
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Broken-hearted” carbon bowl via electron shuttle reaction: energetics and electron coupling†
Gabrielle A.
Leith‡
 ,
Allison M.
Rice‡
,
Brandon J.
Yarbrough
,
Preecha
Kittikhunnatham
,
Abhijai
Mathur
,
Nicholas A.
Morris
,
Megan J.
Francis
,
Anna A.
Berseneva
,
Poonam
Dhull
,
Richard D.
Adams
,
M. Victoria
Bobo
,
Aaron A.
Vannucci
,
Mark D.
Smith
,
Sophya
Garashchuk
and
Natalia B.
Shustova
*
,
Allison M.
Rice‡
,
Brandon J.
Yarbrough
,
Preecha
Kittikhunnatham
,
Abhijai
Mathur
,
Nicholas A.
Morris
,
Megan J.
Francis
,
Anna A.
Berseneva
,
Poonam
Dhull
,
Richard D.
Adams
,
M. Victoria
Bobo
,
Aaron A.
Vannucci
,
Mark D.
Smith
,
Sophya
Garashchuk
and
Natalia B.
Shustova
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA. E-mail: shustova@sc.edu
First published on 8th April 2021
Abstract
Unprecedented one-step C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage leading to opening of the buckybowl (π-bowl), that could provide access to carbon-rich structures with previously inaccessible topologies, is reported; highlighting the possibility to implement drastically different synthetic routes to π-bowls in contrast to conventional ones applied for polycyclic aromatic hydrocarbons. Through theoretical modeling, we evaluated the mechanistic pathways feasible for π-bowl planarization and factors that could affect such a transformation including strain and released energies. Through employment of Marcus theory, optical spectroscopy, and crystallographic analysis, we estimated the possibility of charge transfer and electron coupling between “open” corannulene and a strong electron acceptor such as 7,7,8,8-tetracyanoquinodimethane. Alternative to a one-pot solid-state corannulene “unzipping” route, we reported a nine-step solution-based approach for preparation of novel planar “open” corannulene-based derivatives in which electronic structures and photophysical profiles were estimated through the energies and isosurfaces of the frontier natural transition orbitals.
C bond cleavage leading to opening of the buckybowl (π-bowl), that could provide access to carbon-rich structures with previously inaccessible topologies, is reported; highlighting the possibility to implement drastically different synthetic routes to π-bowls in contrast to conventional ones applied for polycyclic aromatic hydrocarbons. Through theoretical modeling, we evaluated the mechanistic pathways feasible for π-bowl planarization and factors that could affect such a transformation including strain and released energies. Through employment of Marcus theory, optical spectroscopy, and crystallographic analysis, we estimated the possibility of charge transfer and electron coupling between “open” corannulene and a strong electron acceptor such as 7,7,8,8-tetracyanoquinodimethane. Alternative to a one-pot solid-state corannulene “unzipping” route, we reported a nine-step solution-based approach for preparation of novel planar “open” corannulene-based derivatives in which electronic structures and photophysical profiles were estimated through the energies and isosurfaces of the frontier natural transition orbitals.
Introduction
Unzipping nanotubes,1–5 nanosheets,6,7 buckyballs,8–11 or annulenes12–16 is driven by the renewed interest in fundamental understanding and practical access to novel structural transformations,17,18 leading to materials with unique optical and electronic profiles. For instance, cutting and unravelling of nanotubes resulted in nanoribbons having electronic properties that can be varied as a function of their width, and therefore, applied in a variety of electronic devices including field-effect transistors, light-emitting diodes, and transparent conductive electrodes.19,20 In addition, hydrogenation of graphene nanoribbons led to enhanced photoluminescent properties that could pave the way for the development of optically active graphene nanoribbon-based devices.21 However, promotion of selective CC bond cleavage in graphitic materials is challenging,22–24 and although there have been examples of structural changes due to periphery modifications of buckybowls (π-bowls),25–28 ring expansion,22 or opening of the strained π-bowl,29,30 these accounts are overall very limited. Pursuing the route of C–C bond activation in curved π-bowl-containing systems is advantageous as this could lead to addressing challenges such as selective sphere opening for preparation of endohedral fullerenes, shortening carbon nanotubes (CNTs), guest integration within the CNT body, as well as access to a class of materials that has not been prepared through “wet-chemistry, conventional” routes. One strategy to facilitate C–C bond activation is to employ, for instance, strain energy as a variable, release of which could energetically promote such transformations.31 Indeed, as presented in this report, release of strain energy can be the driving force for planarization of the naturally curved buckybowl surface (e.g., C20H10, corannulene), since there is no direct route to cleave a CC bond, except through uncontrollable flash vacuum pyrolysis22,27,29 or addition of a directing group (and a catalyst).32
Although there are numerous reports of catalytic hydrocracking of planar polycyclic aromatic hydrocarbons (PAHs) i.e., increasing the ratio of hydrogen-to-carbon,33–36 there are very few accounts on C–C bond cleavage following the hydrogenation step.37 The literature precedent for C–C bond scission primarily relies on the assistance of transition metal catalysts, high hydrogen pressure, elevated temperatures, or a combination of all three parameters.38–44 Therefore, unexpected C–C bond cleavage (discovered from photophysical studies of alignment of electron donor (corannulene) and acceptor (7,7,8,8-tetracyanoquinodimethane, TCNQ) in the solid state) reported herein led us to probe mechanistic pathways to determine the feasibility for π-bowl planarization and factors that could affect such a transformation including strain energy (Es) and released energy (E′, Scheme 1, see more details in the ESI†). The electron coupling and charge transfer (CT) rates between “open” corannulene (or parent corannulene) and TCNQ were evaluated by applying Marcus theory. In addition to the solid-state reaction, we also offer more “conventional” solution-based nine-step synthetic routes for the preparation of novel “open” corannulene analogs. In the reported findings, we also discuss the electronic structure and photophysical profiles of the synthesized “open” analogs through estimation of their energies and isosurfaces of the frontier highest occupied and lowest unoccupied natural transition orbitals (HONTO and LUNTO).
 | ||
| Scheme 1 (Top) A schematic representation of π-bowl (corannulene) opening through a solid-state route. Single-crystal X-ray structure of (left) corannulene and X-ray structure of (right) 5,6-dimethyl-benzo[ghi]fluoranthene (P-C20H14) (“open” corannulene). (Bottom) Strain energy (Es, purple)31 and released energy (E′, red) as a function of PAH: (left to right) phenanthrene, anthracene, pyrene, coronene, and corannulene. | ||
Results and discussion
The reductive CC bond cleavage and consecutive corannulene planarization to form 5,6-dimethyl-benzo[ghi]fluoranthene (planar corannulene analog (P-C20H14), Scheme 1) was achieved through a one-pot solid-state reaction, in which corannulene (15 mg, 0.060 mmol), TCNQ (an electron shuttle; 14 mg, 0.068 mmol), and zinc powder (a reducing agent; 50 mg, 0.76 mmol) were ground together (further experimental details can be found in the ESI†). After that, the reaction mixture was placed in a glass tube, a drop of hydrochloric acid (proton source) was added, and the glass tube was flame-sealed under dynamic vacuum (4 × 10−5 mbar). Heating the reaction mixture at 200 °C for six days resulted in the formation of dark brown needle-shaped crystals suitable for single-crystal X-ray diffraction analysis (Scheme 1). As shown in Scheme 1, such treatment resulted in planarization of the corannulene bowl through partial hydrogenation and formation of P-C20H14. X-ray crystallographic studies of (C20H10)·(P-C20H14)·(TCNQ) (1) co-crystals revealed that the packing consists of alternating columns of TCNQ and P-C20H14 along the c-axis direction (Fig. S1 and S2†). Furthermore, neither mass spectrometry nor spectroscopic studies identified the presence of any other partially hydrogenated products (Fig. S1–S3†).
To gain insight into a plausible mechanism of such π-bowl opening during the one-pot solid-state synthesis (Scheme 1), we initially tested the hypothesis of whether all components of the reaction mixture were essential to perform the solid-state CC bond cleavage. Our results illustrated that the absence of one of the components of the reaction mixture resulted in either no transformation or formation of (corannulene)2·(TCNQ) co-crystals, previously reported in the literature (CCDC 1037414)45 and also detected in our studies (Fig. S4†). Utilization of a different redox mediator rather than TCNQ (e.g., methyl viologen) did not lead to corannulene opening despite previous reports in which TCNQ and methyl viologen have both been used as electron shuttles in various biological applications.46–49 Variation of synthetic conditions, for instance, replacement of the zinc powder with sodium dithionite50 as a reducing agent did not lead to hydrogenated products (see ESI† for more details).
Utilization of more conventional solution-based routes through heating the same reagents (C20H10/TCNQ/Zn/HCl) in a series of organic solvents was also attempted. We varied the reaction media starting with the solvents possessing low boiling points (e.g., dichloromethane or methanol), transitioning to dichloromethane/water or methanol/water mixtures, and finally attempting heating in the higher boiling glycerol (b.p. = 290 °C) or ethylene glycol (b.p. = 197 °C) to more closely match the reaction temperature (200 °C) of the solid-state synthesis. In all reactions, no evidence of P-C20H14 was detected according to the 1H nuclear magnetic resonance (NMR) spectroscopic or mass spectrometry analysis. Notably, the reported hydrogenation reactions of corannulene typically occurred under relatively harsh conditions (e.g., electron bombardment, alkyllithium reagents, or alkali metals), and even despite them, reactions typically led to hydrogenation of one or two rim CC bonds without carbon–carbon bond cleavage.51–57 Since the developed conditions (C20H10/TCNQ/Zn/HCl) required the presence of zinc, we also probed the Clemmensen reduction that uses zinc amalgam and concentrated hydrochloric acid.58 Mass spectrometry and 1H NMR spectroscopy studies of reaction products detected the presence of only pristine corannulene and did not detect any traces of corannulene hydrogenation. As a logical progression, we surveyed an electrochemical method suitable for arene reductive transformations,59 but proved unsuccessful. Finally, attempts to electrochemically cleave the CC bond by bulk electrolysis were performed in anhydrous N,N-dimethylformamide or acetonitrile for up to two days, but were also not successful. In line with these studies, we probed the reaction conditions previously utilized for the ring-opening of other nonplanar structures such as o-carborane.60–62 For that, we used a triosmium carbonyl complex, Os3(CO)10(NCMe)2; however, no ring-opening of corannulene was observed, while successful o-carborane opening occurred at 150 °C in a nonane reflux.62
Further experimental investigations were pursued to rule out aromaticity stabilization as a main factor by performing reactions with significantly less strained PAHs including pyrene (0.0 kJ mol−1)31 or phenanthrene (0.0 kJ mol−1)63 under experimental conditions similar to those used for the reaction with highly-strained corannulene (101 kJ mol−1;31 see ESI† for more details). As a result, no bond cleavage was detected in any of these systems, and formation of only PAH·TCNQ complexes was observed (e.g., phenanthrene·TCNQ complex (similar to the structure reported in the literature64) or pyrene·TCNQ complex, Fig. S17; see more details in the ESI†). Thus, this experimental evidence suggests that one of the driving forces for the observed solid-state reaction could potentially be a release of energy through buckybowl planarization (Fig. 2a). To prove this hypothesis, we estimated released energy, E′, for PAHs and carbon π-bowls as shown in Scheme 1. For instance, E′ for corannulene was calculated to be 202.0 kJ mol−1 (Fig. S9, B3LYP/6-31+G*, see the ESI for more details†). In contrast, E′ calculated for the PAHs in Scheme 1 was found to be less than 135.7 kJ mol−1 (Table S3†). Therefore, corannulene opening is much more energetically favorable in comparison with the PAHs shown in Scheme 1. A similar statement is also valid for a family of extended π-bowls for which the estimated E′ was even higher than that of corannulene (Scheme S3†).
The estimated enthalpy of the reaction (eqn (1)),
| C20H10 + 2H2 = C20H14 | (1) |
As a next step, we took a closer look at a possible mechanism for π-bowl hydrogenation and C–C bond cleavage. On the basis of our theoretical calculations, experimental results, and literature reports, we hypothesized that the transformation of C20H10 to P-C20H14 occurs in a series of reactions that is first initiated by a sequence of electron and proton transfers in which hydrochloric acid acts as the proton source (Fig. 1). Moreover, probing the strength of the C–C bond revealed a significantly weaker bond (115 kJ mol−1 for C20H12 and 9 kJ mol−1 for C20H12−˙, Fig. 1) than a typical C–C bond in RCH2–CH2R systems, allowing for bond cleavage to occur.65,66 For instance, if R is a substituent on a pyrene or coronene core then the electronic energy of the C–C bond would be approximately 350 kJ mol−1 (Fig. S14†) and 302 kJ mol−1, respectively (Fig. S15†).
 | ||
| Fig. 1 Mechanistic pathway to transform corannulene to P-C20H14. Calculations based on B3LYP/6-31+G* level of theory. | ||
Comprehensive analysis of photophysical data for the obtained crystals of 1 revealed properties that are uncharacteristic of the individual components i.e., corannulene and TCNQ themselves. Based on photoluminescence and epifluorescence microscopy studies, the obtained crystals of 1 exhibited red emission (λmax = 705 nm, λex = 365 nm) in contrast to their constituents (λmax (TCNQ crystal) = undetectable emission and λmax (corannulene crystal) = 490 nm, λex = 365 nm, Fig. S5†). Furthermore, in contrast to diffuse reflectance (DR) profiles of pristine corannulene and TCNQ (Fig. S6†), the appearance of a new red-shifted band (550 nm) in the DR profile of 1 was detected. Based on our theoretical calculations using time-dependent density functional theory (TDDFT), the new band (550 nm) is characteristic of CT complex formation (see the ESI for more details†) that is in line with a previous report on PAHs and TCNQ co-crystals.24 In particular, according to our studies using the B3LYP-D3/6-311+G** level of theory (Fig. 2b), both a bathochromic shift and new band appearance could be attributed to CT66,67 between the HOMO−2 and LUMO of a TCNQ/P-C20H14 “stack” (Fig. 2c). To further shed light on the experimental changes of the emission profile, we examined optical excitations of isolated corannulene, P-C20H14, TCNQ, and the relevant dimers through the TDDFT calculations based on the B3LYP-D3/6-311+G** method. The considered TCNQ/P-C20H14 “stack” is the only species with excitation energies of appreciable strength around 690 nm (1.8 eV; Fig. S7†), which is in agreement with the experimentally observed red emission at λmax = 705 nm (Fig. S5†). The lowest excitations for TCNQ, π-bowl, and P-C20H14 are 413, 288, and 344 nm (3.0, 4.3, and 3.6 eV), respectively (Fig. S7†).
 | ||
| Fig. 2 (a) (Left) Developed route for the preparation of P-C20H14 and (right) a part of the single-crystal X-ray structure of 1 showing an alternating column of TCNQ and P-C20H14. (b) Molecular orbitals of TCNQ/P-C20H14 in 1: HOMO−2 and LUMO, related to electron excitation (transition) from P-C20H14 to TCNQ, respectively. (c) Optical transition strength for TCNQ/P-C20H14 (blue) and TCNQ/C20H10 (gray) calculated using TDDFT/RPA based on the B3LYP-D3/6-311+G** level of theory. | ||
To further probe the idea that CT is more effective in an exclusively planar TCNQ/P-C20H14 “stack” rather than in a TCNQ/C20H10 “stack”, that encounters steric hindrance from the curved surface of the π-bowl,45 we employed Marcus theory68 to compare the electron coupling (that is proportional to CT rate) between TCNQ/C20H10 and TCNQ/P-C20H14 using eqn (2):
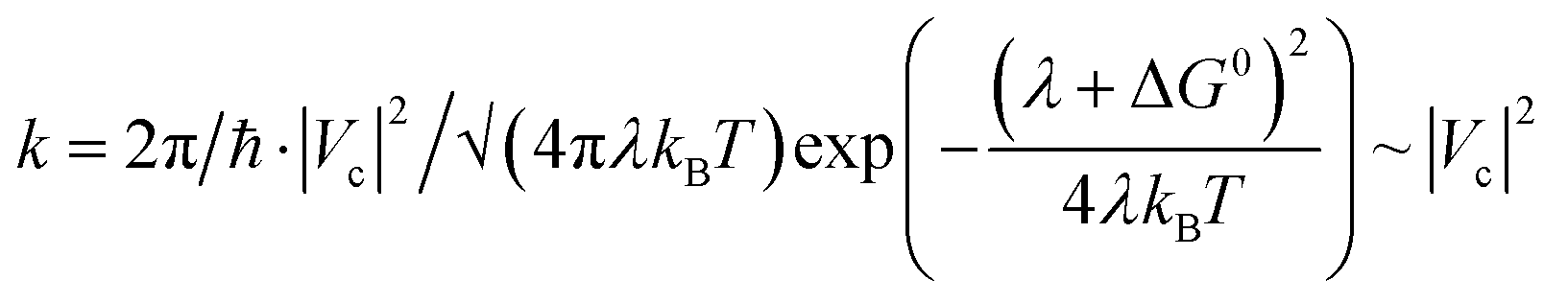 | (2) |
 | ||
| Fig. 3 (Top left) Single-crystal X-ray structure of P-C20H14. (Bottom left) Optical transition strengths computed at the ground state optimal geometry for P-C20H14 in THF (blue) and at the second excited singlet state optimal geometry for P-C20H14 in THF (red). (Right) Energies and isosurfaces of the HONTO and LUNTO of P-C20H14 in the ground and the second singlet excited states. S0 and S2 are the ground and excited states for P-C20H14 of the ground state. S0 and S2′ are the ground and excited state intermediates for the minimum energy geometry of the second excited singlet state. The black solid and wavy arrows indicate absorption (S0 → S2) or emission (S2′ → S0′) and vibrational relaxation (S2 → S2′ and S2′ → S2), respectively. The theory level is TDDFT/RPA based on the B3LYP-D3/6-31+G* method. | ||
As an alternative scalable approach to access a family of “open” corannulene-containing derivatives, we report a solution-phase route (see ESI†).70 Despite a number of required steps (Schemes S1 and S2†), in comparison with the one-step solid-state synthesis, the “solution” approach has some advantages since it does not rely on selection of the specific substrate/electron shuttle/reducing agent system and also provides a scalable route for the synthesis of a library of new planar corannulene-type analogs. The synthetic details for preparation of 5-methylbenzo[ghi]fluoranthene (C19H12, X, Fig. 4a) and 5-ethyl-6-methylbenzo[ghi]fluoranthene (C21H16, X′, Fig. 4b), using this approach, are provided in ESI.† Sublimation of X (Scheme S1†) allowed for the formation of single crystals of X suitable for X-ray diffraction (Fig. S24†). The structure of X′ was confirmed using 1H and 13C NMR spectroscopy and mass spectrometry (Fig. S21†). As in the case of solid-state “open” P-C20H14, both X and X′ structures possess a planar geometry (Fig. S24 and S25†). The emission studies of the prepared X and X′ compounds revealed a red-shifted emission (λmax = 548 nm and 573 nm, λex = 365 nm, respectively, Fig. S26†) in contrast to pristine corannulene (λmax = 490 nm, λex = 365 nm, Fig. S5†). The emission maxima of X and X′ in THF was found to be 479 nm and 502 nm, respectively, (λex = 365 nm) and is hypsochromically shifted compared to the solid-state 548 and 573 nm-centered emission, respectively (λex = 365 nm, Fig. 4a, b, S27, and S28†). In a similar vein to TDDFT calculations of P-C20H14, we determined the optical transitions corresponding to the ground state, first and second excited singlet states of X and X′ (Fig. S27 and S28†). While the electronic transition of the first excited singlet state for both X and X′ did not differ from the optical transitions corresponding to the ground state (351 nm for X and 361 nm for X′), optical transitions for the second excited singlet states were determined to be 395 nm and 412 nm for X and X′, respectively. Electronic transitions corresponding to the second excited singlet state can be associated with the emission profiles that are similar to the experimental data (see the ESI for more details).
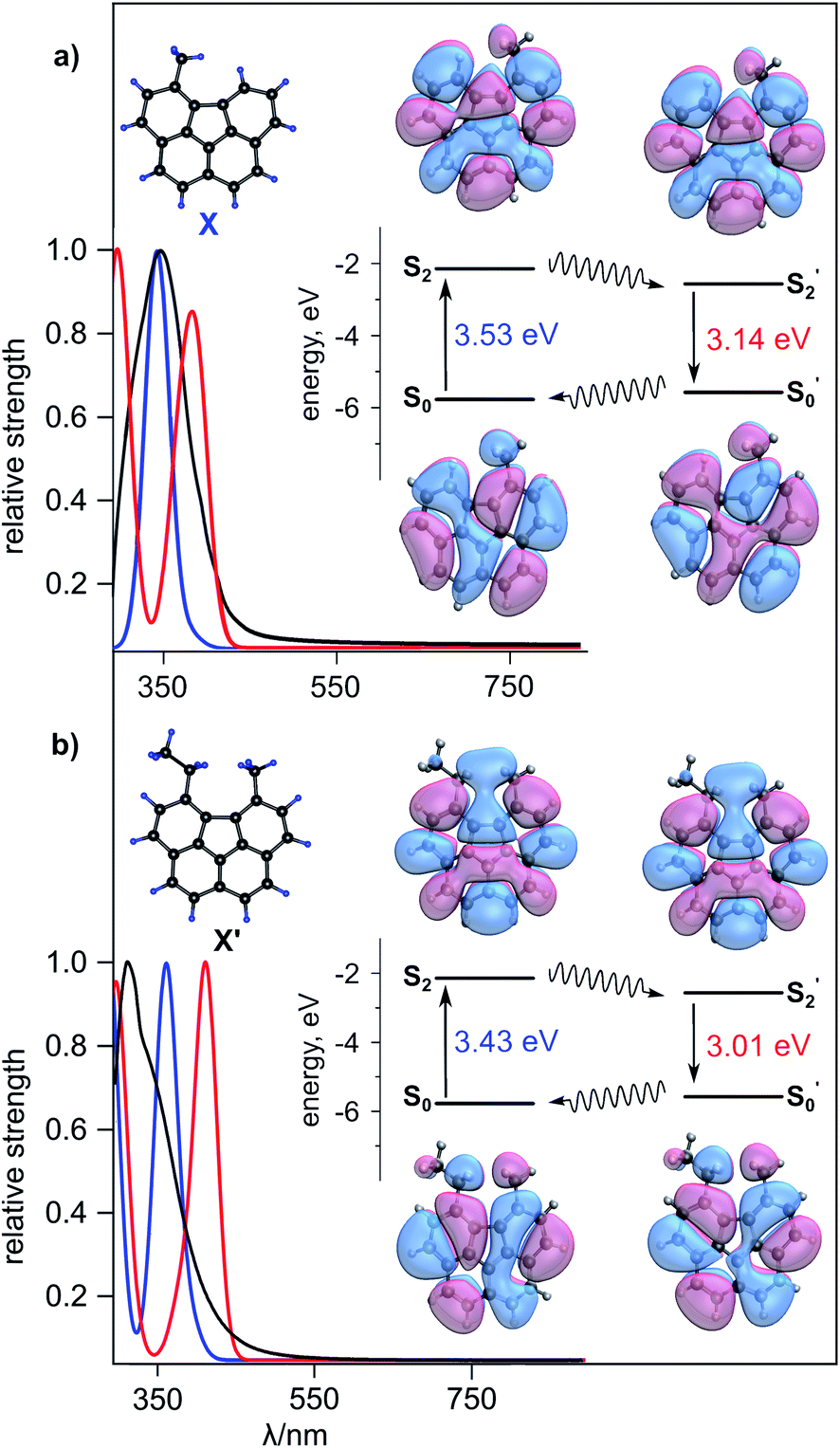 | ||
| Fig. 4 (a) (Top left) Single-crystal X-ray structure of X. (Bottom left) Optical transition strengths computed at the ground state optimal geometry for X in THF (blue) and at the second singlet excited state optimal geometry for X in THF (red). (Right) Energies and isosurfaces of the HONTO and LUNTO of X in the ground and the second singlet excited states. S0 and S2 are the ground and excited states for X of the ground state. S0 and S2′ are the ground and excited state intermediates for the minimum energy geometry of the second excited singlet state. The black solid and wavy arrows indicate absorption (S0 → S2) or emission (S2′ → S0′) and vibrational relaxation (S2 → S2′ and S2′ → S2), respectively. The theory level is TDDFT/RPA based on the B3LYP-D3/6-31+G* method. (b) (Top left) Geometrically optimized structure of X′ based on B3LYP-D3/6-31+G* level of theory. (Bottom left) Optical transition strengths computed at the ground state optimal geometry for X′ in THF (blue) and at the second singlet excited state optimal geometry for X′ in THF (red). (Right) Energies and isosurfaces of HONTO and LUNTO of X′ in the ground and the second singlet excited states. S0 and S2 are the ground and excited states for X′ of the ground state. S0’ and S2′ are the ground and excited state intermediates for the minimum energy geometry of the second excited singlet state. The black solid and wavy arrows indicate absorption (S0 → S2) or emission (S2′ → S0′) and vibrational relaxation (S2 → S2′ and S2′ → S2), respectively. The theory level is TDDFT/RPA based on the B3LYP-D3/6-31+G* method. | ||
Conclusions
To summarize, we report the first example of a unique one-step CC bond cleavage in the traditionally very robust π-bowl occurring via an electron shuttle reaction. Such ring opening is unprecedented in the literature and has not been observed for pristine π-bowls (e.g., corannulene) to date (with the exception of uncontrollable brute force vacuum pyrolysis22). PAH hydrogenation has been previously observed under harsh experimental conditions (e.g., high hydrogen pressure or extreme temperatures above 1000 °C),38–44 therefore the formation of P-C20H14 in a one-pot synthesis under relatively mild conditions is an unexpected and remarkable result. Through employment of Marcus theory, optical spectroscopy, and crystallographic analysis, we estimated the electron coupling between “open” corannulene and a strong electron acceptor, TCNQ. A solution-phase route was employed for preparation of two novel “open” corannulene-based derivatives with the corresponding spectroscopic analysis of their properties experimentally and theoretically. Furthermore, through a combination of theoretical modeling with experimental results, mechanistic studies were undertaken to shed light on possible factors (such as strain energy) that could act as a driving force for the observed π-bowl opening. Our studies highlight the possibility to implement novel synthetic routes for π-bowl transformations, that are drastically different from the conventional approaches toward derivatization of traditional PAHs. Thus, the presented solid-state, solution, and theoretical methodology are the first steps toward understanding possible avenues to prepare barely accessible structures by “unlocking” the corannulene core and application of the latter for molecular electronic development.
Author contributions
G. A. Leith: conceptualization, methodology, formal analysis, investigation, visualization, writing – original draft preparation, and writing – reviewing and editing. A. M. Rice: conceptualization, methodology, formal analysis, investigation, visualization, writing – original draft preparation, and writing – reviewing and editing. B. J. Yarbrough: conceptualization, formal analysis, investigation, and writing – reviewing and editing. P. Kittikhunnatham: methodology, investigation, and writing – reviewing and editing. A. Mathur: methodology, investigation, and writing – reviewing and editing. N. A. Morris: investigation and writing – reviewing and editing. M. J. Francis: investigation and writing – reviewing and editing. A. A. Berseneva: software and writing – reviewing and editing. P. Dhull: investigation, formal analysis, and writing – reviewing and editing. R. D. Adams: investigation, formal analysis, and writing – reviewing and editing. M. V. Bobo: investigation, formal analysis, and writing – reviewing and editing. A. A. Vannucci: investigation, formal analysis, and writing – reviewing and editing. M. D. Smith: resources, formal analysis, visualization, and writing – reviewing and editing. S. Garashchuk: resources, supervision, formal analysis, writing – original draft, and writing – reviewing and editing. N. B. Shustova: conceptualization, resources, writing – original draft preparation, writing – reviewing and editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF CAREER Award (DMR-1553634). N. B. S. thanks the Cottrell Scholar Award from the Research Corporation for Science Advancement, Sloan Research Fellowship provided by Alfred P. Sloan Foundation, the Dreyfus Teaching-Scholar Award supported by the Dreyfus Foundation, and the IAS Hans Fischer fellowship. The computational work (S. G.) is based upon work supported in part by the NSF and SC EPSCoR/IDeA Program under Grants No. CHE-1955768 and OIA-1655740/20-GC03, respectively.Notes and references
- S. V. Rotkin, I. Zharov and K. Hess, AIP Conf. Proc., 2001, 591, 454–457 CrossRef CAS.
- V. S. Thoi, R. E. Usiskin and S. M. Haile, Chem. Sci., 2015, 6, 1570–1577 RSC.
- S.-Y. Zhao, B. Zhang, H. Su, J.-J. Zhang, X.-H. Li, K.-X. Wang, J.-S. Chen, X. Wei and P. Feng, J. Mater. Chem. A, 2018, 6, 4331–4336 RSC.
- M. K. Smith, K. E. Jensen, P. A. Pivak and K. A. Mirica, Chem. Mater., 2016, 28, 5264–5268 CrossRef CAS.
- Z. Zhang, Y. Che, R. A. Smaldone, M. Xu, B. R. Bunes, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2010, 132, 14113–14117 CrossRef CAS PubMed.
- A. Kong, C. Mao, Y. Wang, Q. Lin, X. Bu and P. Feng, J. Mater. Chem. A, 2016, 4, 7305–7312 RSC.
- A. M. Champsaur, J. Yu, X. Roy, D. W. Paley, M. L. Steigerwald, C. Nuckolls and C. M. Bejger, ACS Cent. Sci., 2017, 3, 1050–1055 CrossRef CAS PubMed.
- G. D. Han, A. Maurano, J. G. Weis, V. Bulović and T. M. Swager, Org. Electron., 2016, 31, 48–55 CrossRef CAS.
- O. V. Boltalina, A. A. Popov, I. V. Kuvychko, N. B. Shustova and S. H. Strauss, Chem. Rev., 2015, 115, 1051–1105 CrossRef CAS PubMed.
- A. Hirsch, The Chemistry of Fullerenes, John Wiley and Sons, Inc., 2008 Search PubMed.
- A. M. Rice, E. A. Dolgopolova and N. B. Shustova, Chem. Mater., 2017, 29, 7054–7061 CrossRef CAS.
- X. Jiang, S. D. Laffoon, D. Chen, S. Pérez-Estrada, A. S. Danis, J. Rodríguez-López, M. A. Garcia-Garibay, J. Zhu and J. S. Moore, J. Am. Chem. Soc., 2020, 142, 6493–6498 CrossRef CAS PubMed.
- E. Gonzalez-Rodriguez, M. A. Abdo, G. dos Passos Gomes, S. Ayad, F. D. White, N. P. Tsvetkov, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2020, 142, 8352–8366 CrossRef CAS PubMed.
- A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Y. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed.
- C. Dubceac, A. S. Filatov, A. V. Zabula, A. Y. Rogachev and M. A. Petrukhina, Chem.–Eur. J., 2015, 21, 14268–14279 CrossRef CAS PubMed.
- Z. Zhou, S. N. Spisak, Q. Xu, A. Y. Rogachev, Z. Wei, M. Marcaccio and M. A. Petrukhina, Chem.–Eur. J., 2018, 24, 3455–3463 CrossRef CAS PubMed.
- A. Béziau, S. A. Baudron, G. Rogez and M. W. Hosseini, Inorg. Chem., 2015, 54, 2032–2039 CrossRef PubMed.
- S. A. Baudron and M. W. Hosseini, Chem. Commun., 2016, 52, 13000–13003 RSC.
- I. Martin-Fernandez, D. Wang and Y. Zhang, Nano Lett., 2012, 12, 6175–6179 CrossRef CAS PubMed.
- M. Terrones, ACS Nano, 2010, 4, 1775–1781 CrossRef CAS PubMed.
- B. V. Senkovskiy, M. Pfeiffer, S. K. Alavi, A. Bliesener, J. Zhu, S. Michel, A. V. Fedorov, R. German, D. Hertel, D. Haberer, L. Petaccia, F. R. Fischer, K. Meerholz, P. H. M. Van Loosdrecht, K. Lindfors and A. Grüneis, Nano Lett., 2017, 17, 4029–4037 CrossRef CAS PubMed.
- A. Borchardt, A. Fuchicello, K. V. Kilway, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 1921–1923 CrossRef CAS.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 479, 475–479 CrossRef PubMed.
- I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley and Sons, Inc., Hoboken, 2016 Search PubMed.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- B. M. Schmidt, B. Topolinski, M. Yamada, S. Higashibayashi, M. Shionoya, H. Sakurai and D. Lentz, Chem.–Eur. J., 2013, 19, 13872–13880 CrossRef CAS PubMed.
- L. T. Scott, E. A. Jackson, Q. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107–110 CrossRef CAS PubMed.
- E. Nestoros and M. C. Stuparu, Chem. Commun., 2018, 54, 6503–6519 RSC.
- S. Attar, D. M. Forkey, M. M. Olmstead and A. L. Balch, Chem. Commun., 1998, 1255–1256 RSC.
- T. Hayama, Y. Wu, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2007, 129, 12612–12613 CrossRef CAS PubMed.
- C. H. Sun, G. Q. Lu and H. M. Cheng, J. Phys. Chem. B, 2006, 110, 4563–4568 CrossRef CAS PubMed.
- S. Tashiro, M. Yamada and M. Shionoya, Angew. Chem., Int. Ed., 2015, 54, 5351–5354 CrossRef CAS PubMed.
- J. C. Fetzer, Polycyclic Aromat. Compd., 2007, 27, 143–162 CrossRef CAS.
- S. Yang and L. M. Stock, Energy Fuels, 1996, 10, 1181–1186 CrossRef CAS.
- M. A. Nkansah, A. A. Christy and T. Barth, Polycyclic Aromat. Compd., 2012, 32, 408–422 CrossRef CAS.
- S. C. Korre, M. T. Klein and R. J. Quann, Ind. Eng. Chem. Res., 1997, 36, 2041–2050 CrossRef CAS.
- X.-M. Yue, X.-Y. Wei, S.-Q. Zhang, F.-J. Liu, Z.-M. Zong and X.-Q. Yang, Fuel Process. Technol., 2017, 161, 283–288 CrossRef CAS.
- M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100–1105 RSC.
- R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7346–7355 CrossRef CAS.
- C. T. To, K. S. Choi and K. S. Chan, J. Am. Chem. Soc., 2012, 134, 11388–11391 CrossRef CAS PubMed.
- A. Yamaguchi, N. Mimura, M. Shirai and O. Sato, Sci. Rep., 2017, 7, 46172 CrossRef CAS PubMed.
- X. Kang, G. Luo, L. Luo, S. Hu, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2016, 138, 11550–11559 CrossRef CAS PubMed.
- P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC.
- Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600–614 CrossRef CAS.
- Y. Yoshida, K. Isomura, N. Yuto, H. Kishida and G. Saito, Chem. Lett., 2015, 44, 709–711 CrossRef CAS.
- M. Wang, C. Wölfer, L. Otrin, I. Ivanov, T. Vidaković-Koch and K. Sundmacher, Langmuir, 2018, 34, 5435–5443 CrossRef CAS PubMed.
- F. P. Van der Zee and F. J. Cervantes, Biotechnol. Adv., 2009, 27, 256–277 CrossRef CAS PubMed.
- M. D. Peterson, S. C. Jensen, D. J. Weinberg and E. A. Weiss, ACS Nano, 2014, 8, 2826–2837 CrossRef CAS PubMed.
- L. Striepe and T. Baumgartner, Chem.–Eur. J., 2017, 23, 16924–16940 CrossRef CAS PubMed.
- J. M. Khurana and S. Singh, J. Indian Chem. Soc., 1996, 73, 487–488 CAS.
- A. Sygula, R. Sygula, F. R. Fronczek and P. W. Rabideau, J. Org. Chem., 2002, 67, 6487–6492 CrossRef CAS PubMed.
- S. N. Spisak, G. C. Hoover, Z. Wei, A. V. Zabula, A. S. Filatov and M. A. Petrukhina, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 690–694 CrossRef CAS PubMed.
- E. Shabtai, R. E. Hoffman, P. C. Cheng, E. Bayrd, D. V. Preda, L. T. Scott and M. Rabinovitz, J. Chem. Soc., Perkin Trans. 2, 2001, 129–133 Search PubMed.
- R. M. Chin, B. Baird, M. Jarosh, S. Rassman, B. Barry and W. D. Jones, Organometallics, 2003, 22, 4829–4832 CrossRef CAS.
- R. M. Chin, M. S. Jarosh, J. D. Russell and R. J. Lachicotte, Organometallics, 2002, 21, 2027–2029 CrossRef CAS.
- P. W. Rabideau, Z. Marcinow, R. Sygula and A. Sygula, Tetrahedron Lett., 1993, 34, 6351–6354 CrossRef CAS.
- P. Sundararajan, M. Tsuge, M. Baba, H. Sakurai and Y. P. Lee, J. Chem. Phys., 2019, 151, 044304 CrossRef PubMed.
- T. Nakabayashi and T. Nakabayashi, J. Am. Chem. Soc., 1960, 82, 3900–3906 CrossRef CAS.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838–845 CrossRef CAS PubMed.
- R. D. Adams, J. Kiprotich, D. V. Peryshkov and Y. Onn, Chem.–Eur. J., 2016, 22, 6501–6504 CrossRef CAS PubMed.
- R. D. Adams, E. J. Kiprotich and M. D. Smith, Chem. Commun., 2018, 54, 3464–3467 RSC.
- R. D. Adams, J. Kiprotich, D. V. Peryshkov and Y. O. Wong, Inorg. Chem., 2016, 55, 8207–8213 CrossRef CAS PubMed.
- Y. Nagano and M. Nakano, J. Chem. Thermodyn., 2003, 35, 1403–1412 CrossRef CAS.
- M. A. Dobrowolski, G. Garbarino, M. Mezouar, A. Ciesielski and M. K. Cyrański, CrystEngComm, 2014, 16, 415–429 RSC.
- L. Pauling and L. O. Brockway, J. Am. Chem. Soc., 1937, 59, 1223–1236 CrossRef CAS.
- R. J. Dillon and C. J. Bardeen, J. Phys. Chem. A, 2012, 116, 5145–5150 CrossRef CAS PubMed.
- H. N. Ghosh, J. Phys. Chem. B, 1999, 103, 10382–10387 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and D. O. Cowan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1193–1199 CrossRef.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, computational details, and crystallographic data. CCDC 1874124 (1), 1874126 (C16H10·C12H4N4), 1874125 (1,6,7-trimethylfluoranthene), 1874127 (7-ethyl-1,6,10-trimethylfluoranthene), and 1875444 (5-methylbenzo[ghi]fluoranthene). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06755e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |