
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric hydroalkylation of alkynes and allenes with imidazolidinone derivatives: α-alkenylation of α-amino acids†
Farhad
Panahi
,
Hormoz
Khosravi
 ,
Felix
Bauer
and
Bernhard
Breit
*
,
Felix
Bauer
and
Bernhard
Breit
*
Institut für Organische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg im Breisgau, Germany. E-mail: bernhard.breit@chemie.uni-freiburg.de
First published on 22nd April 2021
Abstract
This work reports a new method for the synthesis of quaternary α-alkenyl substituted amino acids by the enantio- and diastereoselective addition of imidazolidinone derivatives to alkynes and allenes. Further hydrolysis of the imidazolidinone products under acidic conditions afforded biologically relevant amino acid derivatives. This method is geometry-selective (E-isomer), enantio- and diastereoselective, and products were obtained in good to excellent yields. The utility of this new methodology is proved by its operational simplicity and the successful accomplishment of gram-scale reactions. Experimental and computational studies suggest the key role of Li in terms of selectivity and support the proposed reaction mechanism.
Introduction
Alpha quaternary amino acids (α-AAs) not only have a key role in the modification of peptide conformations but also are employed as building blocks for the preparation of biologically and medicinally compounds.1 The introduction of a fourth substituent to the α-carbon (carbon bears the amino and carboxyl groups) of amino acids is a common approach in the asymmetric synthesis of quaternary α-AAs.2 While enantioselective alkylation at α-carbon is well developed, the introduction of unsaturated substituents (aryl and alkenyl) at this carbon has proved more challenging.3Alkenyl α-AAs are important structure motifs to serve as building blocks in synthetic chemistry and drug discovery. These structures have shown interesting biological activities such as antibiotic and anticancer properties.4 Despite of its relevance, the methodologies for the synthesis of enantiopure alkenyl amino acids are limited,5 among them [2,3]-sigmatropic rearrangement of selenimides,6 α-2-tosylethenylation of (S)-2-(pyrrolidin-1-yl)propanoic acid esters,7 and use of alkenylboronic acids are the most used.8 Recently, an intramolecular method for the α-alkenylation of α-AA was reported using diastereoselective rearrangement of an N′-alkenyl urea substrate9 according to the Seebach's “self-regeneration of stereocenters” method (Scheme 1a).10 Thus, considering our extended knowledge on the regioselective addition of different pronucleophiles to allenes and alkynes,11 we envisioned that the direct intermolecular hydroalkylation of alkynes and allenes with imidazolidinone derivatives may provide a new way to obtain quaternary α-alkenyl substituted amino acids (Scheme 1b). This new transformation is highlighted by an increase in the structural complexity of amino acids through the formation of a new C–C bond which generates a new quaternary stereocenter in high diastereoselectivity. Simultaneously, a tri-substituted alkene function is generated with high E-selectivity, itself an important challenge in modern organic synthesis.12 To the best of our knowledge, hydroalkylation of alkynes and allenes with imidazolidinone derivatives toward synthesis of α-alkenylated α-amino acids has not been reported.
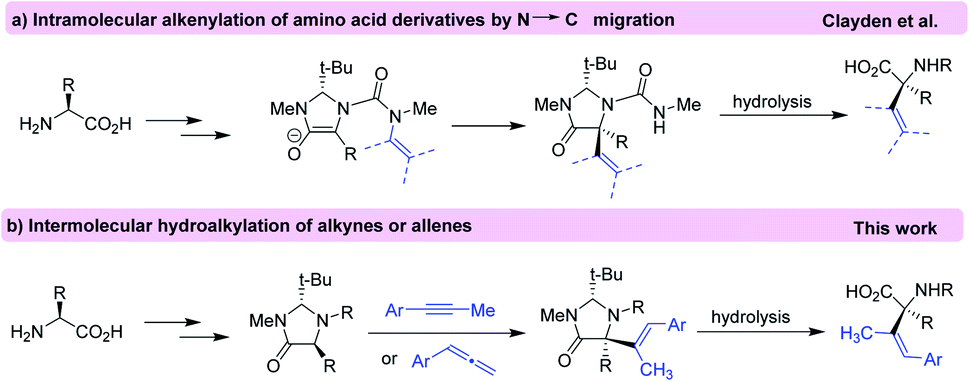 | ||
| Scheme 1 Alkenylation of amino acid derivatives by N′ → C migration (a) and using intermolecular hydroalkylation of alkynes or allenes (b). | ||
Results and discussion
To evaluate the feasibility of this hypothesis, the α-alkenylation of α-AAs via hydroalkylation of alkynes was started using imidazolidinone 1a and alkyne 2a as model substrates (Table 1). An initial experiment with KHMDS (HMDS = hexamethyldisilazane) afforded 73% of desired product 3a with complete diastereoselectivity albeit with a low enantiomeric ratio (er) of 68 : 32 (Table 1, entry 1). Using NaHMDS the yield was slightly improved, but the er decreased to 56 : 44, while the dr maintained >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 2). Remarkably, switching to LiHMDS, 3a was produced in 83% isolated yield in perfect dr and er (Table 1, entry 3).13
:1 (Table 1, entry 2). Remarkably, switching to LiHMDS, 3a was produced in 83% isolated yield in perfect dr and er (Table 1, entry 3).13
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (eq.) | Temp. (°C) | Yieldb (%) | drd | ere |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), M(Li,Na,K) HMDS (0.4 mL, 1.0 M in THF), THF (2.0 mL, 0.08 M). b Yields reported for isolated products. c NMR yield with 1,3,5-trimethoxybenzene (TMB) as reference. d dr was determined using 1H-NMR. e er was determined by chiral HPLC. f Toluene was used as the solvent. g Reaction time was 48 h. | |||||
| 1 | KHMDS (2.0) | 80 | 73 (79)c | >99:1 |
68:32 |
| 2 | NaHMDS (2.0) | 80 | 75 (82)c | >99:1 |
56:44 |
| 3 | LiHMDS (2.0) | 80 | 83 (91)c | >99:1 |
>99:1 |
| 4 | LiHMDS (2.0) | 80 | 78f | >99:1 |
94:6 |
| 5 | LiHMDS (2.0) | 50 | 48 | >99:1 |
>99:1 |
| 6 | LiHMDS (2.0) | r.t. | 25g | >99:1 |
>99:1 |
| 7 | LiHMDS (1.0) | 80 | 23 | >99:1 |
>99:1 |
| 8 | LiHMDS (0.5) | 80 | <5 | >99:1 |
ND |
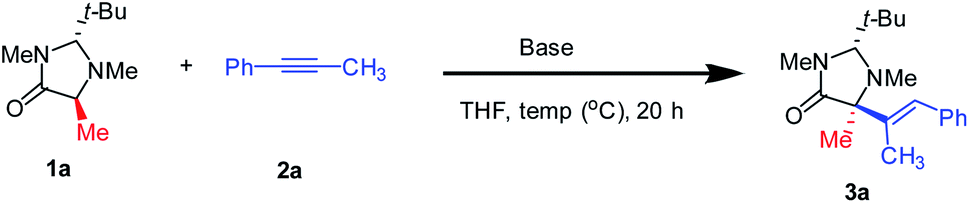
No improvement was observed using other bases proving that LiHMDS is the most efficient base for this protocol (Table 1S†). The solvent screening revealed that THF is the best solvent for this transformation (Table 2S†). In the case of toluene, 78% yield was achieved, however, a slight decrease of the er was observed (Table 1, entry 4). Then, different temperatures were tested finding 80 °C as the optimal (Tables 3S† and 1, entries 5 & 6). It should be noted that the yield dramatically decreased when one equivalent of base was used. Unexpectedly with half equivalent of the base the process did not occur (Table 1, entries 7 & 8). Moreover, the ratio between the starting materials and the base is critical in this process, getting the best results with a 1:1:2 ratio for 1a:2a:base (Table 4S†). With the optimal conditions in hand, we turned our attention to the scope of both amino acids and alkynes (Scheme 2). The scope of imidazolidinone derived amino acids as nucleophile partners were evaluated. Imidazolidinone derivatives from proline, phenylalanine, methionine, leucine, tyrosine, and phenylglycine (as non-natural α-AAs) reacted with different alkynes, affording the products (3a-ai) in good to excellent yields.
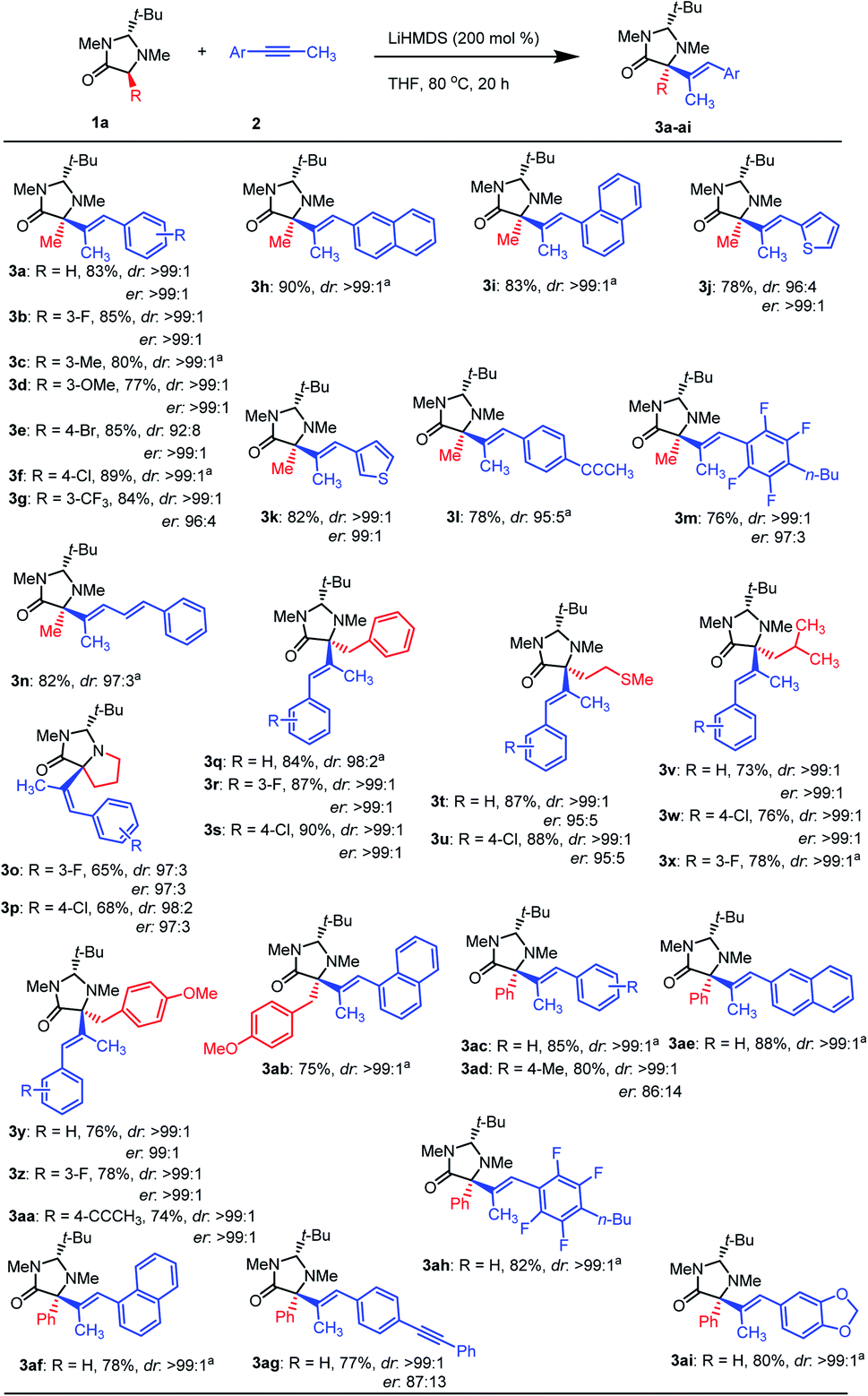 | ||
| Scheme 2 The substrate scope of alkynes and amino acids. Reaction conditions: 1a (0.2 mmol), 2 (0.2 mmol), LiHMDS (0.4 mL, 1.0 M in THF), THF (2.0 mL, 0.08 M), 80 °C, 20 h. Yields reported for isolated products. dr were determined using 1HNMR. aer not determined. | ||
To our delight, a wide range of aryl-methyl alkynes were tolerated affording the corresponding α-alkenylated amino acid derivatives in high yields and perfect dr (Scheme 2). It was observed that alkynes bearing substituents at ortho- (3m,ah), meta- (3b–d,g,o,r,x,z), and para- (3e,f,l,p,s,w,aa,ad,ag) positions of the phenyl ring delivered the corresponding products in high yields and dr.
Alkynes containing naphthyl (3h,i,ab,ae,af), thiophenyl (3j,k) and 1,3-benzodioxole (3ai) worked well under these conditions. Sterically hindered substrates such as ortho-substituted (3m,ah) and 1-naphthyl alkynes (3i,ab,af) gave also products in good yields. Halogen atoms (F, Cl, Br) in different positions (ortho, meta, and para) were tolerated, resulting in the halogenated products in good to excellent yields and high dr, allowing for further functionalizations. No evidence of dehalogenation or benzyne formation was observed for this type of substrates. Only a slight loss of dr was observed for bromo-substituted substrates.
In order to show the versatility and the selectivity of this method, some alkynes with particular functionalities were used. In this sense, the reaction of a polyflouro-substituted alkyne gave 3m and 3ah in high yields and dr. Moreover, a conjugated alkyne ((E)-pent-1-en-3-yn-1-ylbenzene) resulted in product 3n in high yields preserving the configuration of the initial double bond. Product 3ag proved that only aryl-methyl alkyne groups are active substrates in this transformation. Interestingly, when using bis-alkyne (1,4-di-(prop-1-yn-1-yl)benzene) only one functional group reacted, which provide opportunities for new structural diversities (3l,aa). As shown in Scheme 2, the er for all type of amino acids are excellent and only in the case of phenyl glycine due to some racemization because of its acidifying side chain, starting material could not be obtained in high er.3b However, the hydroalkylation process was accomplished in high stereoselectivities.
Stereodivergency can be achieved using either trans- or cis-imidazolidinones which can be easily synthesized from L-amino acids (Scheme 3a).14 In this way, it is possible to access both optical antipodes of the desired functionalized amino acid in high er. In order to highlight the utility of this methodology, a large-scale synthesis (5.5 mmol) of compound 3a was performed with 74% isolated yield in high dr (>99:1) and er (>99 : 1) (Scheme 3b). Hydrolysis of the imidazolidinone products resulted in the release of amino acid derivatives preserving the enantiomeric purity and the alkene geometry (Scheme 3c).15
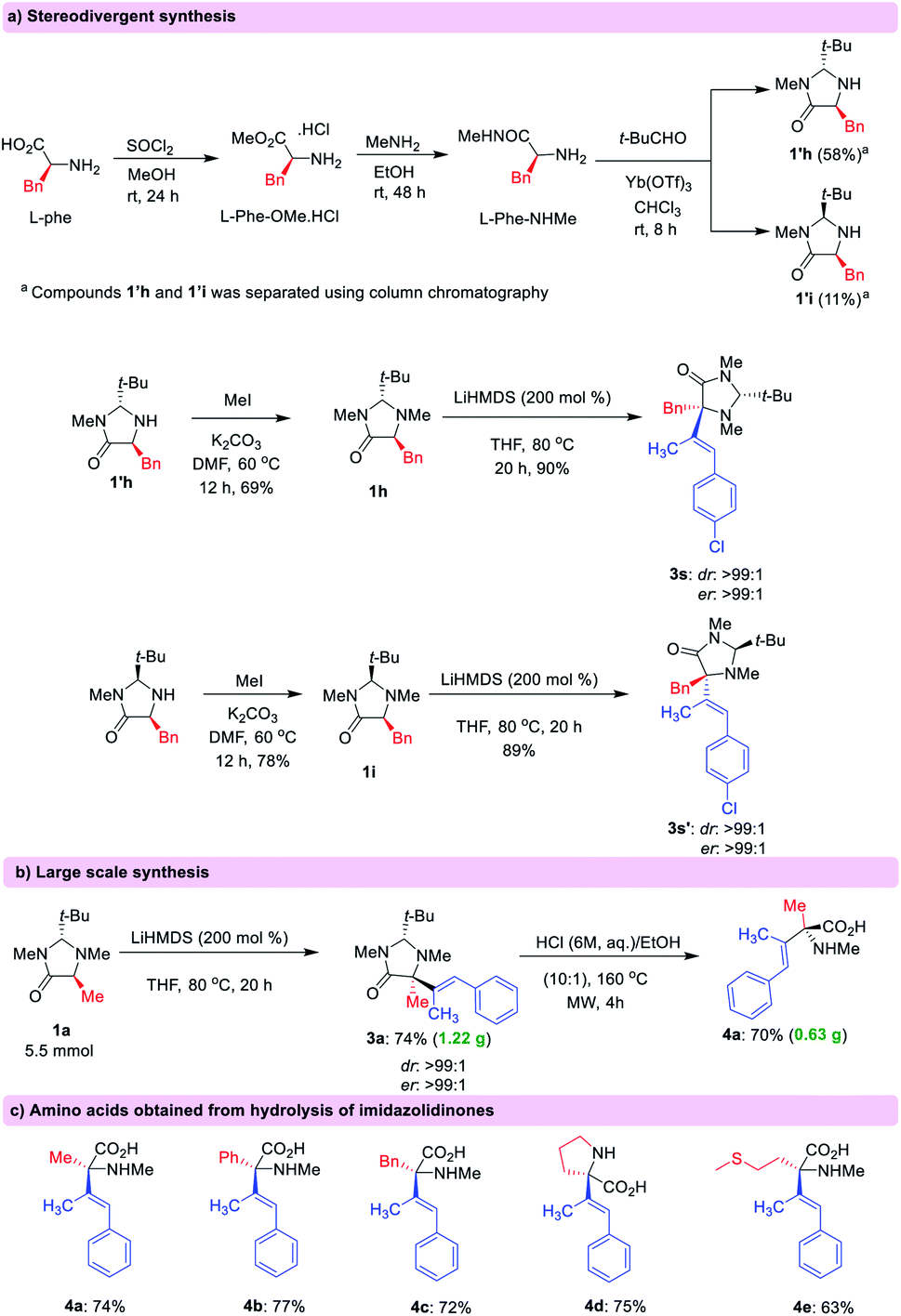 | ||
| Scheme 3 (a) Stereodivergent synthesis of quaternary α-alkenyl substituted imidazolidinone derivatives and their hydrolysis towards α-AAs. (b) Large scale synthesis of α-alkenylated amino acid derivatives. (c) The chemical structure of some α-alkenyl substituted amino acids after hydrolysis of imidazolidinone derivatives. | ||
Considering that the base-promoted isomerization of alkynes to allenes is well-known,16 we assume that the reaction could proceed by the attack of the imidazolidinone enolate to the central carbon of the generated allene.17 This assumption was supported by the requirement of two equivalents of base: one equivalent for the enolate formation and another one for the alkyne to the allene isomerization. Indeed, using 1-phenylallene (4) as the substrate in the presence of one equivalent of base compound 3a was obtained in 85% isolated yield at 80 °C in THF. This shows that allenes can also be used as substrates. Using this modified methodology several imidazolidinone derivatives were converted to the corresponding products (Scheme 4). Similar high yields and stereoselectivities were observed as for the alkyne substrates. Internal allenes also worked with this method (3aj,k). An X-ray crystal structure analysis of a suitable single crystal of 3ac confirmed its relative and absolute configuration (Scheme 4).
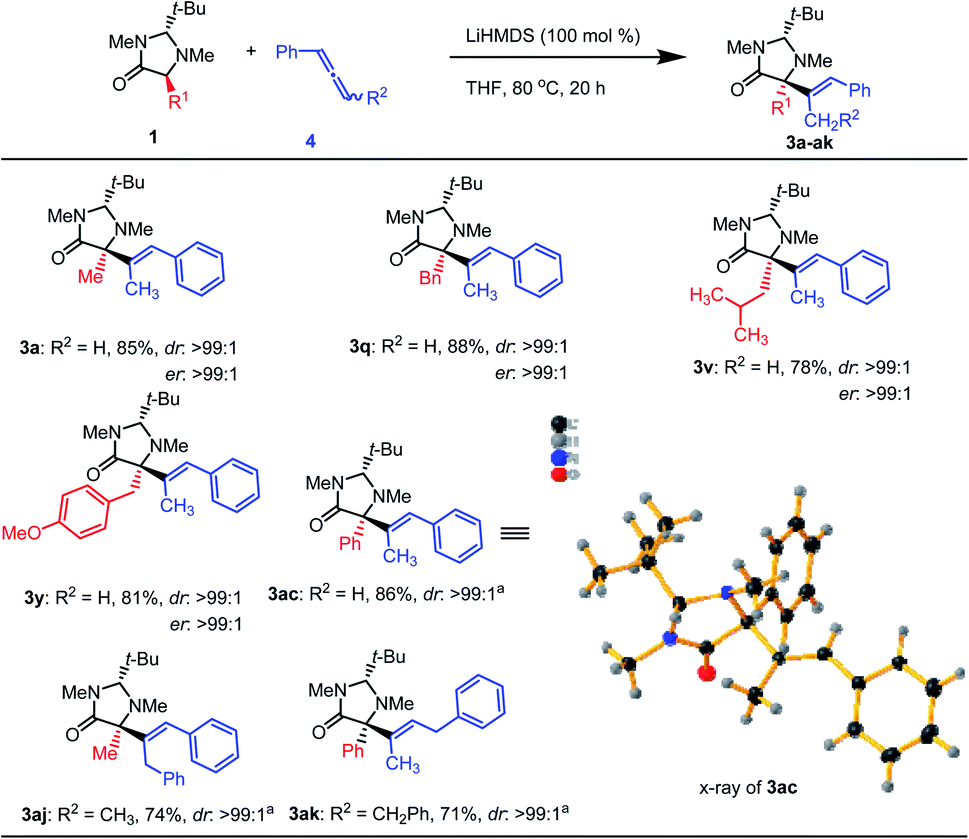 | ||
| Scheme 4 Hydroalkylation of allenes with imidazolidinone derivatives. Reaction conditions: 1a (0.2 mmol), 4a (0.2 mmol), LiHMDS (0.2 mL, 1.0 M in THF), THF (2.0 mL, 0.09 M), 80 °C, 20 h. Yields reported for isolated products. | ||
We propose a plausible reaction pathway for the hydroalkylation of alkynes and allenes with imidazolidinones in Fig. 1a. The first equivalent of the base might initiate the alkyne–allene isomerization process and form monolithiated allene (I). Then, in the presence of a second equivalent of the base I could give a dilithiated allenyl species (II). Based on control experiments this species seems to be the more stable allenyl intermediate.18 Formation of free allene as an electrophile coupling partner in this reaction is possible by protonation of the lithiated allenyl species.19 Also, the deprotonation of the imidazolidinone would afford the corresponding lithium enolate (III). Then, the nucleophilic attack of III to the central allene carbon dictates the alkene geometry anti relative to the aryl group. Finally, protonation of the related carbanion (IV) with HMDS delivers the product.
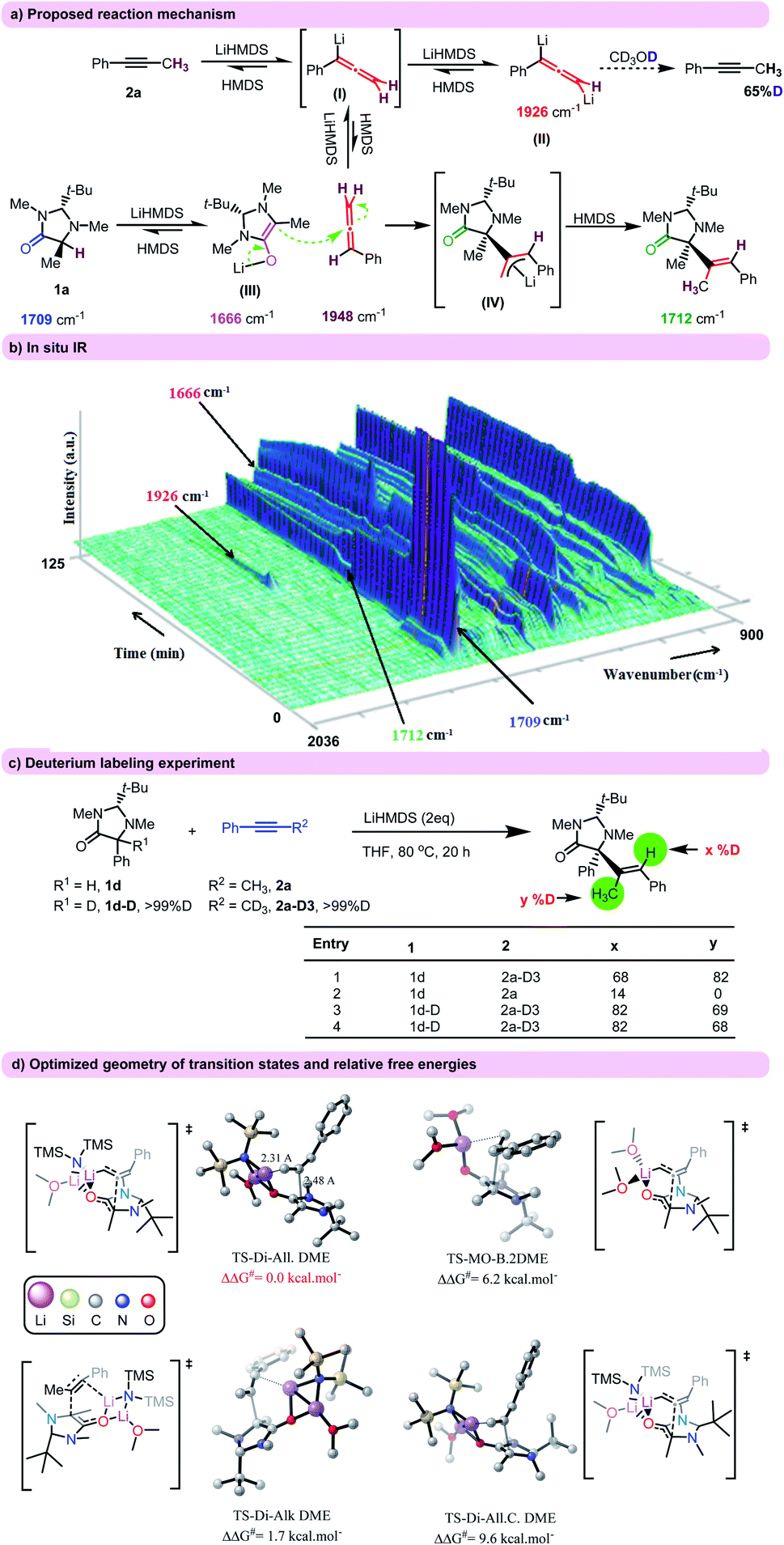 | ||
| Fig. 1 (a) The proposed reaction mechanism. (b) The in situ IR obtained of the reaction between substrates 1a and 2a. (c) Deuterium labeling experiments. (d) Optimized geometry of transition states and relative free energies (in kcal mol−1 are compared with TS-Tr-All. A). All C–H bonds are hidden for simplicity. | ||
Detailed mechanistic investigations were performed in order to support the proposed mechanism. First, the hydroalkylation reaction was evaluated by in situ IR using 1a and 2a as starting materials (Fig. 1b).20 Upon addition of LiHMDS (2 eq.) to a mixture of alkyne in THF at 80 °C, a new absorption band appeared at ∼1926 cm−1. According to literature, this band could be assigned to a dilithiated allenyl species.18a Based on a previous report, n-BuLi (2 eq.) was added to 2a and again the peak at ∼1926 cm−1 was observed proving our hypothesis. This peak disappeared after quenching with CD3OD and no peak was detected in the allene area. The NMR and mass analysis of the IR reaction mixture after quenching with CD3OD showed the regeneration of alkyne 2a with 65% of deuterium labeling in the methyl group (Fig. 1a). These experiments confirm the presence of dilithiated allenyl species II which is in equilibrium with the alkyne, allene, and monolithiated allene moieties.19,20
The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching absorption corresponding to the amide carbonyl group in 1a (∼1709 cm−1) was decayed upon addition of one equivalent of LiHMDS and a new peak at ∼1666 cm−1 was formed which corresponds to the enolate of 1a. Progress of the reaction showed the appearance of a new CO stretching absorption bond which is attributed to the product (∼1712 cm−1).
O stretching absorption corresponding to the amide carbonyl group in 1a (∼1709 cm−1) was decayed upon addition of one equivalent of LiHMDS and a new peak at ∼1666 cm−1 was formed which corresponds to the enolate of 1a. Progress of the reaction showed the appearance of a new CO stretching absorption bond which is attributed to the product (∼1712 cm−1).
Deuterium labeling experiments using substrates 1d and 2a indicated that the deuterium atoms are distributed between two positions of vinylic hydrogen and methyl group (Fig. 1c).20 This deuterium distribution is in agreement with a prior alkyne–allene isomerization and protonation of the carbanion (IV) (Fig. 1a). It should be noted that quenching the reaction with D2O did not change the deuterium incorporation, suggesting that there was no organolithium intermediate present before (Fig. 1c, entry 4). Additionally, the quenching with methyl iodide did not result in the alkylated product.
In order to gain further insight into the reaction mechanism, DFT calculations were carried out.20 Nucleophilic addition of imidazolidinone to alkyne (or its allene counterpart) was considered as the key step in the presence of different aggregations of Li (monomer, dimer, and trimer).21 The optimized structures and relative barrier energies are shown in Fig. 1d, Scheme S1,† using DME (dimethylether) as a model for THF, LiHMDS.3DME for solvated LiHMDS and imidazolidinone 1a and alkyne 2a as substrate. A comparison of the calculated free energies showed that the stability of transition states depends on both the degree of aggregation and solvation. We found in our calculations that in the medium degree of lithium aggregation (dimer), the coordination of solvent to Li (TS-Di-All.DME) is more beneficial than in an unsolvated transition state (Ts-Di-All, Scheme S1†). In addition, for the transition states with less aggregated Li (dimer and monomer) coordination of the solvent seems to be more preferred in comparison to trimer. These results are in agreement with a previous report.22 Computational details show that the medium degree of lithium aggregation (dimer) leads to the most stable transition state. Furthermore, the high difference between the energy of TS-Di-All.DME and TS-Di-All.C.DME (9.6 kcal mol−1) well demonstrates the key role of the tert-butyl group in controlling stereochemistry. More importantly, the geometry of 2a in TS-Di-Alk.DME shows that carbolithiation of alkyne led to the Z product while the E-configuration is the only experimentally observed geometry. The found relative energies of TS-Di-Alk.DME and TS-Tr-Di-All.DME supports the assumption that the reaction is exclusively happening between allene and the enolate.
Conclusions
In conclusion, we have developed a new method for the enantioselective introduction of an alkenyl substituent to the alpha position of amino acids using the diastereoselective hydroalkylation of alkynes and allenes with imidazolidinone derivatives obtained from enantiopure amino acids. This reaction proceeds with complete control of both regio- and diastereoselectivity and allows efficient access to (E)-alkene functionalized quaternary amino acids. The feasibility of this protocol is highlighted by its operational simplicity as well as gram scale application. Further studies in this research field are currently ongoing in our group.Author contributions
The work was conceptualized by FP and BB. FP initiated the project and performed the experiments. HK performed the computational analysis and synthesized some starting materials. FB performed the in situ IR experiments. The first draft of the manuscript was prepared by FP and the final draft was edited by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Alexander von Humboldt Foundation (F.P.) for a postdoctoral fellow-ship. We are grateful to Dr María González Rodriguez for reading the manuscript. We thank Dr Manfred Keller for extensive NMR experimentations and Joshua Emmerich for challenging HPLC separations.Notes and references
- K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS.
- T. Boibessot, D. Bénimélis, P. Meffre and Z. Benfodda, Amino Acids, 2016, 48, 2081–2101 CrossRef CAS PubMed.
- (a) S. Shirakawa, K. Yamamoto and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 838–840 CrossRef CAS PubMed; (b) D. J. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS PubMed; (c) J. Mas-Rosello, S. Hachisu and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 10750–10754 CrossRef CAS PubMed.
- (a) D. B. Berkowitz, W.-J. Jahng and M. L. Pedersen, Bioorg. Med. Chem. Lett., 1996, 6, 2151–2156 CrossRef CAS PubMed; (b) J. P. Scannell, D. L. Pruess, T. C. Demny, L. H. Sello, T. Williams and A. Stempel, J. Antibiot., 1972, 25, 122–127 CrossRef CAS PubMed; (c) M. J. Tisdale, Biochem. Pharmacol., 1980, 29, 501–508 CrossRef CAS.
- (a) A. Rubio and J. Ezquerra, Tetrahedron Lett., 1995, 36, 5823–5826 CrossRef CAS; (b) D. Uraguchi, Y. Ueki, A. Sugiyama and T. Ooi, Chem. Sci., 2013, 4, 1308–1311 RSC.
- A. Armstrong and D. P. G. Emmerson, Org. Lett., 2011, 13, 1040–1043 CrossRef CAS PubMed.
- E. Tayama, T. Igarashi, H. Iwamoto and E. Hasegawa, Org. Biomol. Chem., 2012, 10, 339–345 RSC.
- N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1997, 119, 445–446 CrossRef CAS.
- H. Abas, J. Mas-Rosello, M. M. Amer, D. J. Durand, R. R. Groleau, N. Fey and J. Clayden, Angew. Chem., Int. Ed., 2019, 58, 2418–2422 CrossRef CAS PubMed.
- D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed., 1996, 35, 2708–2748 CrossRef CAS.
- (a) A. M. Haydl, B. Breit, T. Liang and M. J. Krische, Angew. Chem., 2017, 129, 11466 ( Angew. Chem., Int. Ed. , 2017 , 56 , 1312–1325 ) CrossRef; (b) P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef CAS PubMed.
- X.-Y. Lu, M.-L. Hong, H.-P. Zhou, Y. Wang, J.-Y. Wang and X.-T. Ge, Chem. Commun., 2018, 54, 4417–4420 RSC.
- In order to show the key role of Li on enantioselectivity, HMPA (hexamethylphosphoramid) and [12]crown-4 were used as additive to capture the Li and prevent its action in the reaction and interestingly ee was decreased to 73% and 51%, respectively. These experiments suggest the chelation-control role of Li metal via lithium enolate formation in this reaction.
- R. Naef and D. Seebach, Helv. Chim. Acta, 1985, 68, 135–143 CrossRef CAS.
- J. Chatterjee, B. Laufer and H. Kessler, Nat. Protoc., 2012, 7, 432–444 CrossRef CAS PubMed.
- A. S. K. Hashmi, Synthesis of Allenes by Isomerization Reactions, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- D. Hack, M. Blümel, P. Chauhan, A. R. Philipps and D. Enders, Chem. Soc. Rev., 2015, 44, 6059 RSC.
- (a) P. Zargaran, F. F. Mulks, S. Gall, M. Rudolph, F. Rominger and A. S. K. Hashmi, Organometallics, 2019, 38, 1524–1533 CrossRef CAS; (b) N. Alouane, K. Bentayeb, E. Vrancken, H. Gerard and P. Mangeney, Chem.–Eur. J., 2009, 15, 45–48 CrossRef CAS PubMed.
- J. Mateos-Gil, A. Mondal, M. C. Reis and B. L. Feringa, Angew. Chem., 2020, 59, 7823–7829 CrossRef CAS PubMed.
- See ESI for details.†.
- A. J. McNeil and D. B. Collum, J. Am. Chem. Soc., 2015, 127, 5655–5661 CrossRef PubMed.
- S. Popenova, R. C. Mawhinney and G. Schreckenbach, Inorg. Chem., 2007, 46, 3856–3864 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1950247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00240f |
| This journal is © The Royal Society of Chemistry 2021 |