
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium catalyzed β-selective alkylation of vinylpyridines with aldehydes/ketones via N2H4 mediated deoxygenative couplings†
Leiyang
Lv
 *a and
Chao-Jun
Li
*b
*a and
Chao-Jun
Li
*b
aDepartment of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: lvleiyang2008@ruc.edu.cn
bDepartment of Chemistry, FRQNT Center for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 30th December 2020
Abstract
Umpolung (polarity reversal) tactics of aldehydes/ketones have greatly broadened carbonyl chemistry by enabling transformations with electrophilic reagents and deoxygenative functionalizations. Herein, we report the first ruthenium-catalyzed β-selective alkylation of vinylpyridines with both naturally abundant aromatic and aliphatic aldehyde/ketones via N2H4 mediated deoxygenative couplings. Compared with one-electron umpolung of carbonyls to alcohols, this two-electron umpolung strategy realized reductive deoxygenation targets, which were not only applicable to the regioselective alkylation of a broad range of 2/4-alkene substituted pyridines, but also amenable to challenging 3-vinyl and steric-embedded internal pyridines as well as their analogous heterocyclic structures.
Introduction
The pyridine ring is one of the most significant structural subunits that are ubiquitous in a variety of natural products, bioactive compounds, pharmaceuticals and ligands in catalysis.1 The post-elaboration of this privileged scaffold continues to play a vital role to rapidly assemble related functionalized N-containing heterocycles and new molecular architectures.2 To this end, vinyl pyridines have been well adopted as versatile synthons3 in terms of their electrophilic nature, especially in analogous Michael-type additions with external nucleophiles, such as Grignard reagents4 or boronic acids.5 In contrast, the reductive couplings6 of vinyl pyridines with another electrophile have been less explored and remained a challenging study.7 The scarcity of this approach is attributed to three aspects: (1) substantially attenuated activation from the N-heteroaryl group compared with carbonyl or nitrile in Michael acceptors; (2) catalyst poisoning due to the strong coordination of basic nitrogen with a transition-metal catalyst; and (3) the competitive direct reduction of an alkene moiety to ethyl pyridine under reductive conditions. To fill this gap, Krische and co-workers in 2008, made a pioneering contribution in this field, which disclosed the first Rh-catalyzed α-selective reductive C–C coupling of vinyl pyridines with imines under a H2 atmosphere.8As is well-known, carbonyl compounds are versatile, readily-available and naturally abundant feedstocks in synthetic chemistry. In 2012, Lam et al.9 realized an elegant copper-catalyzed α-selective reductive coupling of vinyl pyridines with ketones. The presence of copper-hydride species upon which α-organocopper species was exclusively generated, was responsible for the α-selectivity of the C–C coupling products (Scheme 1a). Later, Ngai,10 and Wang et al.11 independently reported the β-selective reductive coupling of vinyl pyridines with aldehydes via tactfully synergistic Lewis or Brønsted acid/photoredox catalysis (Scheme 1b). The nucleophilic ketyl radical, generated by one electron reduction of electrophilic carbonyl with Hantzsch ester (one electron umpolung), was crucial to warrant C–C bond formation at the β-position of the alkene moiety in a radical process.12 Notably, these seminal reports offered a useful tool to deliver partially reductive (i.e. carbonyl reduced to hydroxyl) products. However, a selective method for the completely reductive couplings (i.e. carbonyl reduced to methylene) of vinyl pyridines with carbonyls is yet to be explored. In addition, only pyridines assembled with 2- and/or 4-alkene substitution (positive charge delocalization at the vinyl group owing to the electron-withdrawing properties of the pyridine ring) were viable substrates. However analogous 3-vinyl pyridine that lacks the conjugate activation effect, even with an acid co-catalyst to lower the olefinic β-carbon LUMO energy, was totally unreactive in the related reactions.13 Moreover, compared with aromatic aldehydes/ketones, the one electron umpolung of aliphatic ones is less accessible due to its higher reduction potential (typically lower than −2 V versus SCE)14 or lacking stabilization from vicinal aromatic rings. Last but not least, examples of reductive coupling of carbonyls with bulky internal vinyl pyridines were rarely reported because of intrinsic steric hindrance. Therefore, the development of a general deoxygenative reductive transformation,15 especially involving challenging 3-vinyl pyridines, and internal pyridines as well as aliphatic aldehydes and ketones to overcome these limitations, would provide novel and complimentary access to synthetically useful products.
 | ||
| Scheme 1 Strategies of reductive couplings of carbonyls with vinylpyridines. | ||
Inspired by the one electron carbonyl umpolung strategy,16 we wondered whether two electron carbonyl umpolung tactics could be used as a solution to realize reductive couplings with vinyl pyridines that were previously unattainable. Based on our previous work regarding N2H4 as the reductant in aryl–aryl/alkyl cross-couplings17 and pinacol couplings,18 as well as N2H4 mediated carbonyl umpolung,19 herein, we wish to report a ruthenium-catalyzed β-selective reductive coupling of vinyl pyridines with aldehydes/ketones using N2H4 as the reducing agent (Scheme 1c). This deoxygenative alkylation methodology exempts the pre-activation of weakly electrophilic vinylpyridines with a Brønsted/Lewis acid, wherein not only 2- or 4 vinyl pyridines but also previously inert 3-vinyl substituted and internal ones as well as other N-containing hetero-aromatic variants were all proved to be eligible electrophiles to couple with umpolung carbonyl compounds including both aromatic and aliphatic aldehydes/ketones.
Results and discussion
To test the feasibility of our hypothesis, we initially chose benzaldehyde (1a) and para-vinyl pyridine (3a) as the model substrates in combination with N2H4 as the reductant (Table 1). After preliminary investigations, we were glad to find that the desired deoxygenative linear product 4aa was attained in 97% yield in the presence of [Ru(p-cymene)Cl2]2 (2.5 mol%), dmpe [1,2-bis(dimethylphosphino)ethane] (5.0 mol%) and 0.5 equivalent of LiOtBu in THF at 80 °C for 12 h (entry 1). Based upon this encouraging result, we then evaluated the parameters that may affect the reaction efficiency. Only trace product 4aa was detected in the absence of the ruthenium catalyst or base (entries 2–3). We observed that the reaction proceeded equally well when [Ru(p-cymene)Cl2]2 was substituted with [Ru(C6Me6)Cl2]2 or Ru(PPh3)3Cl2 (entries 4 and 5) and a slightly decreased efficiency was observed with [RuCl(p-cymene)(dppe)]Cl20 (entry 6), but the yield of 4aa was reduced dramatically with Cp*Ru(cod)Cl or Ru(Cp)(PPh3)2Cl (entries 7 and 8). The catalysts frequently applied in coupling reactions, such as Ni(cod)2 and [Pd(allyl)Cl]2, displayed no catalytic activity in this transformation, probably due to the coordinative complexation of the basic nitrogen of the pyridine (entries 9 and 10). The examination of the ligand revealed that bidentate phosphine dppe [1,2-bis(diphenylphosphino)ethane] gave a comparable result (entry 11), while further increasing the bite angle of the phosphine ligand resulted in diminished efficiency (entries 12 and 13). A base was presumably used to deprotonate the in situ formed hydrazone and its effect was also assessed. KOH showed good reactivity to generate 4aa in 92% yield (enter 14). Relatively weak K3PO4 gave 4aa in moderate yield (entry 15), while K2CO3 or organic base DBU [1,8-diazabicyclo(5.4.0)undec-7-ene] led to poor results (entries 16–17).|
|
||
|---|---|---|
| Entry | Variation from “standard conditions” | 4aa (%) |
| a Reaction conditions: benzaldehyde 1a (0.4 mmol), hydrazine (0.48 mmol), para-vinyl pyridine 3a (0.2 mmol), [Ru(p-cymene)Cl2]2 (2.5 mol%), ligand (5 mol%) and base (0.1 mmol) in THF (0.5 mL) at 80 °C for 12 h under N2 unless otherwise noted. Note that phenyl hydrazone 2a was generated in situ from benzaldehyde and hydrazine without isolation. b The yield of 4aa was determined by 1H NMR using mesitylene as an internal standard and based on 3a (isolated yield of 4aa in parentheses). N.D. = not detected. | ||
| 1 | No change | 97 (95) |
| 2 | No [Ru(p-cymene)Cl2]2 | Trace |
| 3 | No base | Trace |
| 4 | Ru(PPh3)3Cl2 used instead | 96 |
| 5 | Ru(PPh3)3Cl2 used instead | 96 |
| 6 | [RuCl(p-cymene)(dppe)]Cl used instead | 71 |
| 7 | Cp*Ru(cod)Cl used instead | 24 |
| 8 | Ru(Cp)(PPh3)2Cl used instead | 14 |
| 9 | Ni(cod)2 used instead | N.D. |
| 10 | [Pd(allyl)Cl]2 used instead | N.D. |
| 11 | Dppe instead of dmpe | 96 |
| 12 | Dppb instead of dmpe | 84 |
| 13 | Dppf instead of dmpe | 66 |
| 14 | KOH instead of tBuOLi | 92 |
| 15 | K3PO4 instead of tBuOLi | 66 |
| 16 | K2CO3 instead of tBuOLi | 12 |
| 17 | DBU instead of tBuOLi | 10 |
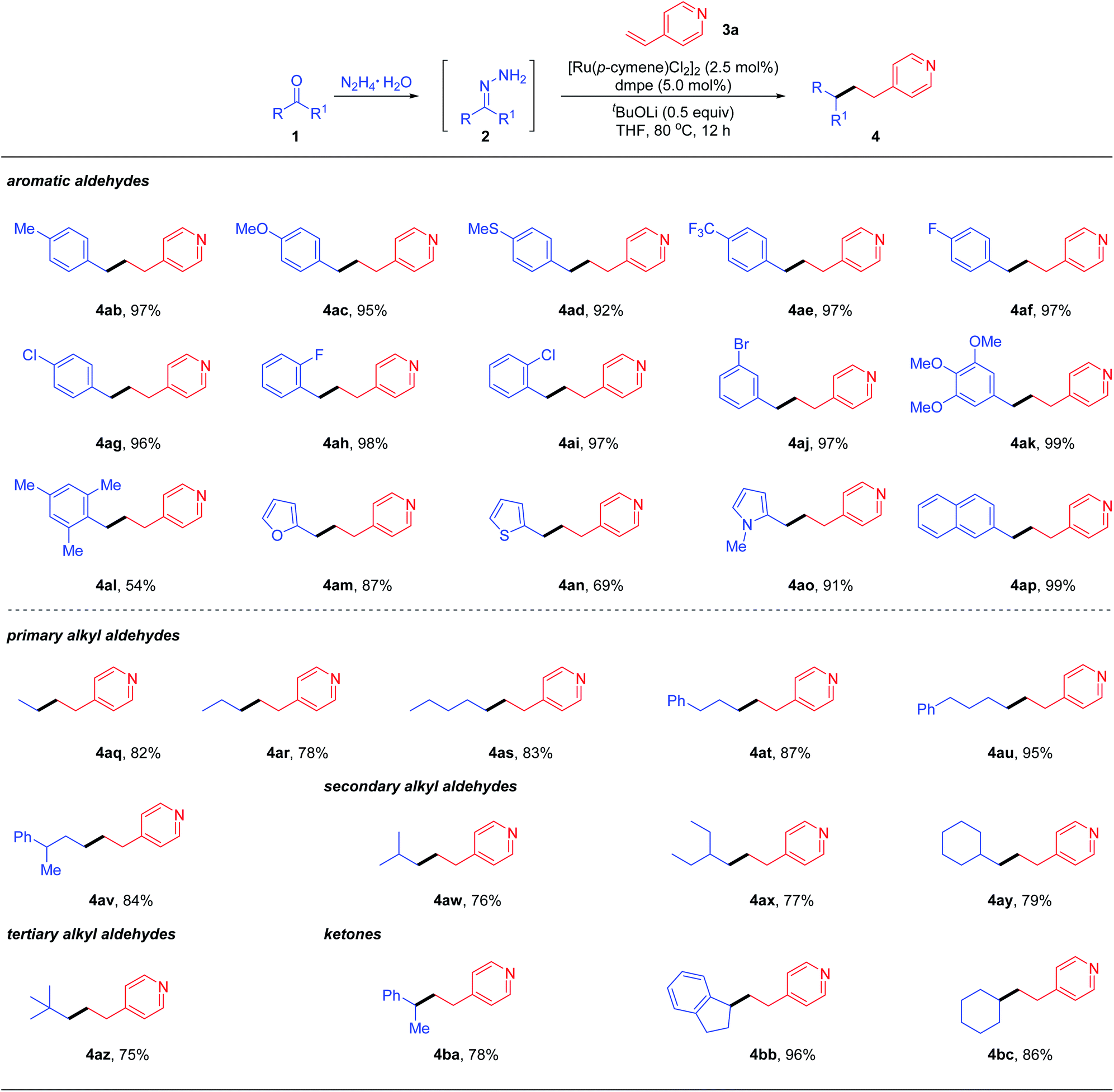
With the optimal reaction conditions obtained, we examined the scope of the reductive couplings with respect to carbonyls. As depicted in Table 2, the reaction provided the deoxygenated products with complete β-selectivity in good to excellent yields.
| a Reaction conditions: aldehyde/ketone 1 (0.4 mmol), hydrazine (0.48 mmol), vinyl heteroarene 3a (0.2 mmol), [Ru(p-cymene)Cl2]2 (2.5 mol%), ligand (5 mol%) and base (0.1 mmol) in THF (0.5 mL) at 80 °C for 12 h under N2. Note that hydrazone 2 was generated in situ from an aldehyde/ketone with hydrazine without isolation; however, the one-pot reaction gave no product. b Yields of isolated products based on 3a. |
|---|
|
For example, aromatic aldehydes bearing either electron-donating or electron-withdrawing substituents at the para-position of aryl rings, smoothly afforded the desired products 4ab–4ae in 92–97% yields. The halogen substituents such as F, Cl and Br, regardless of the substitution patterns, were well tolerated (4af–4aj) without detection of dehalogenative by-products. Electron-rich 3,4,5-trimethoxybenzaldehyde could be readily coupled with 4-vinylpyridines 4ak. The reaction yield was decreased to 54% when sterically hindered mesitaldehyde was tested under the optimal conditions. When hetero-aromatic aldehydes such as furan, thiophene and N-methylpyrrole aldehydes were subjected to the standard conditions, the desired reductive coupling products 4am–4ao were obtained in 69–91% yields. 4ap was also generated efficiently when the aldehyde with extended conjugation (e.g., 1-naphthaldehyde) was tested. As documented in the literature, engagement of ketyl radicals from alkyl aldehydes in reductive couplings via one electron umpolung is largely inhibited unless under harsh conditions, due to its highly negative reduction potential or transient stability. To overcome this limitation as well as to expand the reaction scope, we next evaluated the feasibility of aliphatic aldehydes in the reductive coupling with the two electron umpolung strategy. Gratifyingly, an array of aliphatic aldehydes including primary (1q–1v), secondary (1w–1y) and even bulky tertiary (1z) ones all reacted efficiently and furnished the alkylated pyridine derivatives 4aq–4az in good to excellent yields. Encouraged by these promising results, we attempted to apply more challenging ketones as potential partners in deoxygenative couplings. To our delight, under the standard reaction conditions, acetophenone, 1-indanone and simple cyclohexanone could react smoothly with vinylpyridine, delivering the corresponding products 4ba–4bc in 78–96% yields under the optimal conditions. These results clearly demonstrated the powerful generality and enhanced flexibility of our designed two-electron carbonyl umpolung strategy.
| a Reaction conditions: benzaldehyde (1a, 0.4 mmol), hydrazine (0.48 mmol), vinyl heteroarene 3 (0.2 mmol), [Ru(p-cymene)Cl2]2 (2.5 mol%), ligand (5 mol%) and base (0.1 mmol) in THF (0.5 mL) at 80 °C for 12 h under N2. b Yields of isolated products based on 3. c 0.1 mmol of 2,6-divinylpyridine was used. |
|---|

|
Next, we directed our attention to explore the scope of vinylpyridine coupling partners (Table 3). 2-Vinyl pyridine (3b), 2-methyl-6-vinylpyridine (3c), 2-bromo-6-vinylpyridine (3d) and 2-fluoro-4-vinylpyridine (3e) were converted into the corresponding products 5a–5d in good to excellent yields with complete β-regio-selectivity. Nevertheless, electron-rich 2-methoxy-4-(3-phenylpropyl)pyridine failed to undergo this transformation. Based upon the generality of this deoxygenative alkylation reaction, alkenes bearing other N-heterocyclic aromatic rings such as quinolone, pyrimidine, N-tosyl-pyrrole and benzothiazole were examined accordingly, all affording the desired products 5f–5i in synthetically useful yields. Notably, the deoxygenative coupling method was not limited to activated 2/4-vinyl N-hetero-aromatics; 3-vinylpyridine (3j), 2-(trifluoromethyl)-5-vinylpyridine (3k), and 3-vinylquinoline (3l) as well as 4-vinylisoquinoline (3m), which were challenging in previous Michael additions or coupling reactions due to the lack of conjugation or metal–nitrogen atom coordination activation, were also effective substrates to undergo the deoxygenative coupling reactions. Notably, previous synthesis of these kinds of products mainly relied on transition-metal catalysed cross-coupling reactions involving 3-halopyridines with organometallic reagents.21 Moreover, our preliminary study showed that internal vinylpyridines, such as 4-(prop-1-en-1-yl)pyridine (3n), 4-(hex-1-en-1-yl)pyridine (3o) and 4-(5-phenylpent-1-en-1-yl)pyridine (3p), regardless of cis–trans-isomers, all reacted efficiently and selectively to deliver the β-coupling products 5n–5p. The vinylpyridine with an alkene moiety bearing an α-substituent was totally unreactive (5q). Gratifyingly, the attempt to apply 2,6-divinylpyridine as a reaction partner also proved viable, affording the corresponding bis-alkylation product 5r in 92% yield.
To explore the practicality of this protocol, a gram-scale deoxygenative alkylation of vinylpyridine was performed with a reduced ruthenium catalyst loading (1.0 mol%) and air-stable dppe ligand, and the desired product 4aa was obtained in 89% yield (1.40 g) (Scheme 2a). When N-Ts hydrazone 6 was utilized instead of simple hydrazone 2a to react with vinylpyridine 3a, the corresponding product 4aa was not detected. This result excluded the possibility that hydrazone 2a acted as the benzyl carbene precursor in this deoxygenative coupling reaction (Scheme 2b).22 When deuterated hydrazone 7-d1 was tested with 3a under the standard conditions, no H/D loss or scrambling among the benzylic moiety and other positions was observed (Scheme 2c). This outcome revealed the absence of C–H bond activation of hydrazone or [Ru]–H species generation during the reaction process.23
 | ||
| Scheme 2 Gram-scale and control experiments. | ||
Based on these results and our previous studies,19,23 a possible reaction mechanism was proposed (Scheme 3). Firstly, the catalytically active species A is generated from precatalyst [Ru(p-cymene)Cl2]2 upon ligand dissociation/association with dmpe or dppe. Secondly, hydrazone 2 and vinylpyridine 3 coordinate to the ruthenium center and form a chair-like transition state C, which undergoes intramolecular rearrangement to form a new carbon–carbon bond and the six-membered ring intermediate D simultaneously. Lastly, decomposition of this species via N2 extrusion and protodemetalation releases the final product 4 or 5, and completes the catalytic cycle. An alternative mechanism involving the ruthenium mediated oxidative cyclization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of vinyl pyridine and the CN bond of hydrazone, followed by β-H elimination and reductive elimination cannot be ruled out.8
C bond of vinyl pyridine and the CN bond of hydrazone, followed by β-H elimination and reductive elimination cannot be ruled out.8
 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
We have demonstrated an unprecedented ruthenium-catalyzed β-selective alkylation of vinylpyridines with both aromatic and aliphatic aldehydes/ketones via deoxygenative coupling. A broad range of valuable alkyl pyridine and related N-hetero-aromatic structures were obtained in a highly efficient and selective manner. This N2H4 mediated two-electron carbonyl umpolung process not only solved the challenges of the invalidity of naturally abundant ketones and aliphatic aldehydes as substrates in C–C bond formations via the one-electron umpolung pathway, but also realized selective reductive alkylation of 2/4-alkene substituted pyridines, especially previously challenging 3-vinyl and steric-embedded internal pyridines as well as their analogous heteroarenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J. L.), the Canadian Foundation for Innovation, the FRQNT Centre in Green Chemistry and Catalysis, the Natural Science and Engineering Research Council of Canada and the Killam Research Fellow of the Canadian Council of Arts. The research was also supported by the Fundamental Research Funds (to L. L.) for the Central Universities, and the Research Funds of Renmin University of China.References
- (a) K. C. Majumdar and S. K. Chattopadhyay, Heterocycles in Natural Product Synthesis, Wiley-VCH Verlag, Weinheim, 2011 CrossRef; (b) A.-Y. Guan, C.-L. Liu, X.-F. Sun, Y. Xie and M.-A. Wang, Bioorg. Med. Chem., 2016, 24, 342–353 CrossRef CAS.
- (a) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS; (b) S. L. Rossler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264–9280 CrossRef.
- D. A. Klumpp, Synlett, 2012, 23, 1590–1604 CrossRef CAS.
- (a) R. P. Jumde, F. Lanza, M. J. Veenstra and S. R. Harutyunyan, Science, 2016, 352, 433–437 CrossRef CAS; (b) R. P. Jumde, F. Lanza, T. Pellegrini and S. R. Harutyunyan, Nat. Commun., 2017, 8, 2058 CrossRef. For a review, see: (c) Y. Guo and S. R. Harutyunyan, Beilstein J. Org. Chem., 2020, 16, 1006–1021 CrossRef CAS.
- (a) M. Lautens, A. Roy, K. Fukuoka, K. Fagnou and B. Martín-Matute, J. Am. Chem. Soc., 2001, 123, 5358–5359 CrossRef CAS; (b) J. J. Smith, D. Best and H. W. Lam, Chem. Commun., 2016, 52, 3770–3772 RSC; (c) A. Saxena and H. W. Lam, Chem. Sci., 2011, 2, 2326–2331 RSC.
- L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454–457 CrossRef CAS.
- K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, aah5133 CrossRef.
- V. Komanduri, C. D. Grant and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 12592–12593 CrossRef CAS.
- A. Saxena, B. Choi and H. W. Lam, J. Am. Chem. Soc., 2012, 134, 8428–8431 CrossRef CAS.
- K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS.
- K. Cao, S. M. Tan, R. Lee, S. Yang, H. Jia, X. Zhao, B. Qiao and Z. Jiang, J. Am. Chem. Soc., 2019, 141, 5437–5443 CrossRef CAS.
- K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC.
- (a) H. B. Hepburn and P. Melchiorre, Chem. Commun., 2016, 52, 3520–3523 RSC; (b) X.-Y. Liu, Y.-X. Tan, X. Wang, H. Xu, Y.-H. Wang, P. Tian and G.-Q. Lin, Org. Lett., 2020, 22, 4038–4042 CrossRef CAS; (c) P. Zhang, D. Huang and T. R. Newhouse, J. Am. Chem. Soc., 2020, 142, 1757–1762 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- (a) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 12788–12791 CrossRef CAS; (b) E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS; (c) R.-D. He, C.-L. Li, Q.-Q. Pan, P. Guo, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2019, 141, 12481–12486 CrossRef CAS; (d) Y. Ye, H. Chen, J. L. Sessler and H. Gong, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS; (e) R. Ruzi, K. Liu, C. Zhu and J. Xie, Nat. Commun., 2020, 11, 3312 CrossRef CAS; (f) J. Tang, L. L. Liu, S. Yang, X. Cong, M. Luo and X. Zeng, J. Am. Chem. Soc., 2020, 142, 7715–7720 CrossRef CAS; (g) K. Kang, L. Huang and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 10634–10640 CrossRef CAS.
- (a) L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225–229 CrossRef CAS. For reviews, see: (b) G. A. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307–338 CrossRef CAS; (c) Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CAS.
- L. Lv, Z. Qiu, J. Li, M. Liu and C.-J. Li, Nat. Commun., 2018, 9, 4739 CrossRef.
- Z. Qiu, H. D. M. Pham, J. Li, C.-C. Li, D. J. Castillo-Pazos, R. Z. Khaliullin and C.-J. Li, Chem. Sci., 2019, 10, 10937–10943 RSC.
- (a) H. Wang, X.-J. Dai and C.-J. Li, Nat. Chem., 2017, 9, 374–378 CrossRef CAS; (b) L. Lv, D. Zhu and C.-J. Li, Nat. Commun., 2019, 10, 715 CrossRef. For an account, see: (c) C.-J. Li, J. Huang, X.-J. Dai, H. Wang, N. Chen, W. Wei, H. Zeng, J. Tang, C.-C. Li, D. Zhu and L. Lv, Synlett, 2019, 30, 1508–1524 CrossRef CAS.
- (a) C. Daguenet and P. J. Dyson, Catal. Commun., 2003, 4, 153–157 CrossRef CAS; (b) C. Daguenet, R. Scopelliti and P. J. Dyson, Organometallics, 2004, 23, 4849–4857 CrossRef CAS.
- N. Miyaura, Metal Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, ch. 2, vol. 1 Search PubMed.
- Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC.
- (a) L. Lv, L. Yu, Z. Qiu and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 6466–6472 CrossRef CAS; (b) L. Yu, L. Lv, Z. Qiu, Z. Chen, Z. Tan, Y.-F. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 14009–14013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06586b |
| This journal is © The Royal Society of Chemistry 2021 |