
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring Cu/Al cluster growth and reactivity: from embryonic building blocks to intermetalloid, open-shell superatoms†
Max
Schütz
ab,
Christian
Gemel
ab,
Maximilian
Muhr
 ab,
Christian
Jandl
b,
Samia
Kahlal
c,
Jean-Yves
Saillard
c and
Roland A.
Fischer
*ab
ab,
Christian
Jandl
b,
Samia
Kahlal
c,
Jean-Yves
Saillard
c and
Roland A.
Fischer
*ab
aDepartment of Chemistry, Technical University Munich, Lichtenbergstrasse 4, D-85748 Garching, Germany. E-mail: roland.fischer@tum.de
bCatalysis Research Centre, Technical University Munich, Ernst-Otto-Fischer Strasse 1, D-85748 Garching, Germany
cUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France
First published on 1st April 2021
Abstract
Cluster growth reactions in the system [Cu5](Mes)5 + [Al4](Cp*)4 (Mes = mesitylene, Cp* = pentamethylcyclopentadiene) were explored and monitored by in situ LIFDI-MS and 1H-NMR. Feedback into experimental design allowed for an informed choice and precise adjustment of reaction conditions and led to isolation of the intermetallic cluster [Cu4Al4](Cp*)5(Mes) (1). Cluster 1 reacts with excess 3-hexyne to yield the triangular cluster [Cu2Al](Cp*)3 (2). The two embryonic [Cu4Al4](Cp*)5(Mes) and [Cu2Al](Cp*)3 clusters 1 and 2, respectively, were shown to be intermediates in the formation of an inseparable composite of the closely related clusters [Cu7Al6](Cp*)6 (3), [HCu7Al6](Cp*)6 (3H) and [Cu8Al6](Cp*)6 (4), which just differ by one Cu core atom. The radical nature of the open-shell superatomic [Cu7Al6](Cp*)6 cluster 3 is reflected in its reactivity towards addition of one Cu core atom leading to the closed shell superatom [Cu8Al6](Cp*)6 (4), and as well by its ability to undergo σ(C–H) and σ(Si–H) activation reactions of C6H5CH3 (toluene) and (TMS)3SiH (TMS = tris(trimethylsilyl)).
Introduction
Metal clusters are fascinating chemical objects. Their targeted generation in the gas-phase or in solution as unligated as well as ligand stabilized species has been a “hot topic” of interdisciplinary research for a long time. The famous Schmid cluster [Au55](PPh3)12Cl6, discovered in 1981,3 and the mystery of its true composition and structure may serve as an illuminating example.4Clusters mark the transition from the molecular to the bulk state of matter. The increased understanding of their structural, electronic and physical properties, as well as the development of novel techniques for their synthesis, separation and characterization opened new avenues for cluster chemistry. Advances in computational theoretical analysis revealed interesting connections between electronic structure and reactivity patterns. Examples include small molecule activation (CO, CO2, H2, H2O, etc.) at 3d transition metal 13-atom clusters5 and the reactivity of NO and O2 at Al doped Cu cluster cations.6
Metallic elements dominate the periodic table. Considering the combinatorial space of mixed-metal clusters [M1aM2bMc3…] the enormous and largely unexplored playground becomes obvious since every atom counts in metal cluster chemistry and physics. Likewise, solid-state alloys and intermetallic phases provide rich opportunities for exciting discoveries. They are useful, in particular, as novel materials in heterogeneous catalysis.7
In cluster research, structure–reactivity relationships have mostly been reported for gas-phase clusters or gas-phase generated clusters deposited on a support.8–11 By sputtering or laser ablation of metallic targets under high-vacuum, a rich set of (bimetallic) gas-phase metal clusters is accessible.12,13 Noteworthy, mass-spectrometry is thereby a key tool for the assessment of cluster identities and of reaction products.14–16
We have been attracted to metal clusters as a research objective from the perspective of challenges in organometallic synthesis across the periodic table,17–19 and we are much inspired by linking solid state chemistry of intermetallics with the molecular chemistry of mixed metal clusters.20 There are two categories of wet chemical synthetic strategies for obtaining ligand protected intermetalloid clusters [Mn](R)m (n ≫ m; M = two or more different metal atoms; n = number of metal atoms aggregated in the cluster core; R = hydrocarbon ligand; square brackets denote the cluster core; round brackets denote the core protecting heteroatom-free, all-hydrocarbon ligand shell). Of note, the described nomenclature chosen in this manuscript is different from the IUPAC recommendations for transition metal compounds. However, the authors consider the presented way of formula writing as more intriguing in terms of metal core compositions and valence electron count.
In the “top down” approach, pre-formed ionic cluster subunits are extruded from solid state phases as the parent material by wet-chemical methods with manifold options of cluster core functionalization afterwards. However, control of cluster nuclearity (size) Mn and shape remains very difficult, especially when it comes to bimetallic clusters to be selectively extruded.20–22 Nevertheless, an extremely rich solution chemistry of Zintl clusters has emerged over the past decades.23,24 Many of these clusters were obtained by the reaction of soluble Zintl anions with organic or organometallic precursors.23
The alternative “bottom-up” approach relies on molecular organometallic compounds “M1R1” and “M2R2” (R1, R2 refer to specifically suited all-hydrocarbon ligands; the M/R stoichiometry is not specified here) and the intrinsic reactivity of these precursors against each other (Scheme 1). Modulation of the reaction by the choices of solvent, additives (e.g. H2) and by setting of the extrinsic conditions (temperature, pressure) provides deprotection of the metals from their ligand environments and triggers agglomeration of the released atoms M1 and M2. Eventually, ligated intermetalloid clusters of the general formula [M1aM2b](R1)c(R2)d (n = a + b; m = c + d; n ≫ m) are formed and can be spectroscopically characterized in solution and may even be isolated in pure and single crystalline form. The discovery of the M55 magic number open-shell superatom cluster [Cu43Al12](Cp*)12 of Mackay structure-type may be regarded as a prove of concept. It is a break-through of our work, continued over two decades,17–20 on Hume-Rothery intermetallics inspired metal clusters. The cluster was obtained in toluene solution from the reaction of [Cu]5(Mes)5 with [Al]4(Cp*)4 (abbreviated thereafter as “CuMes” and “AlCp*”; Mes = mesityl = 1,3,5-trimethylbenzene-2-yl; Cp* = C5(CH3)5 = pentamethyl-cyclopentadienyl).2Fig. 1 illustrates the 2-dimensional landscape of ligated binary clusters with Cu/Al as the example. Our research described in this article targets the area in between the n(Al) and n(Cu) axes.
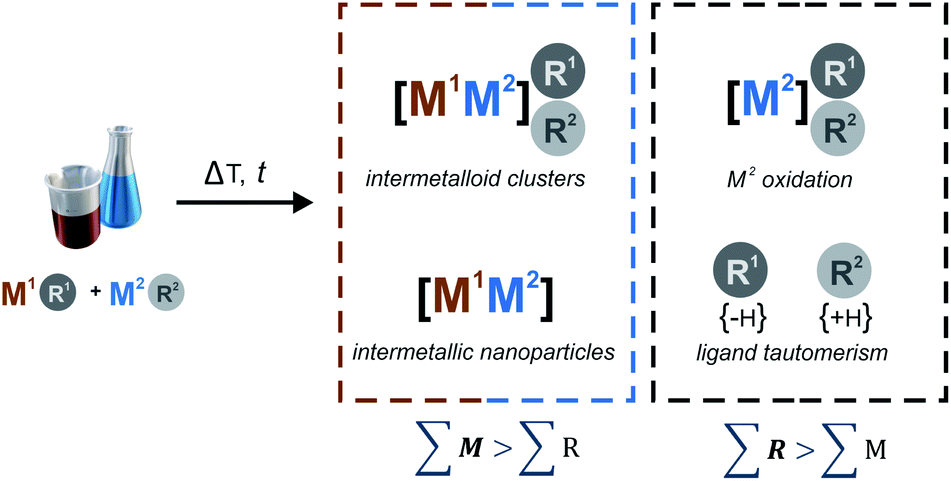 | ||
| Scheme 1 General concept for the bottom-up synthesis of ligated intermetalloid clusters [Mn](R)m (n ≫ m; M = two or more different metal atoms, e.g. M1, M2; n = number of metal atoms aggregated in the cluster core; R = hydrocarbon ligand, e.g. R1, R2; square brackets denote the cluster core; round brackets denote the core protecting ligand shell). Scheme 1 aims to conceptually catch a diversity of elementary reactions of the organometallic precursors M1R1 and M2R2 during cluster formation. Note that the option M1 = M2 is possible. More details and examples are described in the text. | ||
 | ||
| Fig. 1 The two-dimensional landscape of Cu/Al clusters. Only cluster cores are shown for simplicity. The two coordinates n(Cu) and n(Al) localize the so far experimentally characterized heterometallic species, [Cu4Al4](Cp*)5Mes (1), [Cu2Al](Cp*)3 (2), [Cu7Al6](Cp*)6 (3) and [Cu8Al6](Cp*)6 (4) described in this work and [H4Cu6Al6](Cp*)6 and [Cu43Al12](Cp*)12 from our previous reports.1,2 The diagram also shows examples of important homometallic clusters such as [Al4](Cp*)4, [Al50](Cp*)12 (ref. 25), [Cu5](Mes)5 and [Cu30H18](S2P(OnPr)2)12.26 The hydrocarbon ligand shells are omitted for clarity (color code: Cu = orange-brown; Al = blue). | ||
The organometallic cluster synthesis according to Scheme 1, for example targeting specific points in the Cu/Al landscape of Fig. 1, suffers from numerous obstacles and limitations. Usually, a broad product distribution is observed, often with a subtle dependency on small changes in the selection of precursors and reaction conditions. The delicate interplay of multiple elementary reaction steps, reversible and irreversible in nature have to be considered, often of unknown mechanistic details and kinetic parameters.
Despite the accumulated knowledge of organometallic reaction pathways is quite substantial, the outcome of a bottom-up synthesis of ligated intermetallic clusters is typically unpredictable. The successful isolation of chemically pure clusters often follows a trial-and-error approach, unless being based on profound chemical knowledge and intuition.
Rational reaction concepts guiding the choice of organometallic precursors and conditions include ligand exchange and transmetallation, oxidative addition and reductive elimination, homolytic metal ligand bond cleavage, H-abstraction and H-transfer also involving C–H activation and hydrogenolysis of metal ligand bonds. In the specific case of our Hume-Rothery intermetallics inspired organometallic cluster chemistry,17–20 the selected transition metal compounds M1R1, e.g. CuMes, are treated with low valent group 12 and group 13 compounds M2R2 such as ([Zn2]Cp*)2 or AlCp* of Zn(I) and Al(I) oxidation states. Here, the M2R2 component exhibits a unique triple function. It acts as reducing agent for M1R1 and as a sink or reservoir for R1 ligands by formation of oxidized M2 species, e.g. MesZnCp* or Mes2AlCp*, i.e. the formation of M2R22 or M2R23 (and the R1/R2 exchanges derivatives). Secondly, the M2R2 component can also release M2 by R2 transfer and cleavage processes to form alloyed M1/M2 cluster cores. The third function of M2R2 is as capping ligand for trapping [M1aM2b] cores. Our preference of choosing Cp* for R2 is connected with its haptotropic binding mode and flexibility of adopting to different electronic and steric requirements at the M centers. In addition, Cp* contributes to the stabilization of the cluster species by weak interactions (dispersion forces).
The metal precursors M1R1 and M2R2 are typically highly reactive and chemically labile. Thus, homolytic metal–ligand bond cleavage or hydrogenolysis may occur and irreversibly lead to (inter-)metallic nanoparticles (NPs) and organic by-products including tautomers R1,2(−H), R1,2(+H) and dimers R12, R22, R1R2. Scheme 1 aims to conceptually catch this diversity of elementary reactions. The general synthetic concept illustrated in Scheme 1 is of course oversimplified; it yet illustrates fundamental concepts in wet-chemical, bimetallic cluster chemistry. The synthesis of Pd/Ga, Pd/Al, Pd/In, as well as of Cu/Al and Cu/Zn clusters and coordination compounds can be seen as representative examples thereof.1,2,27–32 Noteworthy, Scheme 1 is also applicable for many monometallic cluster species if the possibility of M1 = M2 is taken into account. For example, a plethora of ligated Al clusters is accessible by controlled disproportionation of Al(I)-halide solutions.33 The details of the involved complex reaction networks are different for each given combination of chosen organometallic precursors and conditions. Precise adjustment of all parameters is crucial for a successful and reproducible synthesis of a certain cluster. However, a rational for setting the parameters to obtain a defined cluster and targeting a specific point in the Cu/Al cluster landscape of Fig. 1 is still not there.
Following the synthetic concept of Scheme 1 and based on our results on the coordination chemistry of low valent group 12 and group 13 species M2R2 at transition metal M1 centers we already were able to obtain a range of so-called “nano brass” Cu/Zn clusters of small and medium nuclearities by combining Carmona's [Zn2](Cp*)2 with a variety of Cu(I) complexes as Cu sources. We discovered the embryonic (smallest) building block, the σ-aromatic [CuZn2](Cp*)3,34 and also larger clusters like {[Cu2Zn5](Cp*)5}+ (as [BArF]-salt) or [Cu3Zn4](Cp*)5.30 The isolation of defined Cu/Zn cluster species, especially of larger nuclearities, is a sophisticated task. We are facing low yields, complicated product separation and purification procedures and the information about the nature of species formed in a reaction is often based almost solely on the luck of obtaining single crystals suitable for X-ray diffraction. The very same is true for the related Cu/Al cluster “nano bronze” chemistry. So far, we were able to isolate only two examples, the hydrido M12 cluster [H4Cu6Al6](Cp*)6 and the M55 cluster [Cu43Al12](Cp*)12 mentioned above.1,2 Unfortunately, only small amounts of this latter cluster were obtained by selecting single crystals from the raw product. Intermediates, which certainly exist on the way to this M55 cluster, could neither be isolated nor unambiguously characterized by in situ NMR. However, from the somewhat richer family of isolated and structurally characterized Cu/Zn clusters obtained by a similar chemistry we deduced that there must be an experimental way to shine more light into the dark reaction solutions of the CuMes/AlCp* system.
High resolution mass spectrometry is a powerful method in cluster chemistry and very suitable to establish exact compositions. It is particular versatile for in situ analysis of reaction solutions, which has also been demonstrated to be a suitable method for the analysis of carbonyl cluster mixtures in solution.35–39 However, mass spectrometry has not been widely applied to organometallic cluster chemistry as a tool for monitoring cluster growth, interconversion and the reactivity of clusters towards small molecules. Sample transfer under strict exclusion of air and moisture is needed to avoid decomposition and unwanted parasitic reactions. Also, highly efficient vaporization and smooth ionization techniques are required to yield meaningful and reliable data. Only recently we were able to establish “liquid injection field desorption ionization (LIFDI)” orbitrap mass spectrometry directly coupled to an inert-gas (Ar) reaction chamber (glove box). This instrumentation allowed us to precisely assess the composition of clusters and mixtures of clusters, also directly from the reaction solutions, by detection and analysis of their molecular ion signals and fragmentation patterns.40
With this new coupling of synthesis and analysis we were able to identify and isolate a number of new Cu/Al clusters being accessible from CuMes and AlCp*, quite as we expected. The “embryonic clusters” [Cu4Al4](Cp*)5Mes (1) and [Cu2Al](Cp*)3 (2) were isolated and fully characterized including a detailed bonding analysis on the DFT level of theory. Systematic investigation of cluster growth reactions involving these two building blocks and taking advantage of the direct feedback loop of mass spectrometry of reaction solutions into the parameter optimization of the cluster growth experiment led us to the discovery of the M13/M14 cluster mixture [Cu7Al6](Cp*)6 (3) [HCu7Al6](Cp*)6 (3H) and [Cu8Al6](Cp*)6 (4) (see Fig. 1). Although these three clusters could not be separated from each other, but are accessible as mixtures only, our mass spectrometric approach allows for detailed insight into their reactivity, e.g. C–H and Si–H activation, as a function of the electronic structure (open vs. closed shell).
In this article we wish to introduce the reader to this kind of organometallic cluster chemistry and to our research strategy which aims to systematically harvest fundamental insight from the highly complex, combinatorial chemical space addressed by Scheme 1 and Fig. 1. The article is organized in the following way: First, the principles of organometallic Cu/Al cluster synthesis are described, as discussed in this introduction. Second, we present the analytic characterization and providing evidence of the identity of the newly discovered Cu/Al clusters 1–4 in comparison to the already known ones and in relation to Cu/Al intermetallic solid-state phases. Third, the electronic structure of the clusters, i.e. bonding analysis on the DFT level of theory, is discussed which leads to the fourth, final chapter dealing with the consequences for chemical reactivity. We found that “open shells open doors”. Among the three closely related M13/M14 clusters only the radical species [Cu7Al6](Cp*)6 (3) undergoes facile C–H and Si–H activation reactions with toluene and (TMS)3SiH (TMS = SiMe3). Altogether and based on our new examples presented in this article, we claim a uniquely rich intermetalloid chemistry to be explored.
Results
Principles of cluster growth
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) as well as one metal coordinated mesityl unit. Also the side products AlMes3 (ref. 41) and Mes2AlCp* can be identified in these in situ1H-NMR spectra. Time dependent 1H-NMR spectra indicate the concentration of 1 to be the highest after about 30–80 minutes. As shown in Fig. S1,† only small signals of other Cp* containing species are present at that time, which become more prominent at increased reaction times.
:2:1) as well as one metal coordinated mesityl unit. Also the side products AlMes3 (ref. 41) and Mes2AlCp* can be identified in these in situ1H-NMR spectra. Time dependent 1H-NMR spectra indicate the concentration of 1 to be the highest after about 30–80 minutes. As shown in Fig. S1,† only small signals of other Cp* containing species are present at that time, which become more prominent at increased reaction times.
Observation of Mes2AlCp* and AlMes3 as side products in the formation of 1 is indicative of partial reduction of Cu(I) to Cu(0) by AlCp* accompanied by ligand transfer reactions between Al and Cu. Notably, 1.5 eq. of AlMes3 and Mes2AlCp* are formed per molecule of the [Cu4Al4](Cp*)5(Mes) cluster 1 according to in situ1H-NMR (see ESI, Fig. S1†). A clear stoichiometric reaction would only include formation of one equivalent AlMes3 (Scheme 2), however, we suppose that the thermal instability of 1 (vide infra) leads to a competition between its formation (including formation of AlMes3) and its decay (including formation of Mes2AlCp*). 1 can be interpreted as an aggregate of redox-intermediates occurring in the reduction of Cu(I) and its subsequent coordination by AlCp*. We assume similar mechanisms to occur in the early stages of the synthesis of [Cu43Al12](Cp*)12, as Mes2AlCp* was also identified as a side product this reaction.2
 | ||
| Scheme 2 Synthetic pathways to the new Cu/Al clusters [Cu4Al4](Cp*)5(Mes) (1), [Cu2Al](Cp*)3 (2), and the inseparable mixture of [HCu7Al6](Cp*)6 (3H), [Cu7Al6](Cp*)6 (3), [Cu8Al6](Cp*)6 (4). Cu atoms are illustrated in orange, Al atoms in blue, respectively. For simplicity, only cluster cores are shown. All details of the experimental procedures and analytical characterization data are provided in the ESI.† Upon heating, cluster 1 can be transformed into a mixture of 3, 3H and 4. This mixture can be selectively obtained by a sequence of cluster degradation and growth reactions with additives involving the trimetallic cluster intermediate 2. | ||
Monitoring the reaction by LIFDI-MS analysis (see ESI, Fig. S15†) reveals a variety of intermetallic Cu/Al clusters with the exact composition being dependent on the reaction time (vide infra). However, the molecular ion peak of 1 cannot be detected at all, obviously 1 cannot be ionized without destruction by the LIFDI technique. Also, LIFDI-MS analysis of the isolated single crystals of 1 did not yield a detectable molecular ion signal.
When a toluene solution of 1 is treated with an excess of 3-hexyne (20 °C), a color change from deep-green to orange-brown is observed within a few minutes. The cluster degradation reaction from 1 to 2 is accompanied by consumption of four equivalents 3-hexyne per molecule of 1 as identified by in situ1H-NMR spectroscopy (see ESI, Fig. S2†). After a total reaction time of four days at room-temperature, 2 is clearly identified as main product by 1H-NMR beside a variety of hexyne- and mesitylene containing side-products. LIFDI-MS analysis of the reaction solution reveals [Cu2Al4](Cp*)3(Mes)(Hex)2 (Hex = hexyne = C6H10) to be the main side product of the reaction (see Fig. S16 and Table S2, ESI†).
The presence of [Cu2Al4](Cp*)3(Mes)(Hex)2 as main side product in the synthesis of 2 (vide supra) points towards a cluster degradation mechanism in which 3-hexyne extrudes CuCp* moieties from 1 by stabilizing the electron-deficient, low valent Cu(0) centers through coordination. A detailed analysis of the LIFDI-MS spectra of the reaction reveals a variety of other side products formed (see Table S2, ESI†). Insight into the Cu and Al fate during the reaction can be gained by grouping the reaction products according to their Cu:Al ratio.
Whereas Cu gets mainly incorporated into 2 and some Cu-rich clusters (e.g.3/4 among others), the Al stoichiometry of the reaction is balanced by formation [Cu2Al4](Cp*)3(Mes)(Hex)2 and several other Al-rich clusters and Al-containing coordination compounds. With the two small building blocks 1 and 2 in hand, we set out to explore cluster growth reactions by a detailed study of reaction parameters (see Table S1, ESI†). Obviously, a dynamic composite of three clusters, namely [Cu7Al6](Cp*)6 (3), [Cu8Al6](Cp*)6 (4) and [HCu7Al6](Cp*)6 (3H) is available. Analysis of reaction products was therein mainly conducted using LIFDI-MS analysis. Thereby, the assignment of sum formulas to the peaks was verified by comparison between calculated and experimental isotopic patterns, as exemplary shown in Fig. 3a, b for 4 and 2, in Fig. S21† for 3H, 3D and 3. Subtle differences of only one H atom, e.g. in 3 and its hydride containing counterpart 3H are well recognized by a shift of 1 m/z in the mass spectra (Fig. 3c1/c2).
In conclusion, 2 is therefore clearly an embryonic building block for the formation of larger clusters. Due to its inherent Cu:Al stoichiometry, exact tuning of the amount of Cu(I) added is necessary for the generation of larger Cu/Al clusters.
:0.28) at 75 °C, followed after 4 h by addition of another 0.54 eq. of CuMes and further heating to 75 °C for 18 h (Scheme 2; overall Cu:Al stoichiometry applied 1:1.22). If the reaction of the organometallic sources of Cu and Al is conducted in precisely this way, the product clusters 3 and 4 are the major components in the equilibrated reaction mixture then observed in LIFDI-MS analysis (Fig. S18, ESI†). The reaction is accompanied by formation of Mes2AlCp* and AlMes3 as identified by in situ1H-NMR (Fig. S5†). Notably, compounds 1 and 2 are observed as transient intermediates in the reaction by 1H-NMR analysis before and after addition of the second portion of CuMes, respectively. This underlines their role as building blocks in cluster growth reactions. From these reaction solutions, 3/4 can be obtained as micro-crystalline precipitate after cooling to −30 °C and washing with cold n-hexane in 25% isolated yield. It is noted that the use of slightly less CuMes in this size focused synthesis of 3/4 did not significantly change the molar ratio of 3:4 according to NMR and LIFDI-MS analysis. In this experiment, however, a mixture of 3 and 4 together with the hydridic species 3H is then detected.
Based on these results a selective synthesis of 4 was developed by subjecting the primarily obtained cluster composite 3/4 to different stoichiometric amounts of CuMes. Reaction with 5 molar eq. of CuMes leads to a complete consumption of [Cu7Al6](Cp)6 as identified by in situ LIFDI-MS as well as 1H-NMR (vanishing of the corresponding paramagnetic signal even after short reaction times at room-temperature, see Fig. S6 and S19†). Again, the reaction is accompanied by formation of Mes2AlCp* and AlMes3. Quantitative analysis of the 1H-NMR spectra indicates increasing quantities of 4 being fully consistent with the LIFDI-MS spectra showing 4 as the only intense signal after 5 h reaction time. Indeed, single crystals of 4 were obtained from the filtered reaction solution in very small yield upon cooling to −30 °C. TEM images of evaporated reaction solutions again show the presence of smaller (5 nm) and larger Cu nano particles (ca. 50 nm) (see Fig. S34†). It can be assumed that due to its open-shell character, 3 is more reactive towards and is thus converted to 4, while 4 remains initially unreacted (see Scheme 3 for illustration). However, both clusters 3 and 4 are expected to be susceptible to cluster growth reactions with CuMes and this may lead to larger clusters in the reaction solution which cannot any more be observed by our LIFDI-MS instrumentation at this point of methodological development.
 | ||
| Scheme 3 Illustration of the reaction behavior of the radical species 3 [Cu7Al6](Cp*)6 towards R–H (R = toluyl, silyl) and towards Cu sources. See also Fig. 6 and the text for a rationalization of the radical reactivity of 3 in terms of molecular orbitals. | ||
Characterization of isolated species
 | ||
| Fig. 2 Representation of the molecular structures of the new, fully characterized clusters 1, 2, 3H and 4, as well as of the previously reported clusters [H4Cu6Al6](Cp*)6 (ref. 1) and [Cu43Al12](Cp*)12.2 Color code: Cu = dark orange, Al = blue. The molecular structures in the solid state were obtained by SC-XRD. Thermal ellipsoids are shown at the 50% probability level, hydrogen atoms, co-crystallized solvent molecules and disordered groups are omitted for clarity and ligands are simplified as wireframes. Full details of the structure determination are given in the ESI.† | ||
1 consists of a Cu4Al trigonal bipyramid, (Cu1–Cu4, Al1), whereby the central Cu2 unit is additionally coordinated by two “terminal” (Al2, Al4) as well as one bridging (Al3) AlCp* ligand. Interestingly, to the best of our knowledge, 1 is the first crystallographically characterized compound featuring an AlIMes ligand. Alternatively, a Cu4 butterfly structural motif can be distinguished in the structure of 1, which is known from other molecular Cu clusters like [Cu4Te4](PiPr)4 (ref. 45) and [Cu4](PPh3)2(mt)4 (ref. 46) (mt = 2-mercaptothiazoline). Interestingly, similar structural motifs can also be identified in larger Cu/Al clusters. In this context, the structure of 3H/4 can be described as a superposition of two AlCp* capped Cu-butterfly motifs (see Fig. 4a). An Al atom surrounded by four butterfly-like arranged Cu atoms is also found in the molecular structure of [H4Cu6Al6](Cp*)6,1 while in [Cu43Al12](Cp*)12, AlCp* occupies positions capping Cu3 triangular faces.2 In view of these structural analogies, 1 can be seen as a native “building block” for larger Cu/Al aggregates.
 | ||
| Fig. 3 (a) Comparison of calculated (top) and experimental (bottom) isotopic patterns of 2 as determined by LIFDI-MS. (b) Comparison of calculated (top) and experimental (bottom) isotopic patterns of 4 as determined by LIFDI-MS. (c1/c2) Visualization of the hydride shift of one m/z unit by comparison of calculated mass spectra of 3 and 3H (see Fig. S21† for experimental mass envelopes). (c3) Isotopic pattern (experimental, determined by LIFDI-MS) of a mixture of 3/3H as determined by LIFDI-MS after heating of 3 in toluene (110 °C, 5 days). Fractions of 3 and 3H are illustrated in green and purple, respectively. | ||
 | ||
| Fig. 4 (a) Illustration of the butterfly [Cu4] motif in 1 (left) and 4 (right). (b) Structural evolution of the Cu-kernels in [H4Cu6Al6](Cp*)6, 3H and 4 (from left to right). | ||
Likewise, the embryonic character of 2 is reflected in the appearance of its almost equilateral triangular Cu2Al motif in 1, whereas in larger clusters a direct comparison is difficult due to the lack of CuCp* moieties. In general, the structure of 3H/4 compromises a core of two nested copper tetrahedra embedded into an AlCp* octahedron. Hereby, the outermost Cu positions are only partially occupied (composition according to refined crystal structure: [Cu7.33Al6](Cp*)6), being well consistent with a composite of [HCu7Al6](Cp*)6 and [Cu8Al6](Cp*)6 as determined by LIFDI-MS (vide supra).
In general, the structure of 3H/4 compromises a core of two nested copper tetrahedra embedded into an AlCp* octahedron. Hereby, the outermost Cu positions are only partially occupied composition according to refined crystal structure: [Cu7.33Al6](Cp*)6, being well consistent with a composite of [HCu7Al6](Cp*)6 and [Cu8Al6](Cp*)6 as determined by LIFDI-MS (vide supra).
The hydride could not be located by SC-XRD but DFT calculations allow for an assessment of its binding mode (vide infra). Contrary and as expected, the structure of 4 (SC-XRD data of the isolated pure compound) compromises a fully occupied [Cu8] kernel. The faces of the inner Cu tetrahedron are capped by the Cu atoms of the outer Cu tetrahedron resulting in an overall structural motif, which is well known as “stella quadrangula” from corresponding Hume-Rothery phases like γ-brass (Cu5Zn8) or Cu9Al4.47 We would like to emphasize the structural relationship between the species [H4Cu6Al6](Cp*)6 (dicapped tetrahedron)1 [Cu7Al6](Cp*)6 (triple capped tetrahedron) and [Cu8Al6](Cp*)6 (tetracapped tetrahedron) (see Fig. 4b). Additionally, the tetracapped tetrahedron to tricapped tetrahedron relationship is documented in the literature for homometallic copper clusters, although featuring not Cu(0) but Cu(I).48,49
Triangular motifs are common in all three compounds. Whereas 2 itself is an almost equilateral triangle, in 1, the central triangle of the bipyramid exhibits an angle of 57.75(3)°. The triangular faces of the inner tetrahedron in 3H/4 span almost equilateral triangles with bond angles close to 60°. The outer tetrahedron itself is spanned by almost equilateral triangles.
Elemental analysis of micro-crystalline 3/4 closely matches the values expected for [Cu8Al6](Cp*)6 indicating that the amount of [Cu7Al6](Cp*)6 is below 5 mass%. This is in full agreement with SQUID measurements of the sample, indicating an overall diamagnetic behavior over the whole temperature range with only very small paramagnetic contributions.
Electronic structure characterization
A rationalization of the bonding in 1 can start in considering it as made of six building units, namely one (edge-bridging) Al(η5-Cp*), one Al-Mes, two Cu(η5-Cp*) and two “linear” Cu–Al(η5-Cp*) fragments. Assuming that six electrons are used for Cp* to M bonding (M = Al, Cu) and that the Cu–Al terminal bonds are localized 2-electron bonds, then the Al(η5-Cp*), Al-Mes, Cu(η5-Cp*) and “linear” Cu–Al(η5-Cp*) fragments are 2-, 2-, 0- and 1-electron donors, respectively. Therefore, one is left with 6 electrons for the bonding within the [Cu4Al2] inner core of 1. The M(η5-Cp*) (M = Al, Cu) fragments have only one σ-type hybrid orbital to participate to the bonding. The Cu–R (R = AlCp*) and Al–R (R = Mes) units have three orbitals available. One is a σ-type hybrid and two are π-type np AOs, i.e., “perpendicular” to the M–R bond.50 With such a fragment orbital manifold and looking at the [Cu4Al2] core as an edge-bridged [Cu4Al] trigonal bipyramid, it is easy to predict that there are only two bonding combinations of the fragment frontier orbitals that can be associated with the [Cu4Al] trigonal bipyramid. They are the in-phase combination of the five σ-type hybrid and the π-bonding combination on the [Cu2Al] triangular base of the bipyramid, stabilized by the out-of-phase combination of the two σ-type hybrids of the CuCp* capping units. These orbitals are sketched in Scheme S5† (left side and middle). They contain four of the six cluster electrons. The remaining electron pair is associated with the 2-electron/3-center bonding between the μ2-AlCp* fragment and the Cu–Cu edge it bridges (right side of Scheme S5†). Indeed the “linear” Cu-AlCp* units have enough frontier orbitals to participate to three bonding pairs.This qualitative picture is supported by DFT calculations on 1 and on a simplified and symmetrized model of 1 in which all the methyl groups were replaced by hydrogens (see Fig. 6a). In the real cluster 1 the two “σ-type” cluster MOs of Scheme S5† somehow mix together and some supplementary mixing also occurs with other cluster MOs, in particular with those associated with the terminal Al–Cu bonds. The major computed data for 1 are gathered in Table S6.† There is a good agreement between the optimized geometry and the X-ray structure. The computed 1H- and 13C-NMR chemical shifts match also nicely with their experimental counterparts (Fig. S37 and S38†). From the Wiberg bond indices (WBI) one can see that the strongest Al–Cu bonds are those between Al(Mes) and the two Cu(Cp*) atoms. Cu–Cu bonding appears relatively weak (WBIav = 0.065). As expected, the more positively charged Cu and Al atoms are those bonded to Cp* ligands. It is of note that the Cp* ligands play a crucial role in stabilizing the structure of the very electron-deficient cluster 1. Indeed, when the Cp*s are replaced by simple Cps in the calculation, the structure collapses during the geometry optimization process. The effect of the Cp* ligands is of double nature: their electron-donating ability tends to satisfy as best as possible the electron demand of the cluster core, and they constitute a tight and rather rigid protecting shell around the cluster. Such a shield effect of Cp* ligands around metal cluster cores is already well documented.1,2,18,30
The DFT-optimized structure of 2 is of approximate C2v symmetry, with Cu–Al bonds of 2.294 and 2.295 Å and a Cu–Cu bond of 2.421 Å. These values are expected to be more accurate than their X-ray counterparts (see above). The corresponding WBI values are 0.559, 0.557 and 0.199, respectively. Cu–Cu bonding in 2 is thus much stronger than in 1. The 3-center/2-electron bonding within the triangle is made of the in-phase combination of the unique σ-type hybrid frontier orbital of the three MCp* (M = Cu, Al) constituting fragments and the two electrons provided by the AlCp* units. The corresponding Kohn–Sham orbital is the HOMO−7. It is plotted in Fig. 6b. Thus, 2 is isoelectronic to [Zn2Cu](Cp*)3,34 or more simply, [H3]+. For the sake of comparison, the optimized geometry of the isoelectronic homometallic {[Cu3](Cp*)3}2− hypothetical anion yielded Cu–Cu distances and WBI of 2.389 Å and 0.412, respectively, i.e. stronger Cu–Cu bonding than in 2. The 1H- (2.0 and 1.7 ppm) and 13C-NMR chemical shift (Fig. S37†) computed for 2 are in good agreement with their experimental counterparts.
The rather compact Cu4@Cu4@Al6 shell arrangement of the inner core in 4 suggests looking at it as a superatom51–56 with an electronic structure that can be rationalized within the spherical jellium model.57 Indeed, its number of core bonding electrons (1 × 8 (Cu) + 2 × 6 (AlCp*) = 20) is one of the closed-shell stability “magic” numbers predicted by this model. It corresponds to the 1S2 1P6 1D10 2S2 jellium electron configuration (the 3d(Cu) electrons are not supposed to be involved in this count2). This is confirmed by our DFT calculations on 4 and on a simplified model in which the Cp* ligands were replaced by Cps. In the latter model, ten occupied Kohn–Sham orbitals can be easily identified as the jellium orbitals containing the 20 cluster electrons (Fig. S38†). They clearly derive from the jellium orbitals of the bare [Cu8Al6]6+ core, as illustrated in Fig. S39,† and are only weakly perturbed by the passivating ligand shell.58 The computed HOMO–LUMO gap of 4 is 1.17 eV and the HOMO, which is the weakly bonding 2S orbital (Fig. 6c, left side), lies 0.90 eV above the HOMO−1, which is of 1D nature. This situation suggests that 4 could be easily oxidized, generating a 19-, perhaps 18-electron, species. Relevant computed data are given in Table S7.† The Wiberg indices are consistent with weak and strong Cu–Cu (WBIav = 0.025) and Al–Cu (WBIav = 0.289) bonding, respectively. As for 1 and 2, the computed 1H- (1.8 ppm) and 13C-NMR (115.2 and 12.9 ppm) chemical shifts are in line with their experimental counterparts (vide supra). Removing one of the Cu2/Cu2a second-shell atoms in 4 (see Fig. 2 and 4b), generates the [Cu4@Cu3@Al6](Cp*)6 odd-electron cluster 3, with limited structural changes (see Table S7†) of the optimized geometry and a still rather spherical molecular shape. From the electronic structure point of view, when going from 4 to 3, one loses one 4s(Cu) orbital (involved in cluster bonding) and the electron it contains. The result is a limited weakening of the bonding character in the occupied jellium orbitals. On the other hand, the electron loss generates a paramagnetic 19-electron superatom, with a singly occupied 2S HOMO (Fig. 6c, right side). This situation is somewhat related to that of the 19-electron gas-phase [Cu18]− cluster.59 This SOMO resembles somewhat the HOMO of 4 but is polarized on the capping Cu atom trans to its “missing” congener. It also gets some contribution from three Al atoms (Fig. 6c and Table S7†). The computed value of the 1H-NMR chemical shifts of 3 (1.9 and 1.7 ppm) match approximately with the observed paramagnetic signal of −1.03 ppm. The hypothetical 20-electron anion 3− adopts a rather similar structure with a substantial HOMO–LUMO gap of 1.23 eV. Going from 3 to 3H also restores the closed-shell “magic” 20-electron configuration and provides an additional 1s(H) orbital that can participate to cluster bonding. Two low-energy isomers were found (Fig. 5 and Table S7†). In 3H(a) the hydride occupies the μ3 capping position of the “missing” copper atom in 3, In the isomer 3H(b), which is the most stable by 5 kcal/mol−1, the hydride sits inside the copper cage (H@Cu4@Cu3@Al6). Owing to the elongated shape of the inner Cu4 tetrahedron, the hydride does not occupy its very center, but lies close to the smaller Cu3 triangular face, in a μ3 bonding mode. This configuration exhibits a large HOMO–LUMO gap of 1.81 eV. It is of note that, in both 3H(a) and 3H(b) isomers, the hydride caps the same Cu3 face, but in an outer and inner position, respectively. In both isomers, the hydride NAO charge is substantially negative (−0.35 and −0.58, respectively). The corresponding computed 1H-NMR chemical shifts are also negative (−21.8 and −52.4 ppm, respectively). Test calculations indicate that these extremely shielded values remain stable upon functional and basis set changes. One should also note that all the other 1H-NMR chemical shifts computed for compounds 1–4 agree with their experimentally recorded counterparts (see above and Fig. S37–S39†) and that values of the order of −50 ppm are known.60 In any case, the hydride in 3H(b) (or 3H(a)), is likely to be fluxional, rendering its NMR signal detection problematic.
 | ||
| Fig. 5 The two computed isomers of 3H and their relative free enthalpies G (methyl groups omitted for clarity). The μ3-hydride is shown in black. | ||
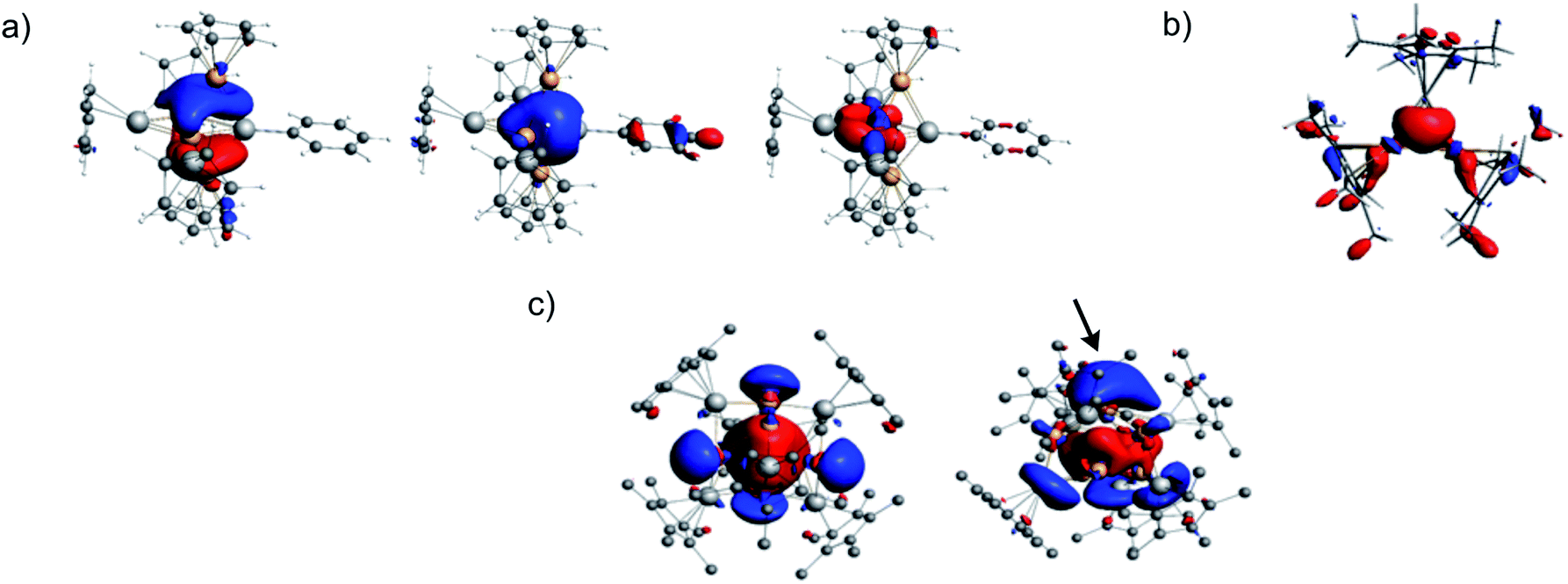 | ||
| Fig. 6 (a) The three occupied Kohn–Sham orbitals containing the six metal–metal bonding electrons in the simplified model for 1, [Cu4Al4](Cp)5(C5H5) (single-point calculation on a symmetrized model of Cs symmetry). (b) The 3-center bonding Kohn–Sham orbital (HOMO−7) associated with the 2-electron/3-center metal–metal bonding in 2. (c) The HOMO of 4 (left) and SOMO of 3 (right). Polarization of the SOMO is marked by an arrow and is attributed to the radical reactivity of 3. | ||
C–H and Si–H activation
When isolated 3/4 is heated in toluene to 110 °C for several days, LIFDI-MS analysis of the solution indicated partial transformation of 3 into 3H (see Fig. 3c3 and Scheme 3). Pentamethylfulvene was identified as an organic reaction product by GC-MS (see Table S5†). However, no reaction at all is observed in toluene-d8 (also no formation of pentamethylfulvene according to 1H-NMR analysis) indicating C–H bond activation of toluene playing a crucial role in the reaction pathway. We suggest a mechanism, in which 3 initially reacts with toluene by H radical transfer forming 3H and toluyl radicals, which intermolecularly C–H activate Cp* ligands in a second reaction step, finally resulting in formation of pentamethylfulvene. The presence of the “deprotected” cluster [Cu8Al6](Cp*)5 as a small peak in LIFDI-MS analysis is consistent with this hypothesis (see Fig. S23, ESI†). It is noted that substantial isotope effects in such H atom transfer reactions from toluene are known in literature.61The reactive open-shell character of 3 is further illustrated by conversion of isolated 3/4 with TTMS (tris(trimethylsilyl)silane) at 110 °C in toluene to a mixture of 3/3H/4 in addition to {[Cu7Al6](Cp*)6(Si(SiMe3)3) – H} and {[Cu8Al6](Cp*)6(Si(SiMe3)3) – H} (see Fig. S24† and Scheme 3). In situ1H-NMR analysis indicates consumption of 3 (see Fig. S13†). Unfortunately, no signal of the hydride species 3H could be observed, probably due the fluctional character of the hydride. In situ1H-NMR and GC-MS analysis of the reaction mixture reveals formation of a variety of polysilanes, such as e.g. hexamethyldisilane, pentamethyldisilane or tetramethyldisilane (see Table S5†), pointing to a radical H-transfer mechanism. When 3/4 is converted with the deuterated analogon (TMS)3SiD, a mixture of 3/3H/3D/4 is observed (see Fig. S26†). Reaction with tetrakis(trimethylsilyl)silane (TMS)4Si yielded a mixture of 3H/3 together with {[Cu8Al6](Cp*)6(SiMe3) – H} as novel species observed in LIFDI-MS analysis (see Fig. S27 and S28†). In this case, (TMS)3SiH together with pentamethylfulvene were detected in GC-MS analysis again pointing towards a radical H-transfer mechanism.
From these results we conclude that the reactive open shell cluster 3 is inclined to C–H and Si–H bond activation reactions with the corresponding closed shell hydride cluster 3H as the thermodynamically stable reaction product. While no C–D activation occurs with toluene-d8 under the conditions applied, Si–D activation is observed, competitively occurring to the C–H activation reactions of toluene and of Si–CH3 groups.
Summary and perspectives
Cluster growth reactions of intermetallic Cu/Al clusters were investigated by means of in situ LIFDI-MS and 1H-NMR spectroscopy. Using this approach, we successfully isolated the thermally labile cluster [Cu4Al4](Cp*)5(Mes) (1) as an early intermediate in the reaction between CuMes and AlCp*. Cluster 1 features the first example of a AlMes fragment, formally Al(I), which is part of a [Cu4Al] trigonal bipyramid. Targeted cluster degradation of 1 with excess of 3-hexyne allows isolation of the triangular, 2-electron cluster [Cu2Al](Cp*)3 (2), the smallest possible member of the Cu/Al cluster family. Cluster growth reactions induced thermally or by specific addition of growth components involving isolated 1 and 2 were investigated by detailed variation of reaction parameters. We thereby discovered the inseparable cluster composite 3/3H/4, which is a mixture of the paramagnetic, open shell superatomic cluster [Cu7Al6](Cp*)6 (3) and its closed shell hydride congener [HCu7Al6](Cp*)6 (3H) as well as the closed-shell cluster [Cu8Al6](Cp*)6 (4), which contains just one Cu core atom more than cluster 3. Only the radical species 3 was shown to be reactive towards C–H and Si–H activation reactions of toluene and (TMS)3SiH, respectively. The stability and low reactivity of the 3H and 4 clusters is in line with their magic 20-electron superatomic counts, although not all superatoms with magic counts are inert. While our results help to understand a variety of chemical processes in the early stage of intermetallic Cu/Al cluster formation, the present study certainly also transcends these immediate outcomes by presenting a methodological approach for the elucidation of reactivity patterns in complex cluster mixtures. Based on LIFDI mass spectrometry and different spectroscopic techniques, the analysis of reaction events in cluster mixtures is rendered possible and efficient without the need to isolate singular clusters. Further work in our group will focus on the selective generation and deeper understanding of larger and more diverse Cu/Al cluster libraries (see Fig. S40† for illustration). Our cluster repertoire in this and previous studies includes mostly small to medium sized TM/E clusters (TM = transition metal, E = group 12/13 metal) with structural frameworks similar to Fig. S40a or b.† Both cluster types are based on a core/shell structure with either the TM (Fig. S42a†)12,13 or the main group metal E (Fig. S40b†)40,41 in the inner core. We could demonstrate on various examples, that the organic protective ligand layer (as represented by the semi-transparent grey shell in Fig. S40†) is in principle either penetrable or partially removable by e.g. hydrogenolysis, protolysis or selective one electron oxidation reactions,7 allowing reactivity studies of such clusters with small organic molecules.13 We also succeeded in the isolation and characterization of the large intermetallic cluster [Cu43Al12](Cp*)12 (ref. 12) with fully ligand protected core/shell type structure as shown in S40c.† However, penetration of the organic ligand shell proved unsuccessful in this example so far, which is basically a consequence of the complete insolubility of this compound. Our long-term strategic goal, however, is the generation of large intermetalloid clusters with random element distribution and open coordination sites (Fig. S40d†) which we hope to set in use for model studies of structure/reactivity relationships in intermetallic materials. We suggest that the basic strategy presented in this work, i.e. the generation as well as reactivity testing of intermetalloid cluster libraries is a promising approach towards this goal. For example, a large cluster library (>10 species) with reproducible composition is observed in reaction solutions obtained from the minimalistic building block [Cu2Al](Cp*)3 (2) and [H6Cu6](L)6 (L = PPh3). We intend to explore these clusters and their reactivity as a function of electronic structure, and we will report on our findings in future publications.Author contributions
Max Schütz: experimental work, single crystal X-ray data acquisition and analysis, manuscript preparation; Christian Gemel: (computerized) data interpretation, manuscript preparation; Maximilian Muhr: initial DFT calculations; Christian Jandl: single crystal data acquisition and analysis; Samia Kahlal: DFT calculations and theoretical bonding analysis; Jean-Yves Saillard: DFT calculations, theoretical bonding analysis and manuscript preparation; Roland A. Fischer: research conception, manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the German Research Foundation (DFG) within a Reinhard Koselleck Project (FI-502/44-1). Prof. T. Fässler (TUM) and Prof. O. Cador (Univ. Rennes) are acknowledged for help with SQUID data acquisition and analysis. Tomanec Ondřej and Prof. Radek Zboril (Regional Center of Advanced Technologies and Materials, 78371 Olomouc, Czech Republic) are acknowledged for their valuable help with HRTEM measurements and EDX data analysis of Cu nanoparticles. Prof. Inoue (TUM) is acknowledged for grateful donation of (TMS)3SiBr. Richard Weininger and Matthias Huber are acknowledged for their help in organometallic precursor synthesis and the conduction of LIFDI-MS measurements. We are grateful to the GENCI (Grand Equipement National de Calcul Intensif) for HPC resources (Project A0050807367).References
- C. Ganesamoorthy, J. Weßing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7943–7947 CrossRef CAS.
- J. Weßing, C. Ganesamoorthy, S. Kahlal, R. Marchal, C. Gemel, O. Cador, A. C. Da Silva, J. L. Da Silva, J. Y. Saillard and R. A. Fischer, Angew. Chem., Int. Ed., 2018, 57, 14630–14634 CrossRef.
- G. Schmid, R. Pfeil, R. Boese, F. Bandermann, S. Meyer, G. Calis and W. Vandervelden, Chem. Ber./Recl., 1981, 114, 3634–3642 CrossRef CAS.
- N. Jian, C. Stapelfeldt, K.-J. Hu, M. Fröba and R. E. Palmer, Nanoscale, 2015, 7, 885–888 RSC.
- V. K. Ocampo-Restrepo, L. Zibordi-Besse and J. L. Da Silva, J. Chem. Phys., 2019, 151, 214301 CrossRef.
- S. Hirabayashi and M. Ichihashi, J. Phys. Chem. A, 2015, 119, 8557–8564 CrossRef CAS PubMed.
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735–765 CrossRef CAS.
- M. Guo, B. Yin, B. Huang, H. Wu and Z. Luo, J. Mater. Chem. C, 2020, 8, 10325–10332 RSC.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- U. Heiz, A. Sanchez, S. Abbet and W.-D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- W. Bouwen, P. Thoen, F. Vanhoutte, S. Bouckaert, F. Despa, H. Weidele, R. E. Silverans and P. Lievens, Rev. Sci. Instrum., 2000, 71, 54–58 CrossRef CAS.
- A. I. Ayesh, N. Qamhieh, S. T. Mahmoud and H. Alawadhi, J. Mater. Res., 2012, 27, 2441 CrossRef CAS.
- D. Cox, K. Reichmann, D. Trevor and A. Kaldor, J. Chem. Phys., 1988, 88, 111–119 CrossRef CAS.
- X.-N. Li, L.-N. Wang, L.-H. Mou and S.-G. He, J. Phys. Chem. A, 2019, 123, 9257–9267 CrossRef CAS.
- X. P. Zou, L. N. Wang, X. N. Li, Q. Y. Liu, Y. X. Zhao, T. M. Ma and S. G. He, Angew. Chem., 2018, 130, 11155–11159 CrossRef.
- R. A. Fischer and J. Weiß, Angew. Chem., Int. Ed., 1999, 38, 2830–2850 CrossRef.
- S. Gonzalez-Gallardo, T. Bollermann, R. A. Fischer and R. Murugavel, Chem. Rev., 2012, 112, 3136–3170 CrossRef CAS PubMed.
- T. Bollermann, C. Gemel and R. A. Fischer, Coord. Chem. Rev., 2012, 256, 537–555 CrossRef CAS.
- K. Mayer, J. Weßing, T. F. Fässler and R. A. Fischer, Angew. Chem., Int. Ed., 2018, 57, 14372–14393 CrossRef CAS PubMed.
- L. Xu and S. C. Sevov, Inorg. Chem., 2000, 39, 5383–5389 CrossRef CAS PubMed.
- S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- R. J. Wilson, N. Lichtenberger, B. Weinert and S. Dehnen, Chem. Rev., 2019, 119, 8506–8554 CrossRef CAS PubMed.
- S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Faessler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed.
- J. Vollet, J. R. Hartig and H. Schnöckel, Angew. Chem., Int. Ed., 2004, 43, 3186–3189 CrossRef CAS PubMed.
- S. K. Barik, S.-C. Huo, C.-Y. Wu, T.-H. Chiu, J.-H. Liao, X. Wang, S. Kahlal, J.-Y. Saillard and C.-W. Liu, Chem.–Eur. J., 2020, 26, 10471–10479 CrossRef CAS PubMed.
- T. Steinke, C. Gemel, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2002, 41, 4761–4763 CrossRef CAS PubMed.
- B. Buchin, T. Steinke, C. Gemel, T. Cadenbach and R. A. Fischer, Z. Anorg. Allg. Chem., 2005, 631, 2756–2762 CrossRef CAS.
- K. Freitag, H. Banh, C. Gemel, R. W. Seidel, S. Kahlal, J.-Y. Saillard and R. A. Fischer, Chem. Commun., 2014, 50, 8681–8684 RSC.
- H. Banh, J. Hornung, T. Kratz, C. Gemel, A. Pöthig, F. Gam, S. Kahlal, J.-Y. Saillard and R. A. Fischer, Chem. Sci., 2018, 9, 8906–8913 RSC.
- M. Schütz, M. Muhr, K. Freitag, C. Gemel, S. Kahlal, J.-Y. Saillard, A. C. Da Silva, J. L. Da Silva, T. F. Fässler and R. A. Fischer, Inorg. Chem., 2020, 59, 9077–9085 CrossRef PubMed.
- C. Gemel, T. Steinke, D. Weiss, M. Cokoja, M. Winter and R. A. Fischer, Organometallics, 2003, 22, 2705–2710 CrossRef CAS.
- A. Schnepf and H. Schnöckel, Angew. Chem., Int. Ed., 2002, 41, 3532–3554 CrossRef CAS PubMed.
- K. Freitag, C. Gemel, P. Jerabek, I. M. Oppel, R. W. Seidel, G. Frenking, H. Banh, K. Dilchert and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 4370–4374 CrossRef CAS PubMed.
- P. J. Dyson, B. F. Johnson, J. S. McIndoe and P. R. Langridge-Smith, Rapid Commun. Mass Spectrom., 2000, 14, 311–313 CrossRef CAS PubMed.
- P. J. Dyson, A. K. Hearley, B. F. Johnson, T. Khimyak, J. S. McIndoe and P. R. Langridge-Smith, Organometallics, 2001, 20, 3970–3974 CrossRef CAS.
- C. P. Butcher, A. Dinca, P. J. Dyson, B. F. Johnson, P. R. Langridge-Smith and J. S. McIndoe, Angew. Chem., Int. Ed., 2003, 42, 5752–5755 CrossRef CAS PubMed.
- C. P. Butcher, P. J. Dyson, B. F. Johnson, T. Khimyak and J. S. McIndoe, Chem.–Eur. J., 2003, 9, 944–950 CrossRef CAS.
- C. Evans, K. M. Mackay and B. K. Nicholson, J. Chem. Soc., Dalton Trans., 2001, 1645–1649 RSC.
- M. Muhr, P. Heiß, M. Schütz, R. Bühler, C. Gemel, M. H. Linden, H. B. Linden and R. A. Fischer, Dalton Trans., to be submitted Search PubMed.
- R. Benn, E. Janssen, H. Lehmkuhl and A. Rufínska, J. Organomet. Chem., 1987, 333, 155–168 CrossRef CAS.
- D. L. Reger and M. F. Huff, Organometallics, 1990, 9, 2807–2810 CrossRef CAS.
- D. W. Macomber and M. D. Rausch, J. Am. Chem. Soc., 1983, 105, 5325–5329 CrossRef CAS.
- R. Drescher, S. Lin, A. Hofmann, C. Lenczyk, S. Kachel, I. Krummenacher, Z. Lin and H. Braunschweig, Chem. Sci., 2020, 11, 5559–5564 RSC.
- D. Fenske and J. C. Steck, Angew. Chem., Int. Ed., 1993, 32, 238–242 CrossRef.
- J. P. Fackler, C. A. López, R. J. Staples, S. Wang, R. Winpenny and R. P. Lattimer, J. Chem. Soc., Chem. Commun., 1992, 146–148 RSC.
- H. Nyman and S. Andersson, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1979, 35, 934–937 CrossRef CAS.
- C. Latouche, S. Kahlal, Y.-R. Lin, J.-H. Liao, E. Furet, C. Liu and J.-Y. Saillard, Inorg. Chem., 2013, 52, 13253–13262 CrossRef CAS PubMed.
- P.-K. Liao, C.-S. Fang, A. J. Edwards, S. Kahlal, J.-Y. Saillard and C. Liu, Inorg. Chem., 2012, 51, 6577–6591 CrossRef CAS PubMed.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital interactions in chemistry, John Wiley & Sons, 2013 Search PubMed.
- S. Khanna and P. Jena, Phys. Rev. Lett., 1992, 69, 1664 CrossRef CAS PubMed.
- S. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Grönbeck and H. Häkkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- Z. Luo and A. W. Castleman, Acc. Chem. Res., 2014, 47, 2931–2940 CrossRef CAS PubMed.
- T. Tsukuda and H. Häkkinen, Protected metal clusters: from fundamentals to applications, Elsevier, 2015 Search PubMed.
- B. Yin and Z. Luo, Coord. Chem. Rev., 2020, 213643 Search PubMed.
- W. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141 CrossRef CAS.
- Z. Luo, A. C. Reber, M. Jia, W. H. Blades, S. N. Khanna and A. Castleman, Chem. Sci., 2016, 7, 3067–3074 RSC.
- B. Yin, Q. Du, L. Geng, H. Zhang, Z. Luo, S. Zhou and J. Zhao, J. Phys. Chem. Lett., 2020, 11, 5807–5814 CrossRef CAS PubMed.
- P. Garbacz, V. V. Terskikh, M. J. Ferguson, G. M. Bernard, M. Kedziorek and R. E. Wasylishen, J. Phys. Chem. A, 2014, 118, 1203–1212 CrossRef CAS PubMed.
- K. A. Gardner and J. M. Mayer, Science, 1995, 269, 1849–1851 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2054079–2054082. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00268f |
| This journal is © The Royal Society of Chemistry 2021 |