESI chapters 1–8†), methodology, project administration, visualization and writing of the original draft. E. A. was responsible for data curation, lead investigation (ESI chapters 1, 4 and 6†), methodology and visualization. D. K. was responsible for conceptualization, data curation, lead investigation (ESI chapters 1 and 4†), methodology, project administration, visualization, supportive supervision and writing, reviewing and editing of the manuscript. S. S. was responsible for conceptualization, data curation, investigation (ESI chapters 1, 2 and 4–6†), methodology and visualization. A. I. was responsible for writing the original draft. L. T and E. O. were responsible for supporting investigation in the synthesis of compounds 18, 19, 32 and 33. G. V. and A. M. were responsible for conceptualization and supervision, funding acquisition, project administration, resources and reviewing and editing of the manuscript.Conflicts of interest
The technology described in the manuscript is part of a pending patent application (EP 19160048.5) by A. M., G. V., E. A., E. D. G., D. K. and S. S.Acknowledgements
E. D. G. is indebted to the BOF-UGent for a PhD grant. The research leading to these results has received funding from the European Union's Seventh Framework Program (FP7/2007–2013)/Marie Curie ITN Grant Agreement No. 316975. The authors thank Complix N.V. for providing the MB23-cys Valentine alphabody. For LC-MS and NMR measurements, we thank Jan Goeman and the NMR Expertise Centre at Ghent University. We thank Dr Tamsyn Montagnon for helpful discussions and comments. We thank Dr Alex Manicardi for scientific discussions in the design of the NAC kinetics experiments and GSH exchange experiments.Notes and references
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- S. B. Gunnoo and A. Madder, Org. Biomol. Chem., 2016, 14, 8002–8013 RSC.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- Y. Zhang, K.-Y. Park, K. F. Suazo and M. D. Distefano, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- A. H. Christian, S. Jia, W. Cao, P. Zhang, A. T. Meza, M. S. Sigman, C. J. Chang and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 12657–12662 CrossRef CAS PubMed.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827–2830 CrossRef CAS PubMed.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798–10801 CrossRef CAS PubMed.
- S. B. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529–553 CrossRef CAS PubMed.
- M. R. Lewis and J. E. Shively, Bioconjugate Chem., 1998, 9, 72–86 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem.–Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Compañón, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jiménez-Osés, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef CAS PubMed.
- D. Kalia, P. V. Malekar and M. Parthasarathy, Angew. Chem., Int. Ed., 2016, 55, 1432–1435 CrossRef CAS PubMed.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11598–11602 CrossRef CAS PubMed.
- M.-A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS PubMed.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781 RSC.
- Q. Luo, Y. Tao, W. Sheng, J. Lu and H. Wang, Nat. Commun., 2019, 10, 142 CrossRef PubMed.
- C. Canovas, M. Moreau, C. Bernhard, A. Oudot, M. Guillemin, F. Denat and V. Goncalves, Angew. Chem., Int. Ed., 2018, 57, 10646–10650 CrossRef CAS PubMed.
- A. Wall, A. G. Wills, N. Forte, C. Bahou, L. Bonin, K. Nicholls, M. T. Ma, V. Chudasama and J. R. Baker, Chem. Sci., 2020,, 11, 11455 Search PubMed.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef CAS PubMed.
- Y. Zhang, C. Zang, G. An, M. Shang, Z. Cui, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2020, 11, 1015 CrossRef CAS PubMed.
- D. Kalaitzakis, M. Triantafyllakis, G. I. Ioannou and G. Vassilikogiannakis, Angew. Chem., 2017, 129, 4078–4081 CrossRef.
- D. Kalaitzakis, M. Triantafyllakis, I. Alexopoulou, M. Sofiadis and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2014, 53, 13201–13205 CrossRef CAS PubMed.
- D. Kalaitzakis, T. Montagnon, I. Alexopoulou and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2012, 51, 8868–8871 CrossRef CAS PubMed.
- T. Montagnon, D. Kalaitzakis, M. Sofiadis and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 180–190 RSC.
- M. Op de Beeck and A. Madder, J. Am. Chem. Soc., 2012, 134, 10737–10740 CrossRef CAS PubMed.
- N. De Laet, E. M. Llamas and A. Madder, ChemPhotoChem, 2018, 2, 575–579 CrossRef CAS.
- E. Antonatou, Y. Verleysen and A. Madder, Org. Biomol. Chem., 2017, 15, 8140–8144 RSC.
- E. Antonatou, K. Hoogewijs, D. Kalaitzakis, A. Baudot, G. Vassilikogiannakis and A. Madder, Chem.–Eur. J., 2016, 22, 8457–8461 CrossRef CAS PubMed.
- D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2015, 54, 6283–6287 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- W. R. Carmody, J. Chem. Educ., 1961, 38, 559–560 CrossRef CAS.
- H. Wu, P. J. LeValley, T. Luo, A. M. Kloxin and K. L. Kiick, Bioconjugate Chem., 2018, 29, 3595–3605 CrossRef CAS.
- J. Desmet, K. Verstraete, Y. Bloch, E. Lorent, Y. Wen, B. Devreese, K. Vandenbroucke, S. Loverix, T. Hettmann, S. Deroo, K. Somers, P. Henderikx, I. Lasters and S. N. Savvides, Nat. Commun., 2014, 5, 5237 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05881e |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |