
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the disulfide bond formation enzymes in epidithiodiketopiperazine alkaloids†
Huan
Liu‡
a,
Jie
Fan‡
 a,
Peng
Zhang
a,
Youcai
Hu
d,
Xingzhong
Liu
a,
Shu-Ming
Li
*b and
Wen-Bing
Yin
*ac
a,
Peng
Zhang
a,
Youcai
Hu
d,
Xingzhong
Liu
a,
Shu-Ming
Li
*b and
Wen-Bing
Yin
*ac
aState Key Laboratory of Mycology, CAS Key Laboratory of Microbial Physiological and Metabolic Engineering, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, P. R. China. E-mail: yinwb@im.ac.cn
bInstitut für Pharmazeutische Biologie und Biotechnologie, Fachbereich Pharmazie, Philipps-Universität Marburg, Robert-Koch-Straße 4, Marburg 35037, Germany. E-mail: shuming.li@staff.uni-marburg.de
cSavaid Medical School, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
dState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, P. R. China
First published on 8th February 2021
Abstract
Epidithiodiketopiperazines (ETPs) are a group of bioactive fungal natural products and structurally feature unique transannular disulfide bridges between α, α or α, β carbons. However, no enzyme has yet been demonstrated to catalyse α, β-disulfide bond formation in these molecules. Through genome mining and gene deletion approaches in Trichoderma hypoxylon, we identified a putative biosynthetic gene cluster of pretrichodermamide A (1), which requires a FAD-dependent oxidoreductase, TdaR, for the irregular α, β-disulfide formation in 1 biosynthesis. In vitro assays of TdaR, together with AclT involved in aspirochlorine and GliT involved in gliotoxin biosynthesis, proved that all three enzymes catalyse not only the conversion of red-pretrichodermamide A (4) to α, β-disulfide-containing 1 but also that of red-gliotoxin (5) to α, α-disulfide-containing gliotoxin (6). These results provide new insights into the thiol-disulfide oxidases responsible for the disulfide bond formation in natural products with significant substrate and catalytic promiscuities.
Introduction
Natural products containing sulfur display remarkable structural diversity and have a wide variety of biological activities.1,2 A special subgroup found in fungi includes epidithiodiketopiperazines (ETPs) featuring a cyclodipeptide (CDP) scaffold, which is decorated by a transannular disulfide bridge. They exhibit a broad range of bioactivities including anticancer, antibacterial, antifungal, antiviral, immunosuppressive and cytotoxic activities.1,3,4 Typically, structural diversity of ETPs possessing different core CDPs is generated by nonribosomal peptide synthetases (NRPSs) and multiple post-assembly modifications.5 Commonly derived from phenylalanine-, tyrosine- and tryptophan-containing CDPs,1 two subtypes of bioactive ETPs carrying disulfide bridges across α, α- or α, β-positions of the diketopiperazine (DKP) skeleton have been identified in diverse fungal species (Fig. 1). Representatively, gliotoxin,6–9 sirodesmin PL,10–14 acetylaranotin,15 dithiosilvatin,16 sporidesmin7,17 and epicorazine18 bear a disulfide bridge across the α and α positions of the DKP skeleton. In contrast, pretrichodermamide A (1),19 gliovirin,20,21 adametizine A,22,23 aspirochlorine as well as its biosynthetic intermediate24,25 and lasiodipline D26 carry an irregular disulfide bridge across corresponding α and β positions (Fig. 1). To date, these unique structures have captured the attention of scientists for their total synthesis27,28 and biosynthesis.1,9,12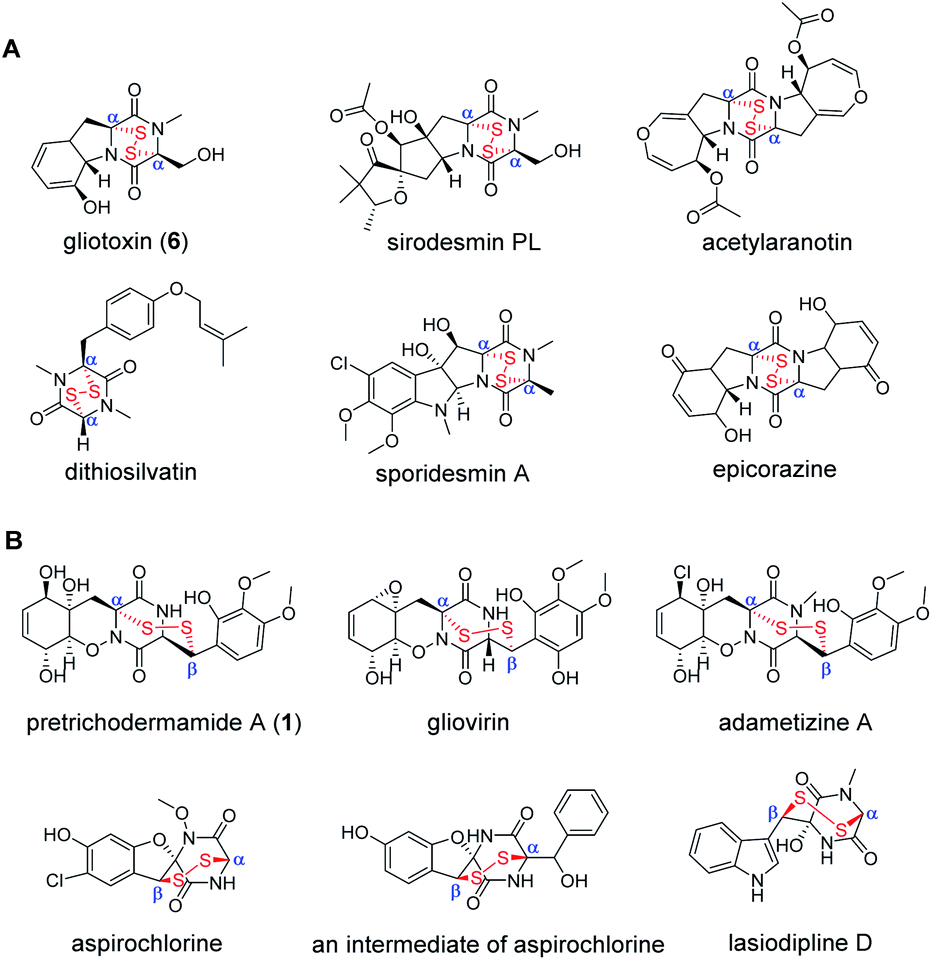 | ||
| Fig. 1 Representative fungal ETPs with an α, α- (A) or α, β- (B) disulfide bond. | ||
Previous studies have elucidated many aspects of the molecular mechanism involved in the biosynthesis of gliotoxin from Aspergillus fumigatus, including cyclo-L-Phe-L-Ser generation by an NRPS enzyme GliP, hydroxylation by cytochrome P450, sulfur moiety incorporation by the addition of glutathione (GSH) and its sequential degradation to the dithiol precursor.5,29,30 A flavin adenine dinucleotide (FAD)-dependent oxidoreductase GliT is responsible for the oxidation of the dithiol group yielding the α, α disulfide-containing gliotoxin.31,32 In addition, biosynthesis of several ETPs including sirodesmin PL from Leptosphaeria maculans and acetylaranotin from Aspergillus terreus was extensively studied by multiple genetic and biochemical approaches.12–15 The halogenated aspirochlorine was also investigated with respect to its crucial roles in the formation of a cyclo-L-Phe-Gly containing ETP from cyclo-L-Phe-L-Phe.24 Although the interesting α, β-disulfide bridge formation was speculated to be catalysed by a GliT homolog, AclT, no experimental evidence was provided.
To identify the enzyme involved in α, β-disulfide bridge formation, we focused on 1 reported in Trichoderma sp. BCC5926, which exhibits antibacterial activity towards Mycobacterium tuberculosis H37Ra.19 The typical α, β-disulfide bridge and 1,2-oxazadecaline moiety of 1 as well as its derivatives have shown to be essential for the observed biological activities.21,22 A biosynthetic route was proposed for gliovirin, a derivative of 1, in Trichoderma virens, based on genome mining and core gene deletion experiments.21 It was speculated that the FAD-dependent oxidoreductase Glv4 or Glv16 was possibly involved in the α, β-disulfide bridge formation. However, data for neither its detailed biosynthesis nor for the α, β-disulfide formation was reported. In this study, we identified 1 from a fungicolous fungus Trichoderma hypoxylon and proposed a possible biosynthetic pathway by genome mining and deletion of two targeted genes. Biochemical investigations proved that a FAD-dependent oxidoreductase, TdaR, was responsible for the α, β-disulfide formation in the biosynthesis of 1. Further in vitro assays demonstrated that three oxidases TdaR, AclT and GliT from different pathways catalysed both α, α- and α, β-disulfide bond formation in the biosynthesis of fungal ETP alkaloids.
Results
Identification of the biosynthetic gene cluster for pretrichodermamide A
To identify the ETP 1, we examined the secondary metabolites of previously reported fungus T. hypoxylon CGMCC 3.17906, because several ETP compounds including trichodermamide A (2), aspergillazine A and aspergillazine C have been identified by our group.33 Spontaneous conversion of 1 to 2 was demonstrated by Seephonkai et al. due to the instability of 1 under alkaline conditions or even in methanol.19 Therefore, we believe that 1 should exist in this strain under controlled culture and extraction conditions. Herein, T. hypoxylon was cultivated in rice medium and the ethyl acetate extracts were analysed on HPLC (Fig. S1 in the ESI†). As expected, one predominant peak with an [M + H]+ ion at m/z 499.0845 was detected in a 14 day-old rice culture, which was subsequently identified as 1 after isolation and structure elucidation by comparing to reported data (Fig. S2, S3 and Table S3†).19 The spontaneous conversion of 1 to 2 was observed, even under cultivation conditions and during isolation (data not shown). This result indicated that 1 was the natural ETP metabolite in T. hypoxylon.To determine the biosynthetic pathway of 1, the genome of T. hypoxylon was sequenced and the draft genome sequence was used for the prediction of putative gene clusters by using AntiSMASH.34 Based on its ETP structural character, 1 should be derived from cyclo-L-Phe-L-Phe (3), which is expected to be assembled by an NRPS.35 One of the eleven NRPS genes T_hypo_11188, termed tdaA, within a large 49.6 kbp cluster (Fig. 2A and Table S4†), contains a T–C–A–T–C domain structure, being similar to the core enzymes AtaP15 and AclP24 in phenylalanine-containing ETP biosynthesis (abbreviations for NRPS domains are as given before35). TdaA shares a sequence similarity of 31% with GliP9 from A. fumigatus and 88% with Glv21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 from T. virens on the amino acid level (Table S4†). To prove its function, tdaA was deleted from the genome by PEG-mediated protoplast transformation using hygromycin B as a selection marker.36 LC-MS analysis revealed complete disappearance of ETP products 1 and 2, as well as the NRPS product 3, indicating that TdaA acts as the core enzyme for the biosynthesis of 1 and 2 (Fig. 2B). To find the candidate gene for the α, β-disulfide bridge formation in 1, further blast analysis was performed using the disulfide bond formation enzyme GliT in the gliotoxin biosynthetic pathway as a probe.32 Two putative FAD-dependent oxidoreductases TdaE (T_hypo_11193) and TdaR (T_hypo_11206) are located in the pretrichodermamide A (tda) cluster. Alignments with known oxidases for thiol-disulfide formation showed the existence of a conserved active binding site CLFC (CXXC) box in TdaR but not in TdaE (Fig. S4†). Therefore, we performed tdaR deletion according to the aforementioned method for tdaA, leading to complete disappearance of 1 and 2, but accumulation of 3 as indicated by the analysis of extracted ion chromatograms (EICs) (Fig. 2B). This proved the involvement of TdaR in the conversion of 3 to 1 and 2. Inspection of the EICs of the culture extracts and structure analysis implied more conversion steps between 3 and 1. Thus, a biosynthetic pathway of 1 and 2 was proposed as shown in Fig. 2C. Briefly, the DKP core is assembled by NRPS TdaA (i), followed by hydroxylations at α and β positions by a putative cytochrome P450 TdaS (ii). After sulfur incorporation and sequential degradation (iii), TdaR catalyses the α, β-disulfide bond formation (iv). Subsequently, further hydroxylations catalysed by multiple cytochrome P450s and methylations catalysed by different methyltransferases (MeTs) lead to the formation of 1 with a 1,2-oxazadecaline moiety and methoxy groups (v). Lastly, spontaneous degradation results in the transformation of 1 to 2 (vi).
21 from T. virens on the amino acid level (Table S4†). To prove its function, tdaA was deleted from the genome by PEG-mediated protoplast transformation using hygromycin B as a selection marker.36 LC-MS analysis revealed complete disappearance of ETP products 1 and 2, as well as the NRPS product 3, indicating that TdaA acts as the core enzyme for the biosynthesis of 1 and 2 (Fig. 2B). To find the candidate gene for the α, β-disulfide bridge formation in 1, further blast analysis was performed using the disulfide bond formation enzyme GliT in the gliotoxin biosynthetic pathway as a probe.32 Two putative FAD-dependent oxidoreductases TdaE (T_hypo_11193) and TdaR (T_hypo_11206) are located in the pretrichodermamide A (tda) cluster. Alignments with known oxidases for thiol-disulfide formation showed the existence of a conserved active binding site CLFC (CXXC) box in TdaR but not in TdaE (Fig. S4†). Therefore, we performed tdaR deletion according to the aforementioned method for tdaA, leading to complete disappearance of 1 and 2, but accumulation of 3 as indicated by the analysis of extracted ion chromatograms (EICs) (Fig. 2B). This proved the involvement of TdaR in the conversion of 3 to 1 and 2. Inspection of the EICs of the culture extracts and structure analysis implied more conversion steps between 3 and 1. Thus, a biosynthetic pathway of 1 and 2 was proposed as shown in Fig. 2C. Briefly, the DKP core is assembled by NRPS TdaA (i), followed by hydroxylations at α and β positions by a putative cytochrome P450 TdaS (ii). After sulfur incorporation and sequential degradation (iii), TdaR catalyses the α, β-disulfide bond formation (iv). Subsequently, further hydroxylations catalysed by multiple cytochrome P450s and methylations catalysed by different methyltransferases (MeTs) lead to the formation of 1 with a 1,2-oxazadecaline moiety and methoxy groups (v). Lastly, spontaneous degradation results in the transformation of 1 to 2 (vi).
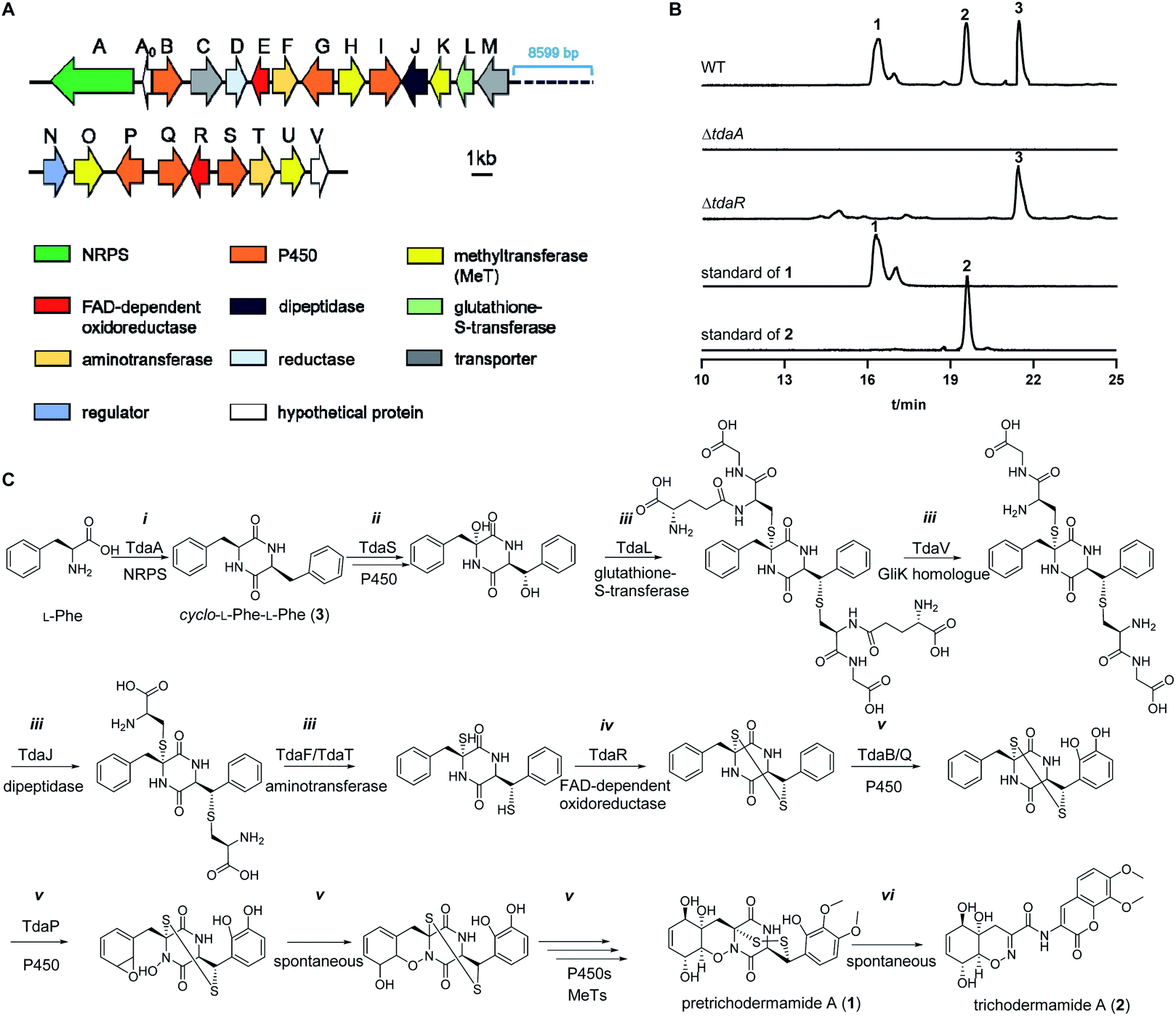 | ||
| Fig. 2 Pretrichodermamide A (1) biosynthetic gene cluster (A), LC-MS analysis of T. hypoxylon strains (B) and the proposed biosynthetic pathway for 1 (C). | ||
TdaR responsible for α, β-disulfide bond formation
To address whether TdaR is involved in the α, β-disulfide bond formation, we performed in vitro biochemical characterisation. Previous studies reported that disulfide bond formation was catalysed by FAD-dependent oxidoreductases from different bacterial and fungal sources.37 Hence, a sequence similarity network of TdaR homologs including 370 FAD-dependent oxidoreductases was generated which led to the identification of four distinct clades at an e-value threshold of 80 (Fig. 3 and S5†).38 TdaR belongs to clade IV which includes GliT and 81 other not yet fully identified fungal enzymes possibly from ETP biosynthetic pathways. Prototypes of the other three clades are DepH (clade I),39 HlmI (clade II)40 and TrxR (clade III)41 from Chromobacterium violaceum, Streptomyces clavuligerus and Escherichia coli, respectively. Interestingly, proteins from clade I, II and IV presumably oxidize small molecules yielding disulfide bonds, while clade III probably acts as reductases for intracellular redox regulation.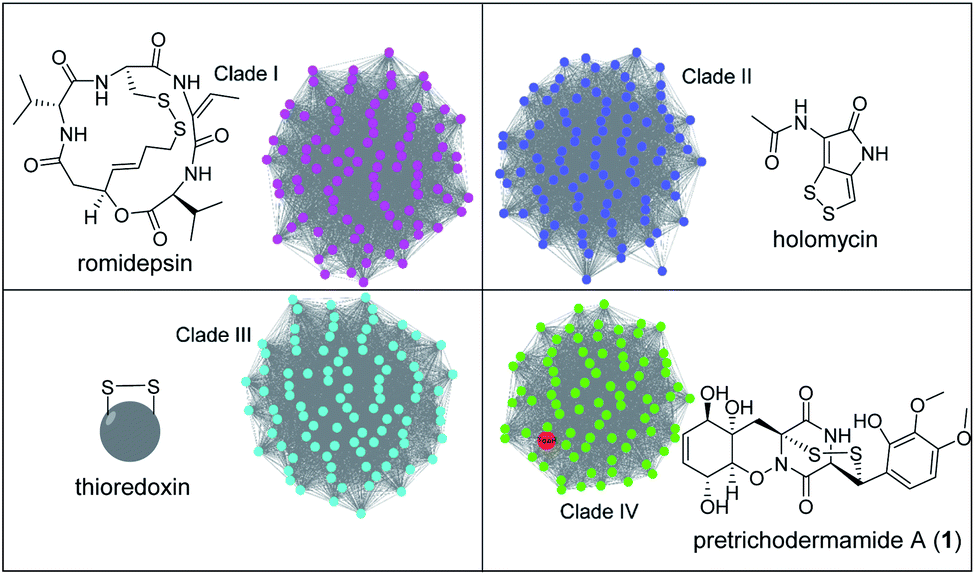 | ||
| Fig. 3 Sequence clustering and phylogenetic analysis of FAD-dependent oxidoreductases involved in thiol-disulfide formation. Four distinct clades are shown in different colors with their representative sulfur-containing products. Romidepsin and holomycin were formed by DepH (clade I) and HlmI (clade II), respectively. TrxR (clade III) is a thioredoxin reductase responsible for the reduction of disulfide. TdaR, together with GliT (involved in gliotoxin biosynthesis) and AclT (involved in aspirochlorine biosynthesis), was located in clade IV with other potential fungal thiol-disulfide oxidases. Amino acid sequences were obtained from the GenBank database. | ||
To investigate its function, the 933 bp long coding sequence of tdaR comprising three exons was cloned from cDNA of T. hypoxylon and overexpressed in E. coli BL21(DE3) cells. The recombinant His6-tagged protein was purified with the aid of Ni-NTA agarose resin to near homogeneity which was confirmed on SDS-PAGE (Fig. 4A), yielding 5.1 mg of purified TdaR per liter of bacterial culture. Due to the instability of the unidentified substrate of TdaR for the α, β-disulfide bond formation, we proposed that this enzyme could also catalyse the conversion of reduced pretrichodermamide A (red-pretrichodermamide A, 4) to 1. Therefore, 1 was chemically reduced to 4 using dithiothreitol (DTT) to provide an appropriate substrate for the enzyme assays (Fig. S6†). HPLC analysis revealed a complete conversion of 0.5 mM 1 to 4 by 1 mM DTT at 30 °C for 30 min (Fig. 5A and S6†). Subsequently, this mixture was directly incubated with 0.1 μM purified TdaR at 37 °C for 30 min. As expected, the enzyme assay led to the formation of 1 which was verified by comparing the retention time with that of an authentic standard (Fig. 5A(i, ii and viiii)). This proved that the FAD-dependent oxidoreductase TdaR was responsible for the α, β-disulfide bond formation in the biosynthesis of 1. Furthermore, the time course of this reaction was examined and monitored by HPLC, indicating that 17% of 0.5 mM 4 was consumed by 0.1 μM TdaR in 5 min (Fig. S7†). However, no conversion of 4 could be observed with heat-denatured TdaR or without O2 in the glove box (Fig. 5A(i and iii)). This indicated a mechanism of α, β-disulfide bond formation similar to GliT-catalysed α, α-disulfide bond formation, which required O2. Accordingly, we proposed that initial transformation occurred in TdaR from the oxidized form (disulfide) to reduced form (dithiol) by the putative CLFC redox system. Subsequently, electron pairs were transferred to FADox from the active site of TdaR. FADred is then reoxidized by O2 to form H2O2, leading to the concomitant production of the disulfide-containing product (Fig. 4B and 6).
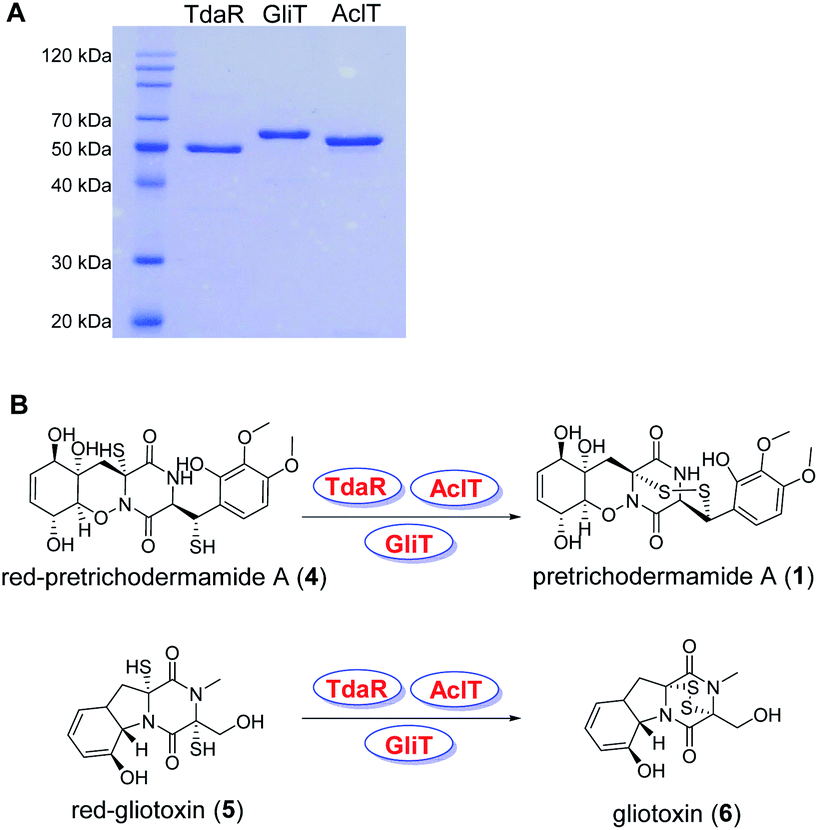 | ||
| Fig. 4 Analysis of TdaR, AclT and GliT on SDS-PAGE (A) and enzyme-mediated disulfide bond formation in the biosynthesis of 1 and 6 (B). | ||
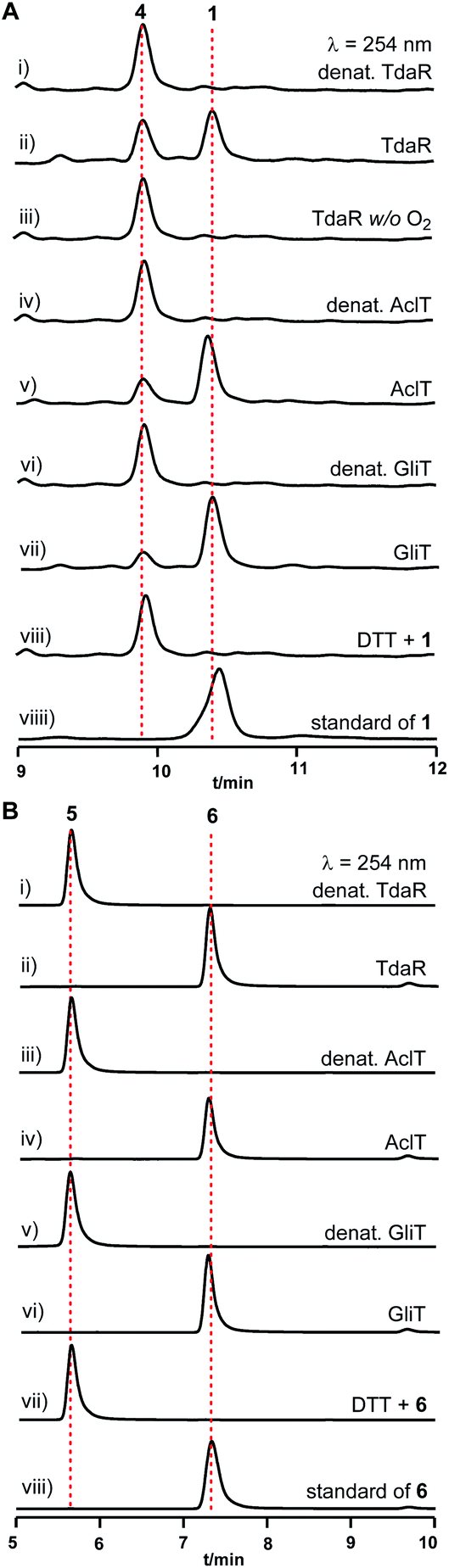 | ||
| Fig. 5 HPLC analysis of enzyme assays of TdaR, AclT and GliT with 4 (A) or 5 (B). UV absorption at 254 nm was illustrated. | ||
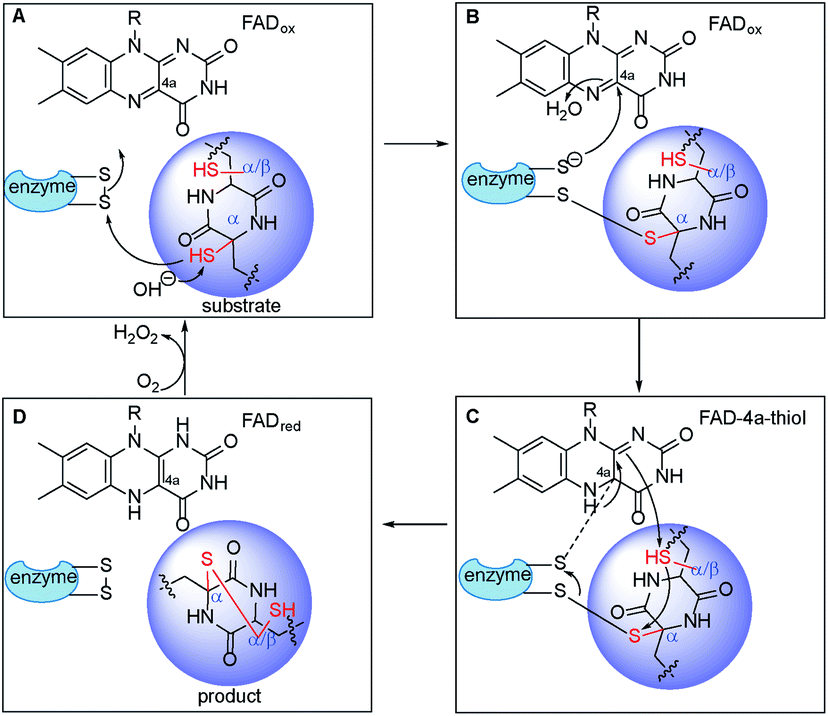 | ||
| Fig. 6 Proposed mechanism of disulfide bond formation in ETP alkaloids by FAD-dependent thiol-disulfide oxidases. A transient mixed-disulfide bond was generated between substrate and enzyme (A and B) leading to a FAD-4a-thiol adduct (C). Subsequently, the disulfide in the ETP alkaloid was formed with concomitant reduction of FAD (D). | ||
To directly confirm the oxidation of TdaR on 4, 4 was purified from the mixture of 1 with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) as a reducing agent on a semi-preparative HPLC (acetonitrile/H2O, 35:65) (Fig. S9†). Incubation of TdaR with the purified 4 revealed a complete conversion of 4 to 1 after incubation at 37 °C for 30 min (Fig. S10A†). However, the formation of 1 was also observed in the negative control containing heat-denatured TdaR and 4, demonstrating the spontaneous disulfide formation. In comparison, disulfide formation in 1 catalysed by TdaR was more efficient than the spontaneous oxidation.
Fungal thiol-disulfide oxidases catalysed both α, α- and α, β-disulfide bond formation
In addition to TdaR, AclT was also predicted to catalyse an α, β-disulfide bond formation in the biosynthesis of aspirochlorine in Aspergillus oryzae.24 However, no detailed investigation on AclT has been reported until now. We presumed that AclT could also catalyse the α, β-disulfide bond formation in the biosynthesis of 1. Therefore, the aclT ORF (945 bp) was amplified from A. oryzae cDNA and cloned into an E. coli expression vector pET28a(+)SUMO. The resulted plasmid pYHL87 was introduced into BL21(DE3) cells for protein overproduction, leading to 11.0 mg of purified AclT per liter of bacterial culture. It showed a yellow color and was confirmed on SDS-PAGE with the predicted size of 50.4 kDa (Fig. 4A). Subsequently, incubation of 4 with 0.1 μM AclT was carried out in a similar way to TdaR. HPLC analysis revealed 1 as the enzymatic product, proving AclT-catalysed α, β-disulfide bond formation (Fig. 5A(iv and v)). Further time course of AclT-catalysed reaction revealed that increasing amounts (18–72%) of compound 4 were consumed by 0.1 μM AclT from 5 min to 30 min (Fig. S7†). In addition, incubation of heat-denatured AclT with the purified 4 also revealed that a trace amount of 1 is formed spontaneously (Fig. S10A†). This was consistent with the enzyme assay of TdaR with 4, indicating that AclT and TdaR performed the same function in the α, β-disulfide bond formation.Notably, bioinformatics study showed that TdaR and AclT involved in the α, β-disulfide bond formation were located in clade IV with several known α, α-disulfide bond-forming enzymes including GliT,9,32 AtaTC15 and SirT12 (Fig. 3 and S5†). The close phylogenetic relationship triggered our interest to test if TdaR and AclT could also catalyse α, α-disulfide bond formation. Thus, reduced gliotoxin (red-gliotoxin, 5) was acquired by co-incubation of gliotoxin (6) with DTT at 30 °C for 30 min or purified from a mixture containing 6 and TCEP (Fig. S6 and S9†). Interestingly, we observed that 5 (0.5 mM) was readily converted into 6 in the presence of TdaR (0.1 μM) or AclT (0.1 μM) after 5 min (Fig. S8†). 5 was completely transformed to 6 in 30 min by both TdaR and AclT (Fig. 5B, S8 and S10B†). These results supported our hypothesis that TdaR and AclT catalyse not only α, β- but also α, α-disulfide bond formation in the biosynthesis of ETP alkaloids. In addition, TdaR and AclT-catalysed α, α-disulfide bond formation with 5 as the substrate was more efficient than the α, β-disulfide bond formation.
In analogy, the coding sequence of GliT, which was proved to catalyse the α, α-disulfide bond formation in the biosynthesis of 6, was cloned into pET28a(+)SUMO and overexpressed in E. coli BL21(DE3). 11.2 mg purified GliT was obtained in a yellow color from 1 liter of bacterial culture and subsequently incubated with 4 and 5 in a similar way to TdaR and AclT. In accordance with the previous study, GliT acts as a FAD-dependent oxidase for the α, α-disulfide bond formation to generate 6 from 5 after 5 min incubation (Fig. 5B and S8†).32 Unprecedentedly, incubation of 4 with GliT also led to the formation of 1, as demonstrated by HPLC analysis (Fig. 5A(vi and vii)). This confirmed that the known α, α-disulfide bond related enzyme GliT also catalysed the α, β-disulfide bond formation. The time course of GliT-catalysed oxidation of 4 was monitored by HPLC, revealing that an increasing amount of 4 ranging from 28–82% was consumed by 0.1 μM GliT from 5 min to 30 min (Fig. S7†).
Discussion
As a ubiquitously distributed element, sulfur is essential for mediating amino acid, peptide, and RNA synthesis in primary metabolites and secondary metabolites.42–44 There are various examples of natural products containing sulfur atoms as key units for biological functions. In the past decades, ETPs have garnered considerable attention regarding their biosynthesis, chemical synthesis and biological application, because the disulfide group contributes promising activities probably by promoting redox cycling, formation of reactive oxygen species and protein conjugation.45,46 Mostly, the sulfur bridge functionality of ETPs is attached to the α, α-positions or α, β-positions of the DKP core in fungi. Significant progress in understanding the biosynthetic pathway of α, α-disulfide-containing 6 has been achieved through genetic and biochemical approaches.5,29,30,32,37 However, fungal α, β-disulfide-containing metabolites have been rarely investigated with respect to their biosynthetic mechanism. Although gene clusters coding for the biosynthesis of α, β-disulfide-containing aspirochlorine and gliovirin have been predicted, the required biochemical functions have remained elusive.21,24In this study, we identified an α, β-disulfide-containing metabolite 1, together with its spontaneous desulfurized product 2, in T. hypoxylon. Genome mining led to the identification of a tda cluster encoding analogues of all aforementioned gliotoxin (gli) cluster enzymes and five additional cytochrome P450s as well as three methyltransferases (Fig. 2A and Table S4†).5 Deletion of the NRPS coding gene tdaA and the FAD-dependent oxidase gene tdaR proved their involvement in the biosynthesis of 1 and 2 from the CDP 3 (Fig. 2B and C). Plausibly, dihydroxylation of 3, α, β-disulfide formation and subsequent hydroxylations as well as methylations will yield the ETP alkaloid 1. Further biochemical investigation of TdaR provided evidence for its involvement in the metabolism of 3 to 1 and proved its crucial role in the formation of the α, β-disulfide bridge (Fig. 4B). Thus, our results first represent an example for understanding the biosynthesis of α, β-disulfide-containing ETP alkaloids, especially the α, β-disulfide formation. Other pretrichodermamide A-like compounds derived from cyclo-L-Phe-L-Phe, including pretrichodermamides,22 gliovirin,21 FA2097,47 aspergillazines,48 peniciadametizines49 and aspirochlorine,24 are very likely also formed in a similar way regarding the installation of the typical α, β-disulfide and a 1,2-oxazadecaline moiety. Therefore, further attempts to mine α, β-disulfide related enzymes would provide evidence for our hypothesis.
Previous study on aspirochlorine biosynthesis has already implied a putative α, β-disulfide related oxidase AclT.24 Herein, we proved that AclT is an analogous oxidase of TdaR catalysing the α, β-disulfide formation. Further biochemical investigations on TdaR and AclT revealed, interestingly, their dual function of catalysing not only the α, β-disulfide formation in 1 but also the α, α-disulfide formation in 6 (Fig. 4B and 5). These results indicated that α, β-disulfide related enzymes also catalysed α, α-disulfide formation, and vice versa. Indeed, enzyme assays confirmed that the α, α-disulfide related GliT was also able to catalyse α, β-disulfide formation in 1. It can be deduced that hydroxylations on the CDP core catalysed by P450 enzymes determine the positions of the two hydroxyl groups and finally the type of the disulfide bond (α, α or α, β).
Taken together, TdaR, AclT and GliT are dual oxidases independently forming the α, α- or α, β-disulfide bridge in fungal ETP biosynthesis. It might well explain why most fungal related FAD-dependent oxidases were located in the same clade IV (Fig. 3), no matter they naturally catalyse α, α- or α, β-disulfide formation. In the phylogenetic analysis of TdaR homologs, the unique clade III, exemplified by TrxR from E. coli, catalysed the reduction of a disulfide bond in thioredoxin.41 In contrast to DepH (clade I) from C. violaceum and HlmI (clade II) from S. clavuligerus involved in intra- or interchenar disulfide bonds, TdaR, AclT and GliT (clade IV) catalysed transannular disulfide bond formation.39,40 They shared a catalytically active CLFC box for redox activation, but different FAD binding sites, i.e. His139 of TdaR and AclT, but Asp139 in GliT (Fig. S4†). Plausibly in analogy to GliT-catalysed disulfide bond formation, fungal oxidases utilized a similar mechanism to install disulfide bonds across α, α- or α, β-positions (Fig. 6).32 A sulfhydryl group in the DKP-containing substrate attacks a disulfide bond in the thioredoxin motif (CXXC) within the oxidoreductase, creating a transient mixed-disulfide bond between the substrate and oxidoreductase (Fig. 6A and B). In this step, FADox receives the electrons at the C4a position, leading to a FAD-4a-thiol adduct. Subsequently, it undergoes attack at the other sulfhydryl group with disulfide formation in the ETP alkaloid and concomitant reduction to FADred (Fig. 6C and D). Eventually, FADred is reoxidized by molecular oxygen instead of NADP+ with the elimination of H2O2. We speculated that the large pocket across the active site and FAD-binding motif of GliT as well as TdaR and AclT50 might result in the broad substrate specificity, making it attractive for further enzyme structure analysis and protein engineering.37
Conclusions
In conclusion, a biosynthetic pathway of pretrichodermamide A (1) was proposed by genetic manipulation in T. hypoxylon and further chemical extract analysis. In vitro biochemical investigations first demonstrated that TdaR and AclT, which are responsible for the α, β-disulfide bond formation in 1 biosynthesis, also catalyse the α, α-disulfide bond formation in 6. Surprisingly, GliT involved in the α, α-disulfide bond formation was proved to catalyse α, β-disulfide bond formation as well. Despite their unrelated fungal sources and different natural metabolite pathways, they share obviously common catalytic ability to produce α, α- or α, β-disulfide-containing products. Our results provided representative examples of fungus-originated thiol-disulfide oxidases catalysing different types of disulfide formation in the biosynthesis of ETP alkaloids. It can be speculated that in the near future more fungus-originated thiol-disulfide oxidases would be discovered which catalyse both α, α- and α, β-disulfide formation, even exhibiting wider specificities towards more substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Rixa Kraut (University of Marburg) for performing LC-MS, and Zhen Cai as well as Xuebing Li (Institute of Microbiology, CAS) for supplying the conditional oxygen-free environment. We also thank the Center for Ocean Mega-Science, Chinese Academy of Sciences. This work was supported by the National Key Research and Development Program of China (2018YFA0900701), National Natural Science Foundation of China (21778075), the Senior User Project of RV KEXUE (KEXUE2019GZ05), Key Research Program of Frontier Sciences, CAS (ZDBS-LY-SM016), Construction of the Registry and Database of Bioparts for Synthetic Biology, CAS (ZSYS-016), the PPP Programmes for Project Related Personal Exchange between the National Natural Science Foundation of China and the German Academic Exchange Service as well as the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, INST 160/620-1).Notes and references
- D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005, 151, 1021–1032 CrossRef CAS.
- T. R. Welch and R. M. Williams, Nat. Prod. Rep., 2014, 31, 1376–1404 RSC.
- K. M. Cook, S. T. Hilton, J. Mecinovic, W. B. Motherwell, W. D. Figg and C. J. Schofield, J. Biol. Chem., 2009, 284, 26831–26838 CrossRef CAS.
- M. Zhu, X. Zhang, X. Huang, H. Wang, K. Anjum, Q. Gu, T. Zhu, G. Zhang and D. Li, J. Nat. Prod., 2020, 83, 2045–2053 CrossRef CAS.
- D. H. Scharf, T. Heinekamp, N. Remme, P. Hortschansky, A. A. Brakhage and C. Hertweck, Appl. Microbiol. Biotechnol., 2012, 93, 467–472 CrossRef CAS.
- G. W. Kirby, G. L. Patrick and D. J. Robins, J. Chem. Soc., Perkin Trans. 1, 1978, 1336–1338 RSC.
- G. W. Kirby and D. J. Robins, The biosynthesis of gliotoxin and related epipolythiodioxopiperazines, Academic Press London, 1980 Search PubMed.
- D. T. Shah and B. Larsen, Mycopathologia, 1991, 116, 203–208 CrossRef CAS.
- D. M. Gardiner and B. J. Howlett, FEMS Microbiol. Lett., 2005, 248, 241–248 CrossRef CAS.
- P. J. Curtis, D. Greatbanks, B. Hesp, A. F. Cameron and A. A. Freer, J. Chem. Soc., Perkin Trans. 1, 1977, 180–189 RSC.
- J.-P. Ferezou, A. Quesneau-Thierry, C. Servy, E. Zissmann and M. Barbier, J. Chem. Soc., Perkin Trans. 1, 1980, 1739–1746 RSC.
- D. M. Gardiner, A. J. Cozijnsen, L. M. Wilson, M. S. Pedras and B. J. Howlett, Mol. Microbiol., 2004, 53, 1307–1318 CrossRef CAS.
- E. M. Fox, D. M. Gardiner, N. P. Keller and B. J. Howlett, Fungal Genet. Biol., 2008, 45, 671–682 CrossRef CAS.
- H.-X. Zou, X. Xie, X.-D. Zheng and S.-M. Li, Appl. Microbiol. Biotechnol., 2011, 89, 1443–1451 CrossRef CAS.
- C. J. Guo, H. H. Yeh, Y. M. Chiang, J. F. Sanchez, S. L. Chang, K. S. Bruno and C. C. Wang, J. Am. Chem. Soc., 2013, 135, 7205–7213 CrossRef CAS.
- N. Kawahara, K. Nozawa, S. Nakajima and K. Kawai, J. Chem. Soc., Perkin Trans. 1, 1987, 2099–2101 RSC.
- R. L. M. Synge and E. P. White, Chem. Ind., 1959, 1546–1547 CAS.
- P. Kleinwaechter, H.-M. Dahse, U. Luhmann, B. Schlegel and K. Dornberger, J. Antibiot., Ser. B, 2001, 54, 521–525 CrossRef.
- P. Seephonkai, P. Kongsaeree, S. Prabpai, M. Isaka and Y. Thebtaranonth, Org. Lett., 2006, 8, 3073–3075 CrossRef CAS.
- R. D. Stipanovic and C. R. Howell, J. Antibiot., 1982, 35, 1326–1330 CrossRef CAS.
- P. D. Sherkhane, R. Bansal, K. Banerjee, S. Chatterjee, D. Oulkar, P. Jain, L. Rosenfelder, S. Elgavish, B. A. Horwitz and P. K. Mukherjee, ChemistrySelect, 2017, 2, 3347–3352 CrossRef CAS.
- R. S. Orfali, A. H. Aly, W. Ebrahim, M. S. Abdel-Aziz, W. E. G. Müller, W. H. Lin, G. Daletos and P. Proksch, Phytochem. Lett., 2015, 11, 168–172 CrossRef CAS.
- Y. Liu, X.-M. Li, L.-H. Meng, W.-L. Jiang, G.-M. Xu, C.-G. Huang and B.-G. Wang, J. Nat. Prod., 2015, 78, 1294–1299 CrossRef CAS.
- Y. Tsunematsu, N. Maeda, M. Yokoyama, P. Chankhamjon, K. Watanabe, K. Scherlach and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14051–14054 CrossRef CAS.
- P. Klausmeyer, T. G. McCloud, K. D. Tucker, J. H. Cardellina and R. H. Shoemaker, J. Nat. Prod., 2005, 68, 1300–1302 CrossRef CAS.
- W. Wei, N. Jiang, Y. N. Mei, Y. L. Chu, H. M. Ge, Y. C. Song, S. W. Ng and R. X. Tan, Phytochemistry, 2014, 100, 103–109 CrossRef CAS.
- J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035–3050 RSC.
- J. Kim and M. Movassaghi, Acc. Chem. Res., 2015, 48, 1159–1171 CrossRef CAS.
- D. H. Scharf, N. Remme, A. Habel, P. Chankhamjon, K. Scherlach, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2011, 133, 12322–12325 CrossRef CAS.
- D. H. Scharf, P. Chankhamjon, K. Scherlach, T. Heinekamp, K. Willing, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 11092–11095 CrossRef CAS.
- J. W. Bok, D. Chung, S. A. Balajee, K. A. Marr, D. Andes, K. F. Nielsen, J. C. Frisvad, K. A. Kirby and N. P. Keller, Infect. Immun., 2006, 74, 6761–6768 CrossRef CAS.
- D. H. Scharf, N. Remme, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 10136–10141 CrossRef CAS.
- J. Sun, Y. Pei, E. Li, W. Li, K. D. Hyde, W.-B. Yin and X. Liu, Sci. Rep., 2016, 6, 37369 CrossRef CAS.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Müller and W. Wohlleben, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef.
- H. Liu, G. Wang, W. Li, X. Liu, E. Li and W. B. Yin, Microbiology, 2018, 164, 769–778 CrossRef CAS.
- D. H. Scharf, M. Groll, A. Habel, T. Heinekamp, C. Hertweck, A. A. Brakhage and E. M. Huber, Angew. Chem., Int. Ed., 2014, 53, 2221–2224 CrossRef CAS.
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 CrossRef.
- C. Wang, S. R. Wesener, H. Zhang and Y.-Q. Cheng, Chem. Biol., 2009, 16, 585–593 CrossRef CAS.
- B. Li and C. T. Walsh, Biochemistry, 2011, 50, 4615–4622 CrossRef CAS.
- G. Waksman, T. S. R. Krishna, C. H. Williams Jr and J. Kuriyan, Mol. Biol., 1994, 236, 800–816 CrossRef CAS.
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS.
- A. D. Keefe, S. L. Miller, G. McDonald and J. Bada, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11904–11906 CrossRef CAS.
- L. Leman, L. Orgel and M. R. Ghadiri, Science, 2004, 306, 283–286 CrossRef CAS.
- G. Chiappetta, S. Ndiaye, A. Igbaria, C. Kumar, J. Vinh and M. B. Toledano, Methods Enzymol., 2010, 473, 199–216 CAS.
- H. A. Scheraga, W. J. Wedemeyer and E. Welker, Methods Enzymol., 2001, 341, 189–221 CAS.
- C. Miyamoto, K. Yokose, T. Furumai and H. B. Maruyama, J. Antibiot., 1982, 35, 374–377 CrossRef CAS.
- R. J. Capon, R. Ratnayake, M. Stewart, E. Lacey, S. Tennant and J. H. Gill, Org. Biomol. Chem., 2005, 3, 123–129 RSC.
- Y. Liu, A. Mándi, X.-M. Li, L.-H. Meng, T. Kurtán and B.-G. Wang, Mar. Drugs, 2015, 13, 3640–3652 CrossRef CAS.
- During the submission of this manuscript, Tsunematsu and co-workers reported a biosynthetic pathway of aspirochlorine from A. oryzae. AclT in this manuscript is identified as AclD in their report. See: Y. Tsunematsu, N. Maeda, M. Sato, K. Hara, H. Hashimoto, K. Watanabe and C. Hertweck, J. Am. Chem. Soc., 2021, 143, 206–213 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06647h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |