
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolarization-induced charge separation in conjugated microporous polymers for efficient visible light-driven C-3 selenocyanation of indoles†
Qiujian
Xie
a,
Yumin
Yang
b,
Weijie
Zhang
a,
Zhu
Gao
a,
Xiaofeng
Li
a,
Juntao
Tang
*a,
Chunyue
Pan
a and
Guipeng
Yu
 *a
*a
aHunan Key Laboratory of Micro & Nano Materials Interface Science, College of Chemistry and Chemical Engineering, Central South University, Lushan South Road 932, Changsha 410083, Hunan, P. R. China. E-mail: gilbertyu@csu.edu.cn; Reynardtang@csu.edu.cn
bQueen Mary University of London Engineering School, Northwestern Polytechnical University, Youyi West Road 127, Xian 710072, Shaanxi, P. R. China
First published on 9th March 2021
Abstract
Conjugated microporous polymers (CMPs) are cost-effective photocatalysts in organic transformations, while they are usually limited by the insufficient separation of photogenerated charges. Here we report a polarization strategy through molecular geometry optimization to promote the charge separation of CMPs. Three CMP photocatalysts with an alternative donor–acceptor skeleton and tunable symmetry were synthesized by the oxidative coupling of bis-carbazoles with electron-deficient bridges (benzene/pyridine/pyrimidine). Simply regulating the polarization of the starting monomers leads to tailorable porosity, photoelectric properties, and photocatalytic activity of the CMPs. They exhibited high efficiency in C-3 selenocyanation of indoles under visible-light and at room temperature, and pyridine-based CMPs with the largest dipole moment gave a yield of up to 94%, superior to their state-of-the-art photocatalyst counterparts. Photo-physical experiments combined with theoretical calculations further supported that the incorporation of the polarized linker introduced an internal electric field, benefitting efficient charge separation. This offered new insight into developing high-performance photocatalysts.
Introduction
Indole derivatives play important roles in organic synthesis and pharmaceutical chemistry.1–3 Specifically, the selenium-containing indole derivatives have attracted broad interest as they are not only pharmaceutical drug candidates but also versatile intermediates for the production of value-added compounds.4,5 Although C-3 selenocyanation is a convenient and efficient route to incorporate selenium moieties into indoles, rare feasible synthetic protocols were presented and most of them required the use of diverse toxic selenocyanate species and heavy-metal catalysts.6–9 Metal-free reagent-mediated selenocyanation, as a green and sustainable process, which has been highlighted as a research trend, unfortunately remained a significant challenge. To the best of our knowledge, the utilization of visible-light-driven C-3 selenocyanation of indoles has not been reported yet. Hence, searching for an alternative photocatalytic process and developing efficient and eco-friendly photocatalysts for selenocyanation of indoles are highly desirable.As a unique class of materials, conjugated microporous polymers (CMPs), combining an extended π-conjugation skeleton with a permanent microporous structure, have recently attracted intensive interest targeting various utilities.10–13 Especially, owing to their excellent chemical robustness and tunable structures,14,15 CMPs offer a valuable platform for developing heterogeneous photoredox catalysts.16 Since the first CMP was reported by Cooper's group in 2007,17 task-specified CMP photocatalysts with functionality-built-in architectures, including rose bengal,18 perylene,19 dipyrrometheneboron difluoride (BODIPY),20 carbazole21 and benzothiadiazole,22,23 have been recently employed in photocatalytic hydrogen evolution and aerobic oxidation reactions. However, within the efforts for pursuing satisfactory performance, a limitation still exists in separating the photo-excited electrons and delivering them into the target regions for producing active species. Regulating the linking pattern of building blocks,24 alternating the composition of donor and acceptor moieties,25 introducing electron-output “tentacles”26 and creating heterojunctions27 have been proved to be efficient strategies. For example, by incorporating electron-rich pyrene and electron-deficient dibenzothiophene-S,S-dioxide into the framework, Zhao et al. found that the polymer with a 3,7-linking pattern (PyDOBT-1) exhibits superior photocatalytic performance in hydrogen generation to that of PyDOBT-2 with a 2,8-linking pattern.28 We also reported a core-tailoring protocol through incorporating electron-rich or deficient units to tailor the photophysical properties of carbazole-based CMPs [CMP-CSUs (s = 5, 6, 7)].29 Although such efficient photocatalytic performance regarding classic transformations has been documented, the detailed boosting mechanism for photocatalytic properties for most CMPs is not well understood.
Herein, we report a polarization strategy to develop CMP photocatalysts consisting of alternating electron-rich (carbazole) and deficient (benzene, pyridine or pyrimidine) units. The polymers were readily prepared through oxidative coupling of precursors with tuned polarity. We envisioned that the strong polarity in electron-deficient units would offer a built-in electric field effect in CMPs, which might facilitate the rapid separation of photogenerated electrons and holes, therefore promoting the photocatalytic activity. To validate this strategy, C-3 selenocyanation of indoles was employed as a model reaction. Significantly, among the three samples, an attractive yield of up to 94% and extensive substrate adaptability were achieved in the reaction catalyzed by pyridine-linked CMP-CSU14 with the largest dipole moment. Compared to the known synthetic protocols, our CMP catalysts function efficiently under mild conditions without the addition of any strong oxidants or toxic additives. The theoretical calculations also revealed that the natural dipole moments induced an internal electric field to force the migration of photogenerated electrons and holes in an opposite orientation, therefore enhancing the photogenerated charge separation. Moreover, the excellent stability of these metal-free heterogeneous photocatalysts enabled a robust recycling capability with well-retained performance even over five consecutive runs, which is not available for the other known catalysts for selenocyanation.
Results and discussion
Three carbazole monomers named 9,9′-(benzene)-bis(9H-carbazole), 9,9′-(pyridine-2,5-diyl)-bis(9H-carbazole), and 9,9′-(pyrimidine-2,5-diyl)-bis(9H-carbazole) were synthesized by the nucleophilic displacement chemistry of 1,4-dibromobenzene, 2,5-dibromopyridine and 2,5-dibromopyrimidine, respectively. The carbazole CMPs (termed CMP-CSU5, CMP-CSU14, and CMP-CSU15) were obtained as insoluble solid powders via a FeCl3-catalyzed oxidative polymerization in anhydrous chloroform (CHCl3) under a nitrogen atmosphere in high yields (85–90%)30 (Fig. 1 and Scheme S1†). The chemical structure of the synthesized CMP-CSUs was proved by Fourier transform infrared (FT-IR) and 13C cross-polarization/magic-angle-spinning solid-state nuclear magnetic resonance (13C CP/MAS NMR) measurements. Partial FT-IR spectra demonstrated the disappearance of absorption at around 721 cm−1 (disubstituted phenyl ring of carbazole) as well as the emerged signals in the range of 750–810 cm−1 for the C–H vibration of the disubstituted carbazole ring, confirming the successful coupling (Fig. 2a and S1–S3†).31 In a typical 13C CP/MAS NMR spectrum of CMP-CSU14, the peaks at 135 and 133 ppm were attributed to the aryl carbons adjacent to the carbazole and pyridine nitrogen atom, and that at 124 ppm to the carbon of substituted phenyl carbons, further revealing the oxidative chemistry32–34 (Fig. S4†). The scanning electron microscopy (SEM) measurements show that all CMP-CSUs have an irregular and aggregated morphology with a particle size in the range of 100–300 nm (Fig. S5†). The powder X-ray diffraction (PXRD) analysis suggested that all the collected polymers are amorphous materials (Fig. S6†). The porosity features of the obtained polymers were acquired from nitrogen (N2) sorption isotherms (77 K) (Fig. S7†) and the corresponding results are collected and summarized in Table S1.† According to the International Union of Pure and Applied Chemistry classification, our polymers displayed type I combined with type II nitrogen isotherm features and showed a hierarchically porous structure combining mesoporosity with certain microporosity.35 All three samples gave considerably high Brunauer–Emmett–Teller (BET) specific surface area (exceeding 500 m2 g−1) and the highest BET surface area combined with a pore volume of 0.79 cm3 g−1 was acquired for CMP-CSU5 (benzene-linked), which is in line with the previous reports.29 CMP-CSU15 gave the lowest BET surface area (528.6 m2 g−1), possibly due to the low reactivity of its precursor (PymDCz) in the oxidative coupling reaction possibly due to the electrophilicity of pyrimidine.36 Based upon NL-DFT calculations, the pore-width-distributions of CMP-CSUs were centered at 1.0–1.7 nm and 2.0–5.0 nm (Fig. S8†). The hierarchical porous structure with over 71% mesoporosity appears to be an ideal feature for organic transformations involving large-sized substrates or products in photocatalytic reactions.37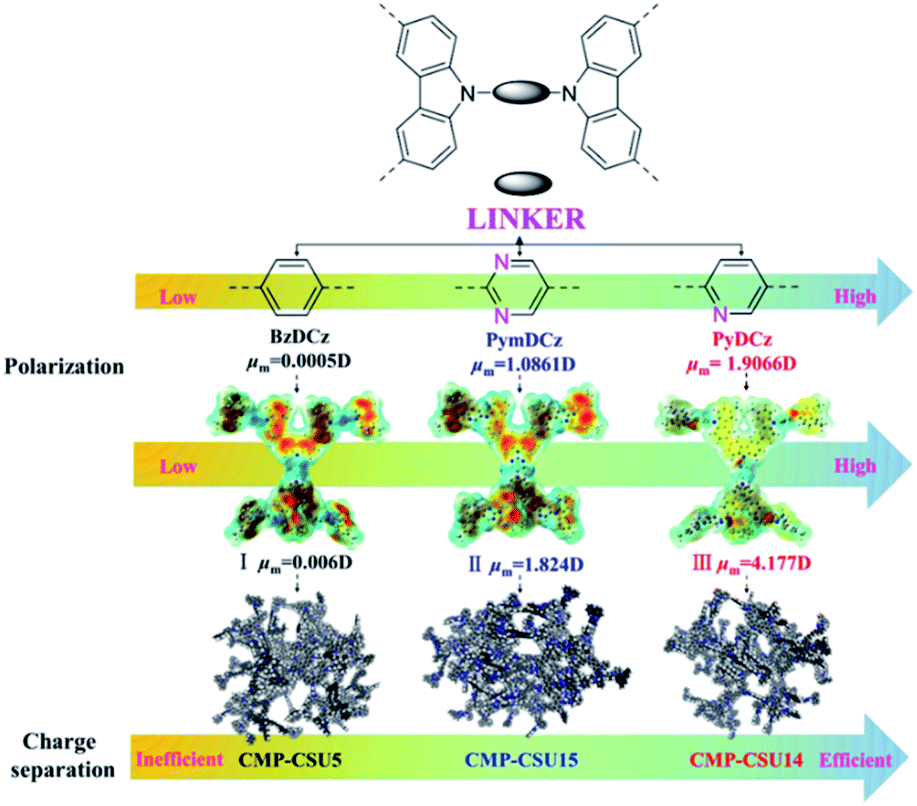 | ||
| Fig. 1 The design strategy of the corresponding monomers and oligomers of CMP-CSUs with polarized structures ((I) oligomer for CMP-CSU5, (II) oligomer for CMP-CSU15, and (III) oligomer for CMP-CSU14). | ||
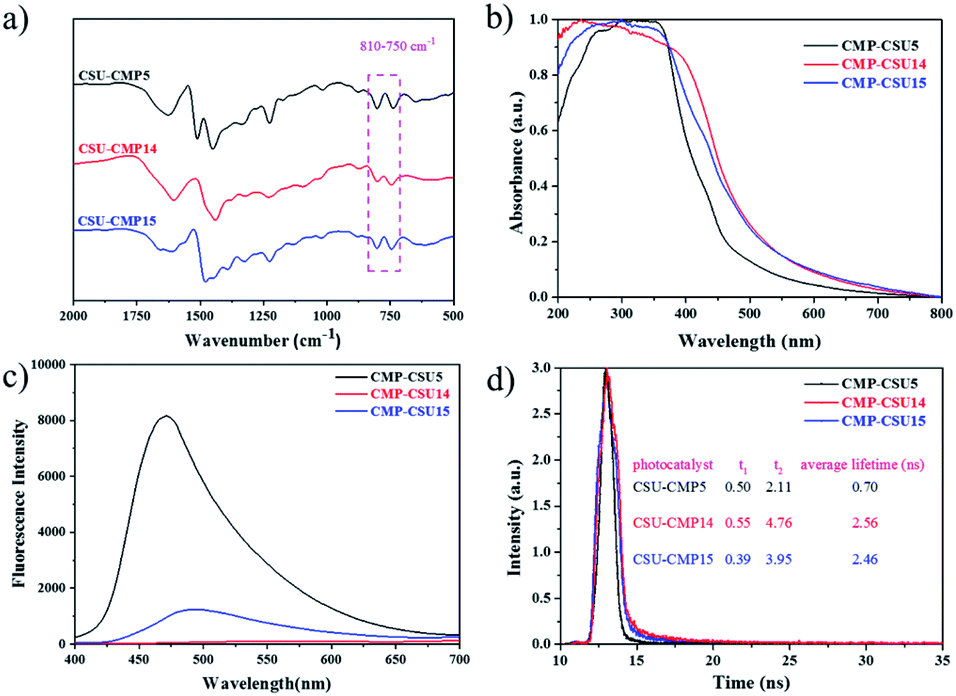 | ||
| Fig. 2 (a) Partial FT-IR spectra of CMP-CSUs; (b) normalized solid state UV/Vis absorption spectra of CMP-CSUs; (c) photoluminescence spectra (PL) of CMP-CSUs; (d) photoluminescence decay traces of CMP-CSUs. | ||
The light harvesting properties and optical band gaps of the as-synthesized materials were assessed by diffuse reflectance spectrum (DRS) measurements. Both CMP-CSU14 and CMP-CSU15 exhibited relatively broad absorption from 200 to 800 nm (Fig. 2b), which indicates that the absorbed light of collected polymers could cover a wide range of the visible region.38 Notably, CMP-CSU5 had a maximum absorption at 350 nm, and a distinct blue-shift of the absorption peak compared with the other two polymers with a different electron-deficient linker. The optical band gaps were derived from the transformed Kubelka–Munk theory (Fig. S9†). As for CMP-CSU5, CMP-CSU14 and CMP-CSU15, the energy levels of the lowest unoccupied molecular orbital (LUMO) were recorded to be at −0.845, −0.834 and −0.822 V vs. SCE, respectively, and thus can reduce oxygen to generate O2˙− (−0.57 V vs. SCE) (Fig. S10†).39 The relative production of O2˙− in CMP-CSUs has been compared by the oxidation experiment with N,N,N′,N′-tetramethyl-1,4-phenylenediamine (TMPD) (Fig. S11†). The intensity variations in UV/Vis absorption spectra verified the superior O2˙− production rate of CMP-CSU14 compared to CMP-CSU5 and CMP-CSU15. Accordingly, the calculated highest occupied molecular orbital (HOMO) energy levels were far beyond the level of E(SeCN−/˙SeCN) (where SeCN− represents the selenocyanate anion), which appears to be sufficient to trigger the selenocyanation reaction (Fig. S12 and S13†).
Photoluminescence (PL) spectroscopy was employed to elucidate the charge carrier migration and separation behavior of the samples. The PL intensity of CMP-CSU14 was quenched dramatically in comparison to that of CMP-CSU5 and CMP-CSU15, indicating the effective charge transfer and the inhibited charge carrier recombination40 (Fig. 2c). The rate of charge transfer was further investigated by electrochemical impedance spectroscopy (EIS). Among the three samples, the pyridine-bridged CMP-CSU14 exhibited the smallest radius in the Nyquist plots (Fig. S14†), indicating the lowest charge transfer resistance as well as the highest charge mobility.41 The highest photocurrent under light irradiation was observed for CMP-CSU14 (Fig. S15†), which suggests the most effective separation of photo-induced electrons and holes as well as the fastest charge mobility.42 Moreover, the production of transient photocurrent observed for our CMP-CSUs is highly reversible and would repeat over five straight ON/OFF irradiation runs, indicative of high photoelectrochemical stability. To quantify the lifetimes of the carrier behavior during the photocatalytic process, photoluminescence decay measurements (Fig. 2d) were conducted, and the average photoluminescence lifetimes were calculated using the following equation: τaverage lifetimes = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). The average photoluminescence lifetimes of CMP-CSU5, CMP-CSU14, and CMP-CSU15 were measured to be 0.7 ns (τ1 = 0.50 ns, τ2 = 2.11 ns), 2.46 ns (τ1 = 0.39 ns, τ2 = 3.95 ns), and 2.56 ns (τ1 = 0.55 ns, τ2 = 4.76 ns), respectively (Table S2†).43 The prolonged decay lifetime of CMP-CSU14 implies its slower charge recombination and faster electron transfer than the others, and alternately it could provide more photo-induced carriers, indicative of improved intrinsic activity during the photocatalytic processes.44
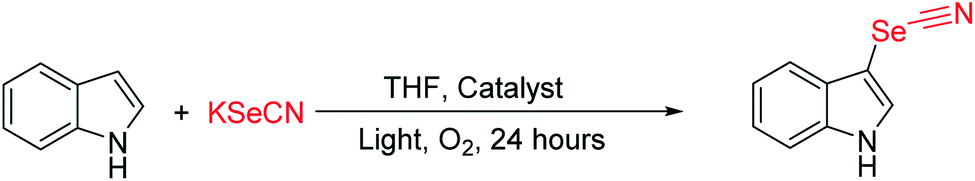
On the basis of the promising photoredox properties of the three CMP polymers, the possibility for their photocatalytic transformation was investigated in C–H functionalization for indoles. The C-3 selenocyanation of indoles was selected as the model reaction due to its great importance in constructing valuable pharmaceutical drug candidates (Fig. S16†). By choosing 1H-indole as a model substrate, potassium selenocyanate (KSeCN) as the SeCN− source, and molecular oxygen as a green oxidant, the photo-reaction conditions were screened and optimized under visible-light (Tables 1 and S3†). A yield of 94% for the target product was obtained by using CSU-CMP14 with tetrahydrofuran (THF) as the medium upon visible-light irradiation for at least 24 h (Table 1, entry 1). However, decreased yields (41% and 87%) were obtained when the catalyst was replaced by CMP-CSU5 and CMP-CSU15, respectively (Table 1, entries 2 and 3). To illustrate the influence of surface area of CMP-CSU14 and CMP-CSU15, the reaction time was extended to 36 hours to fully make sure they reached the equilibrium state. The yields of the target product were measured to be 95% and 86% when catalyzed by CMP-CSU14 and CMP-CSU15. To better understand the relevance of surface area on the indole C-3 selenocyanation activity, 1H-indole and CMP-CSUs were transferred to a transparent reaction tube and the mixture was stirred in the dark (Fig. S17†). The lowest adsorption rate for CMP-CSU5 and the fast abdsorption rate for CMP-CSU15 were revealed, demonstrating that the surface area is not the key factor for determining the photocatalytic activities. We speculated that the intense difference was presumably ascribed to the different charge separation and transfer efficiency of the photocatalysts. The performance of the known state-of-the-art photocatalysts was also investigated under identical conditions (Table 1). Note that only moderate yields were detected when organic dyes such as fluorescein were applied (Table 1, entry 6). Negligible yields or barely no products were found under the catalysis of g-C3N4, TiO2 and MoS2 (Table 1, entries 7–9).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | Conversion (%) |
| a Reaction conditions: 1H-indole (0.2 mmol), KSeCN (0.4 mmol, 2 equiv.), catalyst (10 mg), tetrahydrofuran (THF 2 mL), RT = 25 ± 2 °C, 24 hours, O2 (∼0.1 MPa), using a 14 W LED lamp (0.20 W cm−2) as the light source. b 1H NMR yield (using mesitylene as an internal standard). c No catalyst. d No reaction. | |||
| 1 | CMP-CSU14 | 94 | 100 |
| 2 | CMP-CSU15 | 87 | 98 |
| 3 | CMP-CSU5 | 41 | 55 |
| 4 | Fluorescein | 48 | 60 |
| 5 | g-C3N4 | 24 | 43 |
| 6 | TiO2 | 5 | 15 |
| 7 | MoS2 | Trace | Trace |
| 8c | — | NRd | — |
Electron paramagnetic resonance (EPR) tests were carried out to further explore the possible mechanism of the selenocyanation of C-3 indoles. When 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was employed as a superoxide radical (O2˙−) scavenger, the intensity of the EPR signal (magnetic field of 319–326 mT) of CMP-CSU14 was obviously enhanced under light-irradiation, indicating the effective production of O2˙− upon photo-excitation (Fig. S18†). Singlet oxygen (1O2) was also captured by 2,2,6,6-tetramethylpiperidine (TEMP) under light irradiation in the presence of CMP-CSU14, and the signals showed a moderate enhancement compared to that in the dark (Fig. S19†). To further investigate the effect of the photogenerated electron–hole pairs and intermediates during C-3 selenocyanation of indoles, different scavengers were charged into the reaction mixture under light irradiation (Fig. S20†). In the presence of a hole scavenger (KI), a decreased yield of 81% was obtained, which illustrated that the holes participate in the reaction but do not play a critical role. The addition of benzoquinone that captures superoxide effectively led to a significantly low yield (15%). By adding a singlet oxygen scavenger (sodium azide), the corresponding product was obtained with a moderate yield of 42%. However, a negligible yield (8%) was acquired when 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) was utilized to quench the radical. The control experiments indicate that the reactive oxygen species, i.e. O2˙− and 1O2 play vital roles in the C-3 selenocyanation of indoles. Based on these results, a putative reaction mechanism for indole C-3 selenocyanation was proposed (Fig. 3). Under light irradiation, CMP-CSU14 was triggered to form CMP-CSU14* and quenched by oxygen to generate both superoxide radical (O2˙−) and CMP-CSU14+via a singlet electron transfer (SET) process. Via the procedure of energy transfer of the triplet electrons, the generated singlet oxygen (1O2)45 would participate in the reaction to oxidize the intermediate B. The photoredox cycle was completed by a single electron transfer (SET) between SeCN− and CMP-CSU14+, which generated SeCN˙ and CMP-CSU14, respectively. Next, the produced holes (CMP-CSU14+) and reactive oxygen species (O2˙− and 1O2) oxidized the selenocyanate anion to the selenocyanate radical, and a radical intermediate (B) would be generated under the attack of the selenocyanate radical. Subsequently, the intermediate B was further oxidized to intermediate C, and afforded the terminal product (3A) by giving out a proton.
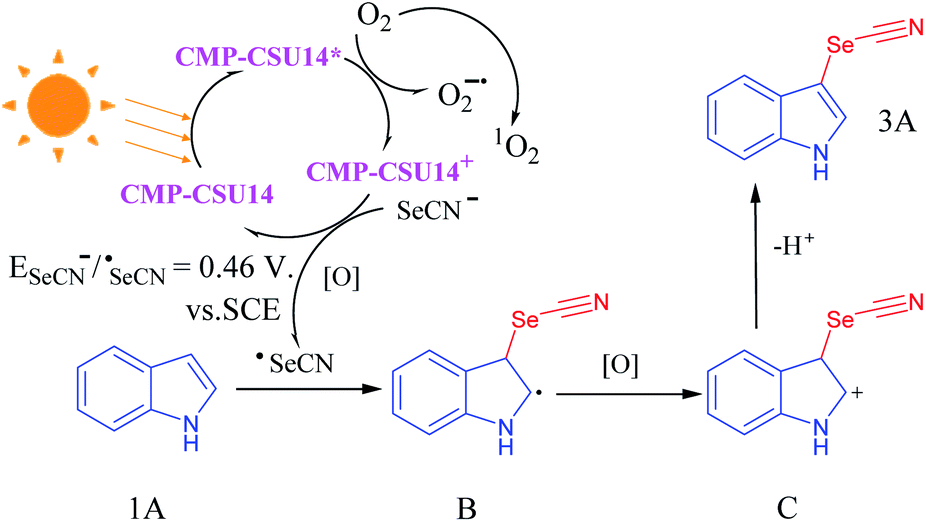 | ||
| Fig. 3 Proposed mechanism for the C-3 selenocyanation of indoles. | ||
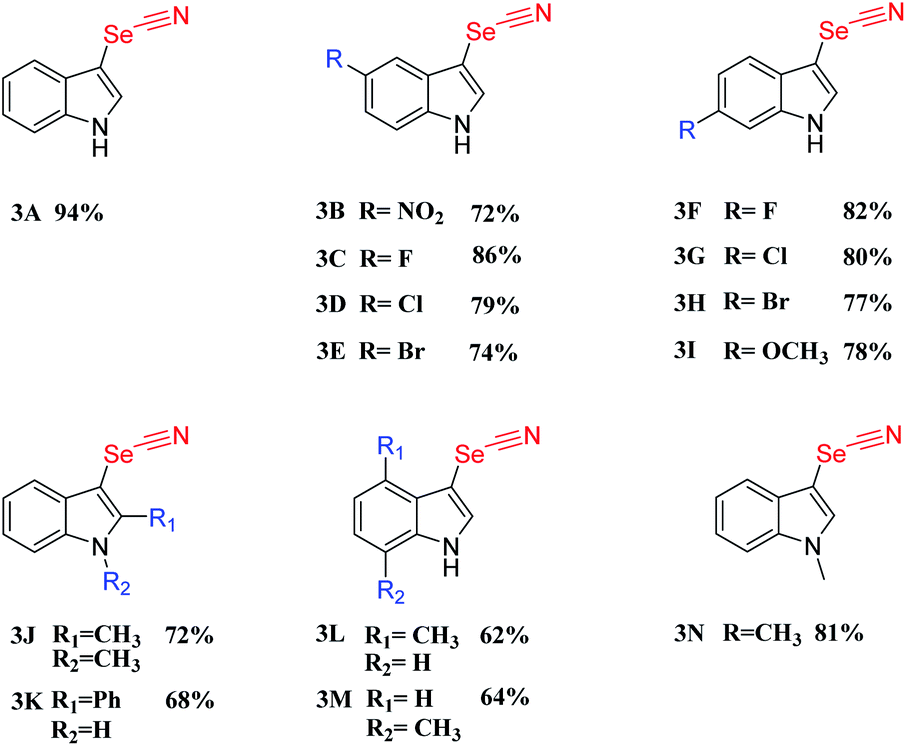
After establishing the standard conditions for the C-3 selenocyanation of indoles, the general applicability and selectivity of selenocyanation for diverse indole substrates were investigated. Moderate to good yields were obtained when incorporating electron-withdrawing indole derivatives such as 6-fluoro, 6-chloro, and 6-bromo substituted ones (Table 2, 3B–H). However, when electron-donating groups were anchored, the indole derivatives furnished the target products in lower yields, which may indicate that the electron-donating characteristic has negative effects on the C-3 selenocyanation (Table 2, 3I–M). When a methyl or phenyl unit was incorporated at the C-2 position, corresponding product yields decreased to 72% or 48%, respectively (Table 2, 3J–K). This may suggest that the steric hindrance and the electron-donating effect at the C-2 position also attenuated the photocatalytic performance. For those derivatives with N-substituted groups, like N-methyl-indole (Table 2, 3N), the C-3-selenocyanation product was afforded in a yield of 81%. CMP-CSU14 was selected as a representative photocatalyst to demonstrate the recycling capability. Our CMP photocatalyst could be re-employed at least five times without substantial decline of photocatalytic efficiency (Fig. S21†). Meanwhile, the structural features of the catalyst such as primary skeleton connectivity and morphology were retained, with limited changes in the FT-IR spectra along with the SEM images after the photocatalytic cycles (Fig. S22 and S23†).
|
|
|---|
| a Reaction conditions: indoles (0.2 mmol, 1 equiv.), KSeCN (0.4 mmol, 2 equiv.), CMP-CSU14 (10 mg), THF (2 mL), RT = 25 ± 2 °C, 24 hours, O2 (∼0.1 MPa), using a 14 W LED lamp (0.20 W cm−2) as the light source. 1H NMR yield (using mesitylene as an internal standard). |
|
To gain deeper insights into the deciding factor for the attractive photocatalytic performance, the geometry of the starting monomers and oligomeric precursors was optimized by density functional theory (DFT) calculations (B3LYP functional, 6-31G(d) basis set).46 As a consequence of the selected nitrogen doping, changes in the dihedral angle between the central aryl plane and the carbazole unit could be expected. The optimal geometry of these monomers exhibited distinct twisted degrees and dipole moments (Fig. 4a–c and S24†). BzDCz shows a slightly higher value (54.5° and 54.5°) compared to pyridine-based PyDCz (35.7° and 55.2°) and PymDCz (0.07° and 59.2°). It is noted that PyDCz possesses the largest dipole moment (1.65522 debye), followed by a decreasing trend with PymDCz of 0.90407 debye and BzDCz of 0.00002 debye. In addition, the polarization of CMP-CSUs was further revealed by their corresponding oligomers (Fig. S25†). With the introduction of nitrogen atoms, the dipole moment of the oligomeric precursors was gradually increased from 0.006 debye (for CMP-CSU5) to 4.177 debye (for CSU-CMP14). The polarization of CMP-CSUs would also be identified by the fluorescence spectroscopic measurements.47 The shifts of the maximum emission wavelength were detected for the polymers with diverse natural dipole moments. As depicted in Fig. 4d, a minimum shift of 36 nm of CMP-CSU5 was found in ethyl acetate compared to that in hexane. A slight shift could be attributed to the weakest dipole moment of CMP-CSU5 with minimum polarization. In contrast, for CMP-CSU15, a distinct shift of emission maxima was demonstrated with an increase over 52 nm in ethyl acetate relative to that in hexane (Fig. 4e). Significantly, the shift of emission maxima was up to 68 nm for CMP-CSU14 with the largest dipole moment (Fig. 4f). We speculated that the dipole moment will induce an internal electric field that was considered to be useful for electron–hole separation upon photoexcitation.48 Considering the precursor (PyDCz) of CMP-CSU14, the orientation of the induced internal electric field is opposite to that of the natural dipole moments (Fig. 5a).49 Therefore, the separation of photogenerated electrons and holes in an opposite orientation is effectively under the force of the internal electric field.
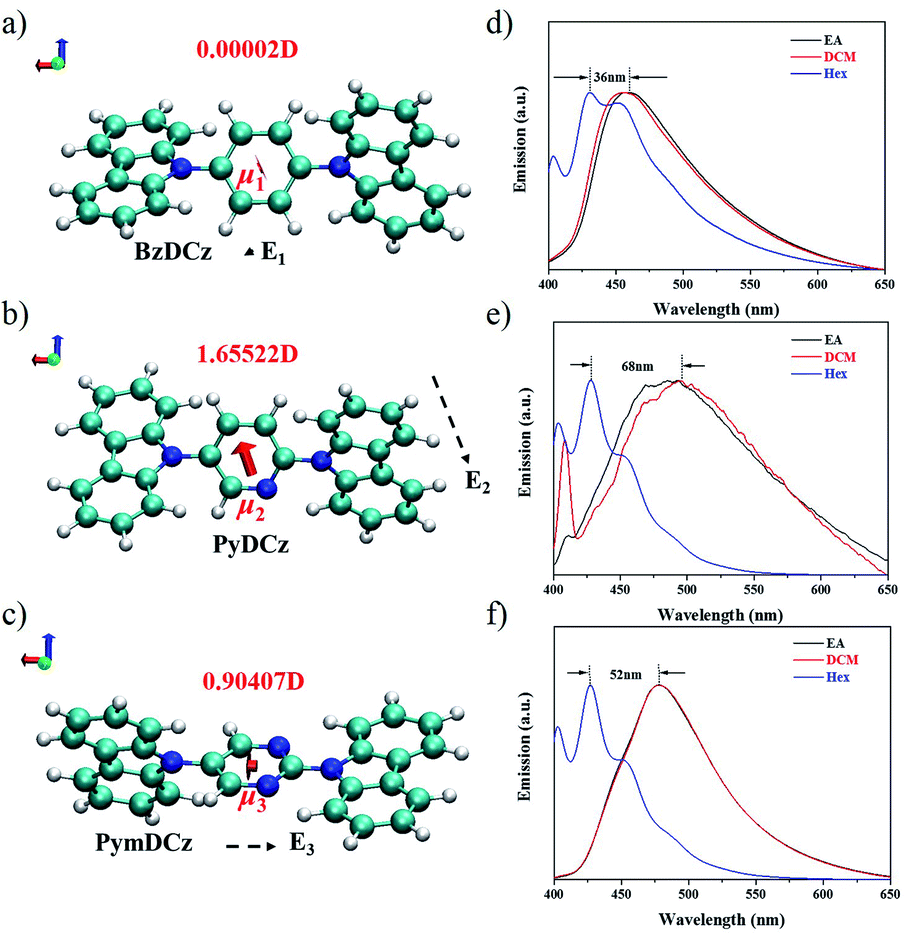 | ||
| Fig. 4 Internal electric field (E1, E2, E3), configuration and natural dipole moment of (a) BzDCz, (b) PyDCz, and (c) PymDCz; PL emission spectra of (d) CMP-CSU5, (e) CMP-CSU14, and (f) CMP-CSU15 in different polar solvents (each suspension contains 0.2 mg sample and 4 mL solvent; EA-ethyl acetate; DCM-dichloromethane; Hex-hexane). | ||
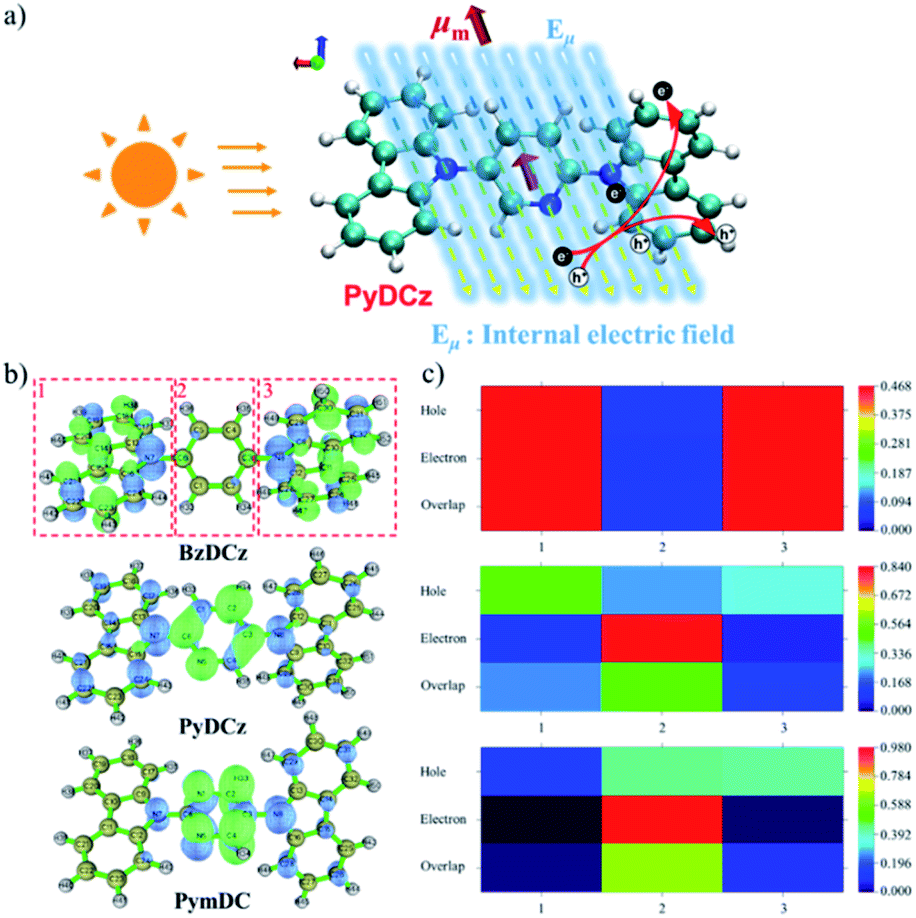 | ||
| Fig. 5 (a) The mechanism of the internal electric field for the separation of photogenerated electrons and holes; (b) electron and hole distribution of excited monomer fragments (green represents electrons; blue represents holes); (c) transition density matrix heat map of electron and hole contribution distribution of excited monomer fragments. | ||
To further understand the electron–hole pair separation in the monomers of CMP-CSUs, electronic transitions among the monomers were calculated based on time-dependent density functional theory (TD-DFT) calculations (CAM-B3LYP functional, 6-31G(d) basis set).50 The electronic transitions of the monomers were obtained by using a multifunctional wavefunction analyzer (Multiwfn).51–53 To investigate the electron and hole distribution, the optimized geometries of the three monomers in the excited state were divided into three fragments (Fig. 5b). In BzDCz, the electrons and holes are mainly distributed in the carbazole part, indicating the lowest efficiency of photoexcited separation. However, for the other two monomers, the electrons are mainly distributed in the electron-deficient part (pyridine and pyrimidine) and the holes are spread over the carbazole units, demonstrating the efficient electron–hole separation. Transition density matrix heat map (TDM-HM) of excited monomer fragments revealed that the electron transferred from carbazole to the electron-deficient unit pyridine or pyrimidine (Fig. 5c and S26a†). Notably, the degree of electron–hole separation is higher for PyDCz compared to that of PymDCz, which could illustrate the superior photocatalytic performance of CMP-CSU14 over CMP-CSU15. Since the nitrogen atom (N5) only distributes electrons in the electron deficient part (pyridine) in the excited state (Fig. S26b†), we inferred that the nitrogen atom could also act as an electronic output site of CMP-CSU14 for transferring the electrons to the substrate.
Conclusions
In summary, we have demonstrated a structural design of conjugated microporous polymer catalysts based on regulating the dipole moment of the starting monomers or precursors. Photophysical characterization as well as DFT calculations revealed that the polarization induced internal electric field has a crucial impact on the photocatalytic activity of the obtained networks. Remarkably, the pyridine-bridged CMP-CSU14 was found to be a promising heterogeneous photocatalyst with the highest catalytic performance for the C-3 selenocyanation of indoles, being an attractive alternative to traditional metal-based catalysts. The high photocatalytic efficiency of CMP-CSU14 could be attributed to the effective separation of the photo-induced electron–hole pairs and the stronger generation ability of active oxygen species. This study also provides a possible strategy to precisely tailor the optoelectronic properties of CMPs, which not only offers new insight into developing high performance organic photocatalysts, but also opens the gate for extending the ideas to other photocatalytic organic transformations.Experimental
Chemicals
2,5-Dibromopyridine, 2,5-dibromopyrimidine, 1,4-dibromobenzene, 9H-carbazole, copper(I) iodide and 1,10-phenanthroline were purchased from Aldrich and used without purification. Nitrobenzene (PhNO2) and anhydrous ferric chloride (FeCl3) were obtained from Aladdin Industrial Corporation (Shanghai, China). Other solvents such as dichloromethane (DCM), petroleum ether (PE), ammonia solution and tetrahydrofuran were obtained from commercial sources and used without further purification. The purification of chloroform (CHCl3) was conducted by distillation over calcium hydride (CaH2) and stored over 4A molecular sieves prior to utilization.General procedure for C-3 selenocyanation of indoles based on CMP-CSU14
Indoles (0.2 mmol), potassium selenocyanate (KSeCN) (0.4 mmol, 2 equiv.), tetrahydrofuran (2 mL) and CMP-CSU14 (10 mg) were transferred to a transparent reaction tube. The reaction mixture was degassed and protected with an O2 balloon (∼0.1 MPa), and then stirred under irradiation with a light source (a 14 W blue LED, 0.20 W cm−2, distance app. 8 cm) for 24 hours. After completion of the reactions, the corresponding yields were determined with an internal standard (mesitylene). The obtained residue was purified by column chromatography on silica gel to obtain the target product (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM = 10:1–1:2).
:DCM = 10:1–1:2).
Author contributions
Guipeng Yu, Chunyue Pan, and Juntao Tang proposed the idea and designed the experiments. Qiujian Xie performed the experiments and carried out the structural characterization. Weijie Zhang helped to analyse the results of photocatalytic experiments. Zhu Gao helped with the structural characterization analysis. Xiaofeng Li helped to synthesize the materials. Yumin Yang performed the DFT calculations. Qiujian Xie, Juntao Tang, and Guipeng Yu wrote the manuscript with contributions from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 21674129, 51873232 and 21636010). We also acknowledge the NMR measurements by the Modern Analysis and Testing Centre of Central South University.Notes and references
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357–4416 CrossRef CAS PubMed.
- A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed.
- T. Jensen, H. Pedersen, B. Bang-Andersen, R. Madsen and M. Jørgensen, Angew. Chem., Int. Ed., 2008, 47, 888–890 CrossRef CAS PubMed.
- G. Mugesh, W. W. Du Mont and H. Sies, Chem. Rev., 2001, 101, 2125–2179 CrossRef CAS PubMed.
- C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255–6285 CrossRef CAS PubMed.
- E. E. Aynsley, N. N. Greenwood and M. J. Sprague, J. Chem. Soc., 1965, 2395–2402 RSC.
- A. Krief, W. Dumont and C. Delmotte, Angew. Chem., Int. Ed., 2000, 39, 1669–1672 CrossRef CAS PubMed.
- G. W. Parshall, J. Am. Chem. Soc., 1966, 88, 704–708 CrossRef CAS.
- H. Rheinboldt and E. Giesbrecht, J. Am. Chem. Soc., 1949, 71, 1740–1741 CrossRef CAS.
- W. Liu, S.-D. Jiang, Y. Yan, W. Wang, J. Li, K. Leng, S. Japip, J. Liu, H. Xu, Y. Liu, I.-H. Park, Y. Bao, W. Yu, M. D. Guiver, S. Zhang and K. P. Loh, Nat. Commun., 2020, 11, 1633 CrossRef PubMed.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- G. Mukherjee, J. Thote, H. B. Aiyappa, S. Kandambeth, S. Banerjee, K. Vanka and R. Banerjee, Chem. Commun., 2017, 53, 4461–4464 RSC.
- J.-X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- C. M. Aitchison, M. Sachs, M. A. Little, L. Wilbraham, N. J. Brownbill, C. M. Kane, F. Blanc, M. A. Zwijnenburg, J. R. Durrant, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 8744–8756 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J. X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS.
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, Adv. Mater., 2020, 32, 1–6 Search PubMed.
- M. Liras, M. Pintado-Sierra, M. Iglesias and F. Sánchez, J. Mater. Chem. A, 2016, 4, 17274–17278 RSC.
- J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS.
- D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC.
- B. Grimm, C. Risko, J. D. Azoulay, J.-L. Brédas and G. C. Bazan, Chem. Sci., 2013, 4, 1807–1819 RSC.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181–191 RSC.
- Z. A. Lan, W. Ren, X. Chen, Y. Zhang and X. Wang, Appl. Catal., B, 2019, 245, 596–603 CrossRef CAS.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J. X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- W. Zhang, J. Tang, W. Yu, Q. Huang, Y. Fu, G. Kuang, C. Pan and G. Yu, ACS Catal., 2018, 8, 8084–8091 CrossRef CAS.
- Q. Chen, D. P. Liu, J. H. Zhu and B. H. Han, Macromolecules, 2014, 47, 5926–5931 CrossRef CAS.
- C. Gu, Y. Chen, Z. Zhang, S. Xue, S. Sun, K. Zhang, C. Zhong, H. Zhang, Y. Pan, Y. Lv, Y. Yang, F. Li, S. Zhang, F. Huang and Y. Ma, Adv. Mater., 2013, 25, 3443–3448 CrossRef CAS PubMed.
- J. Luo, X. Zhang, J. Lu and J. Zhang, ACS Catal., 2017, 7, 5062–5070 CrossRef CAS.
- Y. Yuan, H. Huang, L. Chen and Y. Chen, Macromolecules, 2017, 50, 4993–5003 CrossRef CAS.
- R. R. Zhang, Q. Yin, H. P. Liang, Q. Chen, W. H. Luo and B. H. Han, Polymer, 2018, 143, 87–95 CrossRef CAS.
- Y. Zhi, S. Ma, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, Appl. Catal., B, 2019, 244, 36–44 CrossRef CAS.
- C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef CAS.
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590–596 CrossRef CAS PubMed.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- R. Li, Z. J. Wang, L. Wang, B. C. Ma, S. Ghasimi, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2016, 6, 1113–1121 CrossRef CAS.
- L. Cui, J. Song, A. F. McGuire, S. Kang, X. Fang, J. Wang, C. Yin, X. Li, Y. Wang and B. Cui, ACS Nano, 2018, 12, 5551–5558 CrossRef CAS PubMed.
- D. Ma, J. W. Shi, Y. Zou, Z. Fan, X. Ji, C. Niu and L. Wang, Nano Energy, 2017, 39, 183–191 CrossRef CAS.
- X. Men, H. Chen, K. Chang, X. Fang, C. Wu, W. Qin and S. Yin, Appl. Catal., B, 2016, 187, 367–374 CrossRef CAS.
- W. Zhang, S. Li, X. Tang, J. Tang, C. Pan and G. Yu, Appl. Catal., B, 2020, 272, 118982 CrossRef CAS.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- C. Xu, Y. Zhang, F. Pan, W. Huang, B. Deng, J. Liu, Z. Wang, M. Ni and K. Cen, Nano Energy, 2017, 41, 308–319 CrossRef CAS.
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed.
- F. Chen, H. Huang, L. Guo, Y. Zhang and T. Ma, Angew. Chem., Int. Ed., 2019, 58, 10061–10073 CrossRef CAS PubMed.
- H. Huang, S. Tu, X. Du and Y. Zhang, J. Colloid Interface Sci., 2018, 509, 113–122 CrossRef CAS PubMed.
- J. D. Smith, A. M. Jamhawi, J. B. Jasinski, F. Gallou, J. Ge, R. Advincula, J. Liu and S. Handa, Nat. Commun., 2019, 10, 1837 CrossRef PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- X. Mu, J. Wang and M. Sun, J. Phys. Chem. C, 2019, 123, 14132–14143 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06951e |
| This journal is © The Royal Society of Chemistry 2021 |