
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active benzimidazolium sulfonamides as cationic thiolating reagents for reductive cross-coupling of organic halides†
Weigang
Zhang
,
Mengjun
Huang
,
Zhenlei
Zou
,
Zhengguang
Wu
 ,
Shengyang
Ni
,
Lingyu
Kong
,
Youxuan
Zheng
,
Yi
Wang
* and
Yi
Pan
,
Shengyang
Ni
,
Lingyu
Kong
,
Youxuan
Zheng
,
Yi
Wang
* and
Yi
Pan
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, Collaborative Innovation Center of Advanced Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yiwang@nju.edu.cn
First published on 22nd December 2020
Abstract
Redox-active benzimidazolium sulfonamides as thiolating reagents have been developed for reductive C–S bond coupling. The IMDN-SO2R reagent provides a bench-stable cationic precursor to generate a portfolio of highly active N–S intermediates, which can be successfully applied in cross-electrophilic coupling with various organic halides. The employment of an electrophilic sulfur source solved the problem of catalyst deactivation and avoided odorous thiols, featuring practical conditions, broad substrate scope, and excellent tolerance.
The high frequency of sulfur-containing moieties in natural products,1 bioactive molecules,2 pharmaceuticals,3 organic materials,4 fragrances5 and asymmetric catalysis as chiral catalysts/ligands6 has triggered the best endeavours for the selective construction of C–S bonds. The conventional cross-coupling of thiols with aryl halides generally relies on the conversion of mercaptans to thiolates by means of transition-metal catalysis7 (such as Pd,8 Cu,9 and Ni10) and other metals,11 although these efforts were plagued by several drawbacks. The strong coordination of thiolates to metals often leads to catalyst deactivation and displays low efficiencies. Therefore, high catalyst loading, specific ligands, excessive heating and strong bases are often required to facilitate this transformation (Scheme 1a, left). Recent development using photochemical12 and electrochemical13 induced thiol radicals as a sulfur source could avoid the problem of catalyst poisoning, although restricted substrate scope was displayed (Scheme 1a, middle). Despite the progress made for C–S bond construction,14 the longstanding issues that exist in the above-mentioned strategies should not be overlooked.
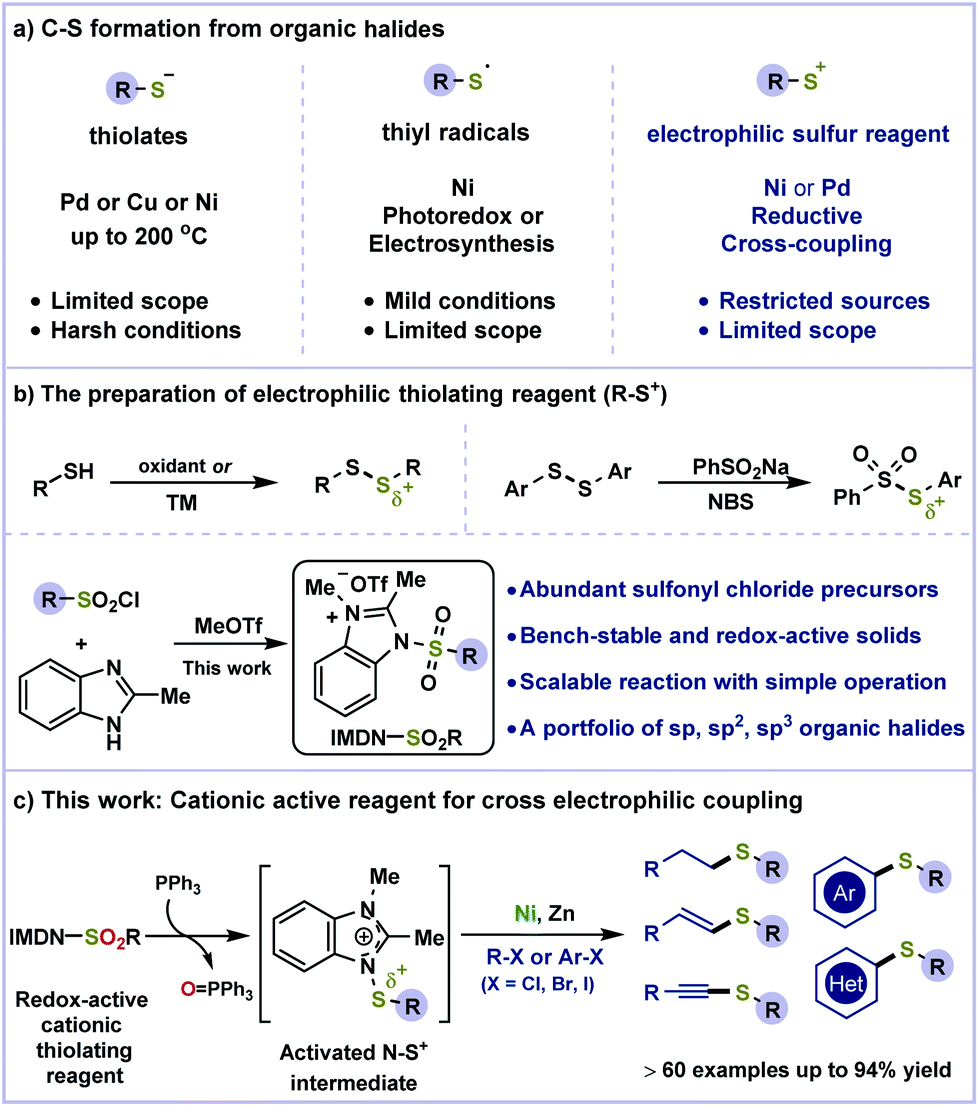 | ||
| Scheme 1 Origin of the reaction design. (a) C–S formation from organic halides. (b) The preparation of the electrophilic thiolating reagent (R–S+). (c) This work: cationic active reagent for cross electrophilic coupling. | ||
Compared to classical cross-coupling processes, the nickel-catalyzed reductive cross-coupling of two electrophilic partners has emerged as a powerful tool for the replacement of air- and moisture-sensitive organometallic reagents.15 The cross electrophilic coupling for the construction of C–S bonds is more challenging due to the lack of sulfur sources and homo-coupling of organic halides (Scheme 1a, right). Several examples of reductive thiolation16 have been described using thiol derivatives as S+ sources (Scheme 1b). We speculated that readily accessible sulfonyl chloride as an electrophilic sulfur source could avoid the use of highly toxic thiols and significantly the substrate scope.17 However, the direct reduction of sulfonyl chloride inevitably resulted in dimerization to disulfide.17b Putatively, activated by an electron-deficient heterocycle such as imidazole, sulfonyl chloride could be assembled into a bench-stable cationic reagent (Scheme 1b). Benzimidazolium sulfonamides would be better electron acceptors18 and easily reduced by PPh3 to generate a highly reactive N–S+ species in situ. The positive charge of this intermediate is delocalized on both nitrogens of imidazole. Followed by the cleavage of the weak N–S bond (BDE ≈ 70 kcal mol−1),19 the electrophilic sulfur species can be captured by metal catalysts for cross-coupling. Herein, we have described a redox-active benzimidazolium sulfonamide reagent (IMDN-SO2R) for Ni-catalyzed reductive coupling of organic halides for a portfolio of C(sp)–S, C(sp2)–S and C(sp3)–S bond formations (Scheme 1c). This strategy avoids the formation of disulfide by-products and the use of organometallic reagents, which facilitates purification and enhances the functionality tolerance.
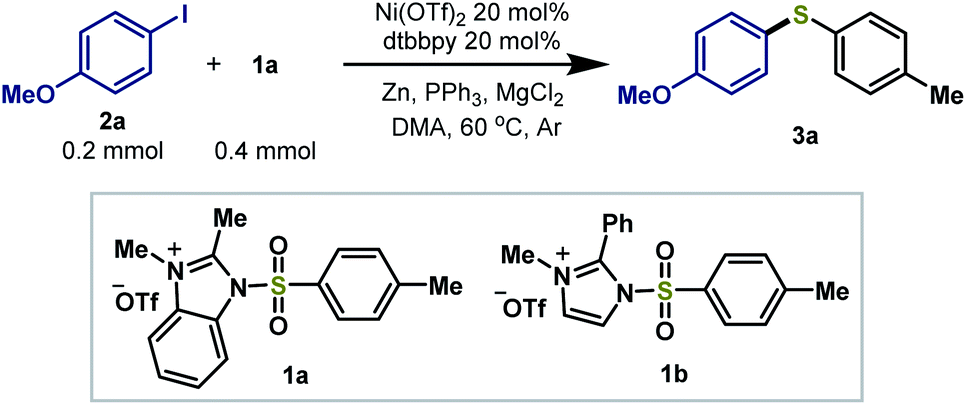
To determine the suitable conditions for the reductive coupling of the cationic reagent with organic halides, we first studied the reductive thiolation of p-iodo-methoxybenzene (2a) and 1a (2 equiv.) with a survey of Ni catalysts in the presence of dtbbpy (20 mol%), PPh3 (2.5 equiv.), Zn powder (3.0 equiv.) and MgCl2 (2.0 equiv.) in DMA at 60 °C (Table 1). Ni(OTf)2 as a catalyst was able to promote the reductive process to afford the desired aryl thioether product 3a in 92% isolated yield (entry 1). When using Ni(cod)2, Ni(acac)2, Ni(OAc)2·4H2O and Ni(PCy3)2Cl2, lower yields were obtained (entries 2–5). Decreasing the 1a loading to 1.5 equiv., the yield of 3a reduced to 75% (entry 6). Switching 1a to a 2-phenyl substituted imidazolium sulfonamide reagent 1b, similar yields were achieved (entry 7). When decreasing both Ni(OTf)2 and dtbbpy loading to 10 mol%, the yield reduced to 75%. In the absence of Ni(OTf)2 or Zn powder, no desired product was observed (entries 9–10). Switching Zn powder to an organic reductant tetrakis-(dimethylamino)ethylene (TDAE), still a low conversion of the reaction was observed (entry 11). The results demonstrated that no organozinc reagents were generated. In the absence of the ligand, MgCl2 or PPh3, the reaction resulted in low yields, indicating that these ingredients were essential for this catalytic system (entries 12–14). Thus, the optimized conditions were selected for further investigation of the reaction scope.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Variation | Yielda/% | Entry | Variation | Yielda/% |
| 1 | None | 96 (92)b | 8 | 10 mol% [Ni] | 83 |
| 2 | Ni(acac)2 | 42 | 9 | No Ni(OTf)2 | nd |
| 3 | Ni(cod)2 | 63 | 10 | No Zn | nd |
| 4 | Ni(OAc)2·4H2O | 21 | 11 | TDAE for Zn | 21 |
| 5 | Ni(PCy3)2Cl2 | 57 | 12 | No dtbbpy | 46 |
| 6 | 1.5 equiv. 1a | 75 | 13 | No MgCl2 | 39 |
| 7 | 2.0 equiv. 1b | 88 | 14 | No PPh3 | 20 |
The generality of the reductive thiolation was evaluated under the optimized conditions (Scheme 2). A wide range of aryl iodides containing either electron-withdrawing or electron-donating functionality were tolerated, delivering products with methoxy (3a, 3c), trifluoromethoxy (3b), methyl (3d), acetyl (3e), ester (3f), boronate (3g), and phenyl (3h) groups in good to excellent yields (63–94%). 5-Iodobenzo[d][1,3]dioxole and 2-iodofluorene also furnished the corresponding adducts (3i, 3j) in 91% and 82% yields, respectively. Notably, aryl iodides bearing sensitive groups such as amine (3k) and hydroxyl (3l) were engaged in the cross-coupling to forge the C–S bond in good yields. Aryl bromides (3a, 3m) were also found compatible. The scope of the heteroaryl halide coupling partner was also explored. Various five- and six-membered heterocycles, including thiophene (3n), pyrazole (3o), quinoline (3p), carbazole (3q), indole (3r), isothiazole (3s), benzofuran (3t), benzothiophene (3u), benzothiazole (3v), pyrimidine (3w), pyrazine (3x), and pyridine (3y, 3z) derived heteroaryl bromides and iodides were treated with 1a to produce the corresponding sulfides in good yields. In the cases of relatively unreactive organic chlorides, the corresponding coupling products (3aa–3cc) could be obtained in low yields under the standard conditions. Subsequently, we assessed the scope of aryl sulfonamides. Reagents 1c–1k containing electron-withdrawing and electron-donating substitutions on the benzene ring afforded the desired products in good to excellent yields (3dd–3ll). Sterically hindered imidazolium sulfonamides 2,4,6-trimethylated 1j and 2,4,6-triisopropylated 1k showed good compatibility in this reaction to give the corresponding products 3kk and 3ll in high yields.
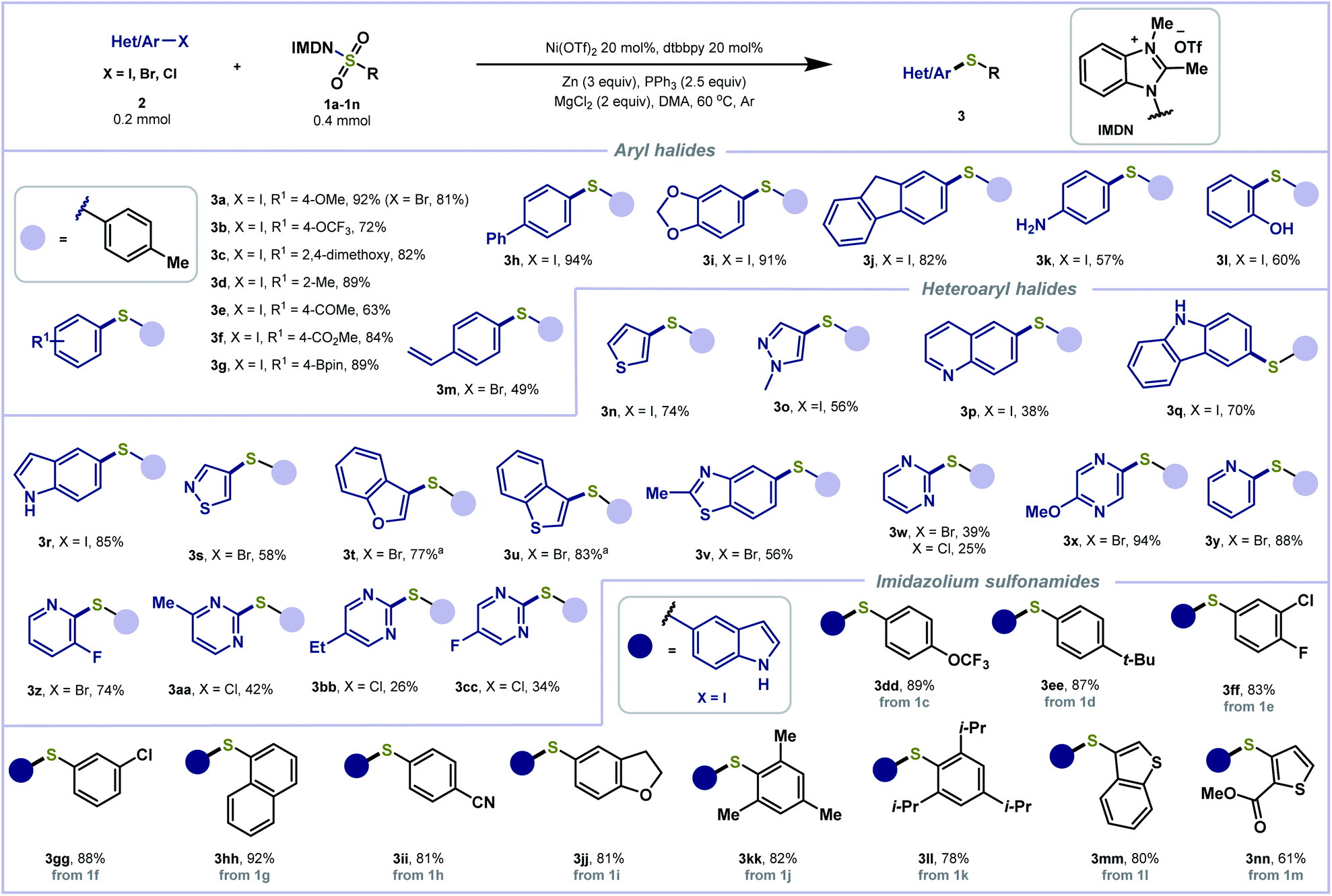 | ||
| Scheme 2 Substrate scope of (hetero)aryl and benzimidazolium sulfonamides. aReaction was performed at 80 °C. | ||
The more challenging aliphatic halides have also been examined with the redox-active reagent 1a (Scheme 3). Primary and secondary alkyl halides yielded alkyl sulfides (5a–5k and 5l) in good to excellent yields. No dimerization side-product was observed. Functionalities including esters (5b, 5i, and 5j), sulfide (5c), alkene (5f), acetal (5h) and ether (5k) are tolerated. Some sensitive functional groups including silyl ether (5e) and organoboronate (5g) were well tolerated, provided in 75% and 57% yields, respectively. Alkenyl halides were also tolerated to afford 7a and 7b in good yields. In addition, alkynyl bromides were employed for reductive thiolation with benzimidazolium sulfonamides 1a and 1c–1i to afford the corresponding C(sp)–S bond coupling products (9a–9h) in moderate to good yields.
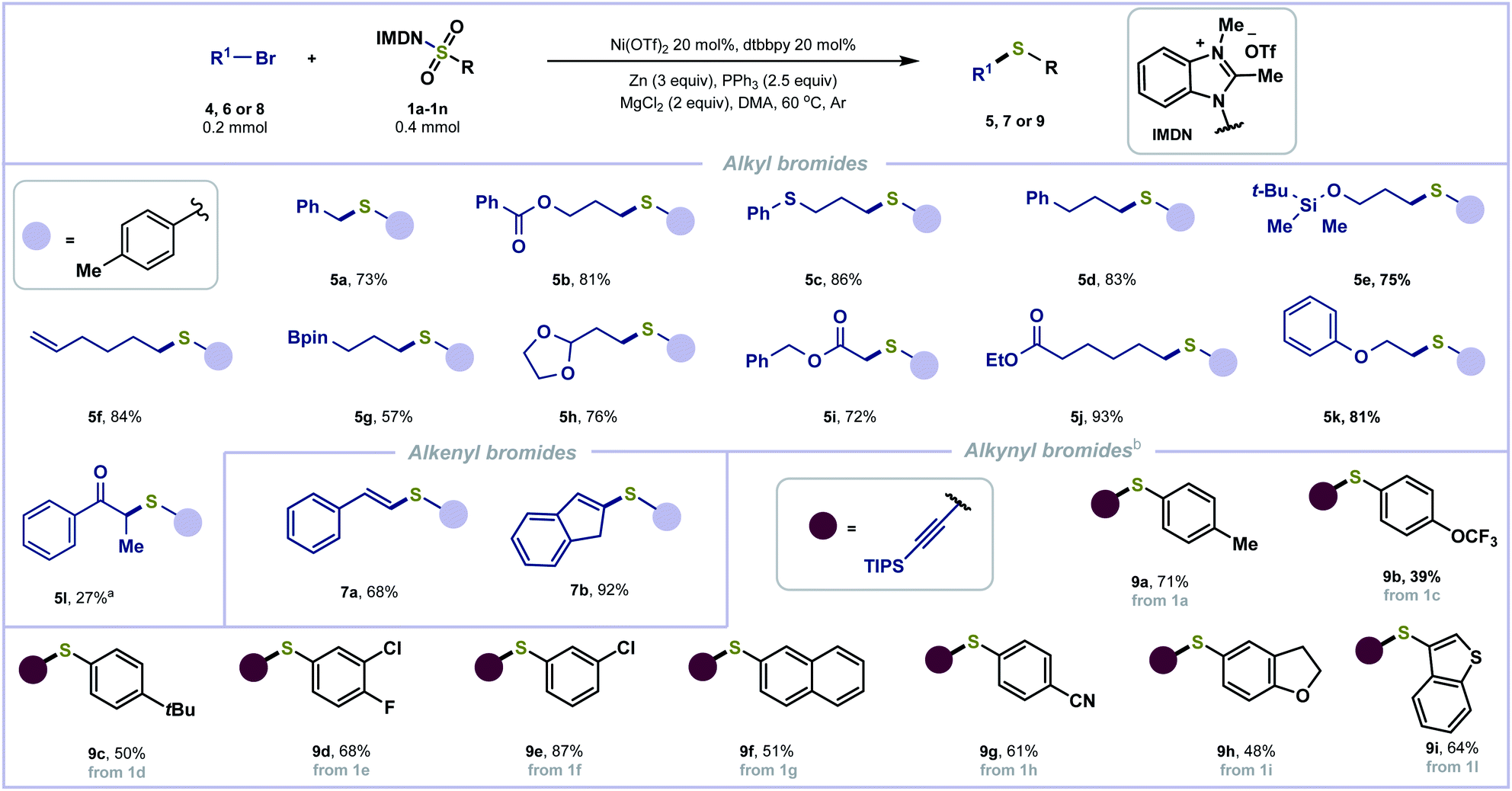 | ||
| Scheme 3 Substrate scope of alkyl, alkenyl and alkynyl bromide. aNi(PCy3)2Cl2 instead of Ni(OTf)2 and without dtbbpy. b4-(Trifluoromethyl)pyridine (0.2 mmol) and THF (2 mL) were used instead of dtbbpy and DMA. | ||
To demonstrate the synthetic potential of this cross-electrophile coupling, a gram scale reaction was performed with 1a and p-iodo-methoxybenzene 2a under the standard conditions (Scheme 4a). The reductive thiolation was also applied in the late-stage modification of biologically active molecules and synthesis of pharmaceutically active molecules. D-Glucose (10a), (+)-α-tocopherol (10b), rosin amine (10c), and 17a-methyl-drostanolone-derived (10d) alkyl halides were compatible to afford thiolated products in good yields (Scheme 4b). Treating benzimidazolium sulfonamide 1n with piperazine-derived iodobenzene 11 generated the thiolated intermediate 12 and deprotection with TFA afforded anti-depressive vortioxetine 13 (ref. 20) in 50% overall yields (Scheme 4c).
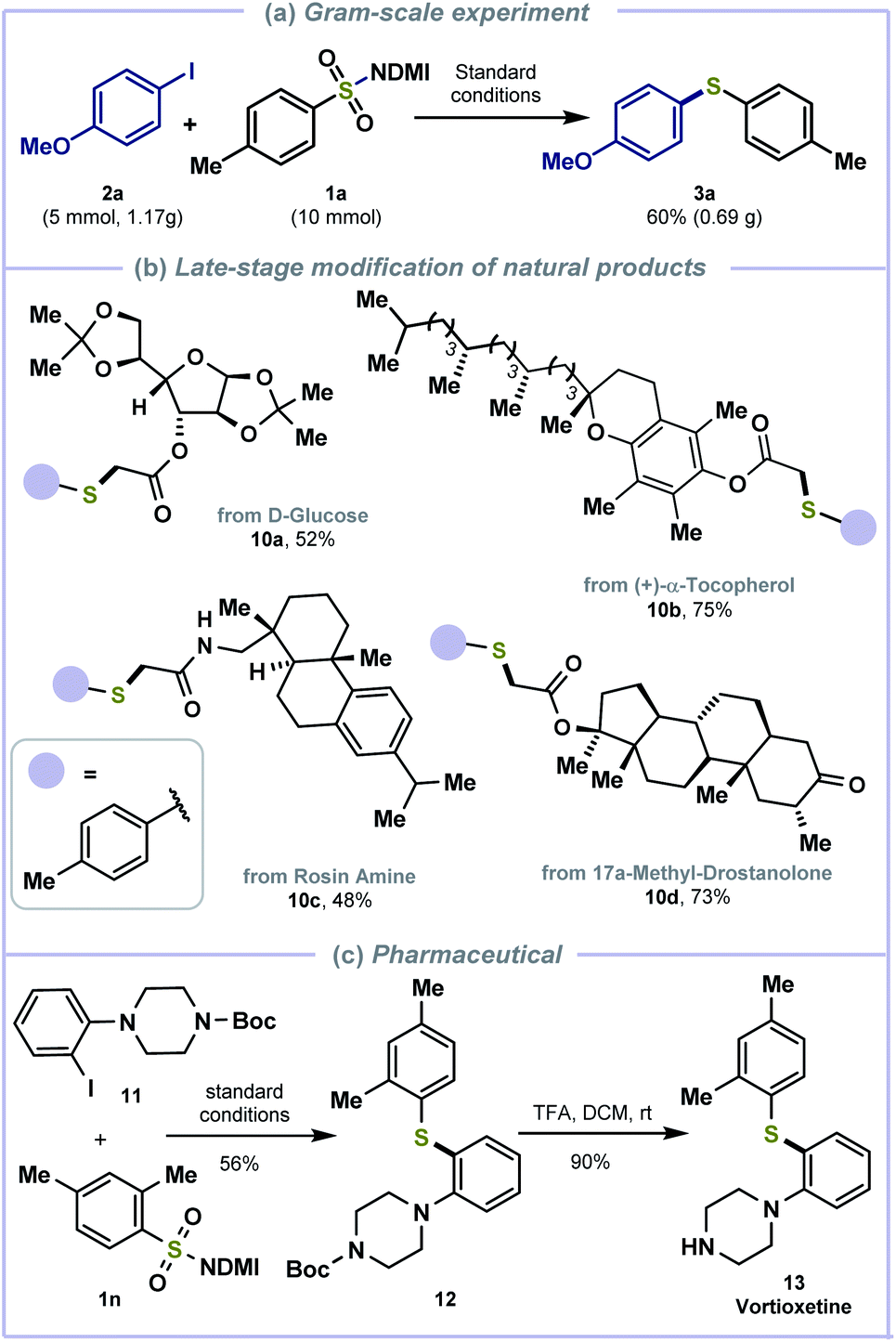 | ||
| Scheme 4 Further transformations. (a) Gram-scale experiment. (b) Late-stage modification of natural products. (c) The synthesis of vortioxetine using our protocol. | ||
To investigate the mechanism for this reductive thiolation, a series of control experiments have been carried out. First, the treatment of benzimidazolium sulfonamide 1a with PPh3 furnished the key N–S+ intermediate Int-I and Ph3P = O, which were confirmed by 1H NMR, HRMS data and 31P NMR (Scheme 5a, see the ESI†). A mixture of Int-I and 2a was able to afford 3a in 80% yield under the standard conditions, indicating that the reaction did go through this route (Scheme 5b). Furthermore, the key nickel-complex 14 was prepared and reacted with Int-I to furnish thioether 3d in 15% yield (Scheme 5c). Finally, sulfone 15 was used in the absence of benzimidazolium sulfonamide 1a and 2a under standard conditions. No reduced product 3a was detected, which indicates that the PPh3 reduction occurs before the cross-coupling (Scheme 5d). On the basis of the experimental results and previous reports,16c,g a plausible mechanism for the reductive thiolation of organic halides is proposed (Scheme 5e). Initially, the reduction of the Ni(II) salt by zinc affords the active Ni(0) catalyst A, which undergoes oxidative addition into the C–X bond of organic halides to give the intermediate R–Ni(II)X B. The following reduction of B forms R–Ni(I) intermediate C.15b,16g Then C and Int-I undergo stepwise single-electron transfer with the possibility of radical trapping within a solvent cage to afford intermediate D before the generation of R–Ni(III)-SAr E and benzoimidazole residue 16. Finally, the reductive elimination of E furnishes the desired thioether product and Ni(I) species F which is reduced by zinc to facilitate the next catalytic cycle.
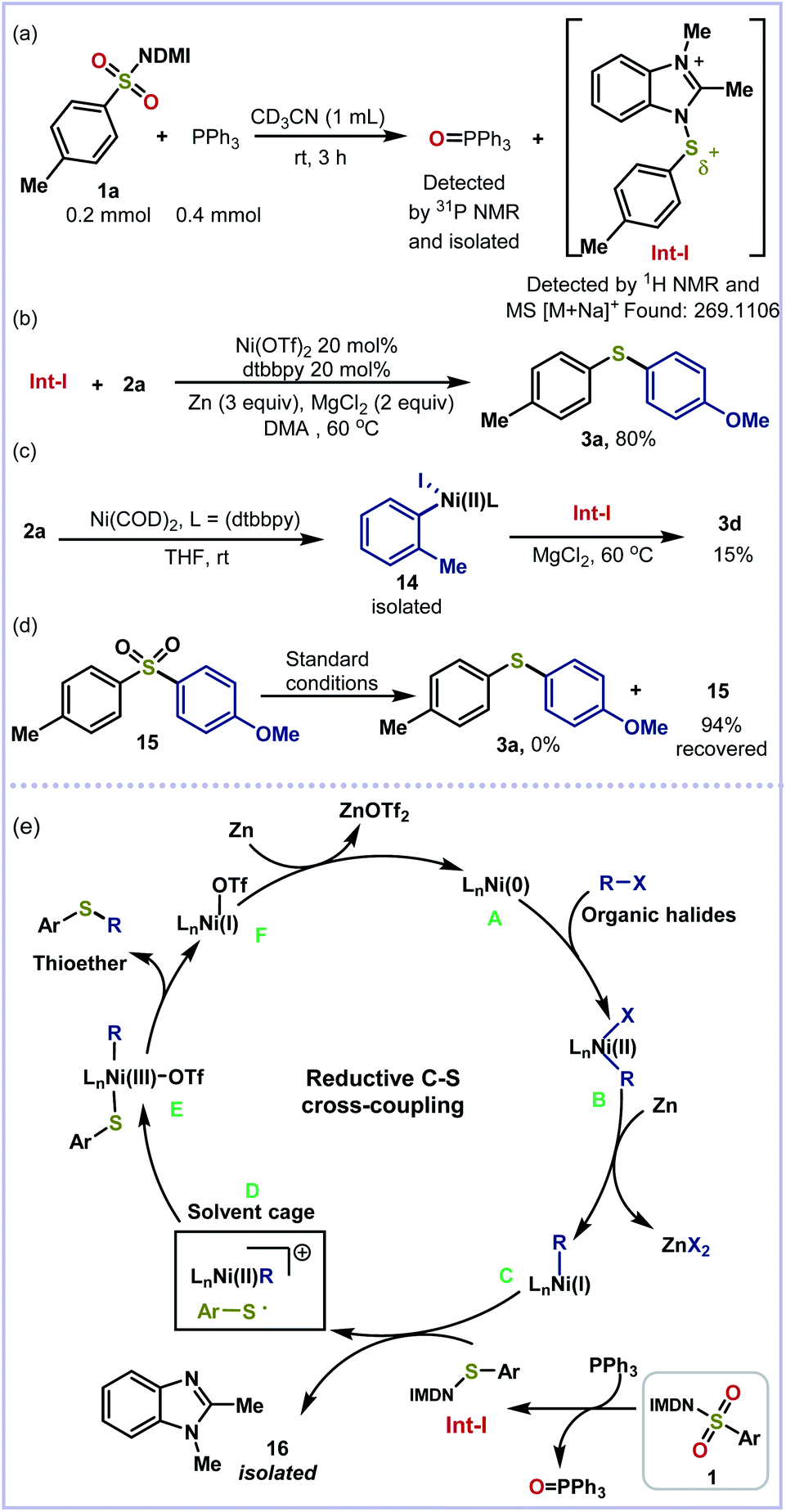 | ||
| Scheme 5 Mechanistic studies and proposed mechanism. | ||
Conclusions
In summary, we have developed a universal reductive thiolation protocol of organic halides with benzimidazolium sulfonamides. This bench-stable crystalline reagent is readily achievable in large quantities from abundant sulfonyl chloride. The key design of this redox-active reagent is the cationic nature. The reduction potential of sulfonyl group can be improved due to the cationic nature and in favor of in situ generating electrophilic N–S+ reagent by the reduction of triphenylphosphine. This approach features practical conditions, broad substrate scope, and excellent functional group tolerance for the incorporation of sulfur-containing moieties into organic molecules. Further study of the cationic reagent IMDN-SO2R is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professor Shaolin Zhu is gratefully acknowledged for helpful discussion. This work was supported by the National Natural Science Foundation of China (No. 21772085 and 21971107) and the Fundamental Research Funds for the Central Universities (No. 020514380220, 020514380131, 020514913412, and 020514913214). We also thank the Collaborative Innovation Center of Advanced Microstructures and Jiangsu Provincial Key Laboratory of Photonic and Electronic Materials at Nanjing University for support.Notes and references
- (a) N. Wang, P. Saidhareddy and X. F. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC; (b) K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS.
- (a) R. A. Bragg, S. Brocklehurst, F. Gustafsson, J. Goodman, K. Hickling, P. A. MacFaul, S. Swallow and J. Tugwood, Chem. Res. Toxicol., 2015, 28, 1991–1999 Search PubMed; (b) F. Y. Zhu, Y. M. Wang, M. C. He, Z. H. Yan and S. Lin, Chinese J. Org. Chem., 2019, 39, 1175–1180 Search PubMed.
- (a) M. H. Feng, B. Q. Tang, H. L. Steven and X. F. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS; (b) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS; (c) L. Liu, J. E. Stelmach, S. R. Natarajan, M. H. Chen, S. B. Singh, C. D. Schwartz, C. E. Fitzgerald, S. J. O'Keefe, D. M. Zaller, D. M. Schmatz and J. B. Doherty, Bioorg. Med. Chem. Lett., 2003, 13, 3979–3982 CrossRef CAS; (d) G. Liu, J. R. Huth, E. T. Olejniczak, R. Mendoza, P. DeVries, S. Leitza, E. B. Reilly, G. F. Okasinski, S. W. Fesik and T. W. von Geldern, J. Med. Chem., 2001, 44, 1202–1210 CrossRef CAS.
- (a) H. Iino, T. Usui and J. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS; (b) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS; (c) T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura and J. Takeya, Adv. Mater., 2013, 25, 6392–6397 CrossRef CAS.
- M. Wang, C. H. Wang and X. F. Jiang, Chinese J. Org. Chem., 2019, 39, 2139–2147 CrossRef CAS.
- (a) S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS; (b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133–5209 CrossRef CAS; (c) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611–620 CrossRef CAS.
- (a) C. F. Lee, Y. C. Liu and S. S. Badsara, Chem.–Asian J., 2014, 9, 706–722 CrossRef CAS; (b) C. C. Eichman and J. P. Stambuli, Molecules, 2011, 16, 590–608 CrossRef CAS; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS; (d) E. Alvaro and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7858–7868 CrossRef CAS; (e) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS; (f) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS; (g) T. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef.
- (a) N. Velasco, C. Virumbrales, R. Sanz, S. Suarez-Pantiga and M. A. Fernandez-Rodriguez, Org. Lett., 2018, 20, 2848–2852 CrossRef CAS; (b) M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem.–Asian J., 2014, 20, 2459–2462 CAS; (c) M. A. Fernández-Rodríguez, Q. L. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180–2181 CrossRef; (d) M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS.
- (a) B. Movassagh and Z. Hosseinzadeh, Synlett, 2015, 27, 777–781 CrossRef; (b) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548–9552 CrossRef CAS; (c) H. L. Kao and C. F. Lee, Org. Lett., 2011, 13, 5204–5207 CrossRef CAS; (d) H. Zhang, W. Cao and D. W. Ma, Synth. Commun., 2007, 37, 25–35 CrossRef CAS; (e) L. Rout, T. K. Sen and T. Punniyamurthy, Angew. Chem., Int. Ed., 2007, 46, 5583–5586 CrossRef CAS; (f) C. G. Bates, R. K. Gujadhur and D. Venkataraman, Org. Lett., 2002, 4, 2803–2806 CrossRef CAS.
- (a) K. D. Jones, D. J. Power, D. Bierer, K. M. Gericke and S. G. Stewart, Org. Lett., 2018, 20, 208–211 CrossRef CAS; (b) S. Jammi, P. Barua, L. Rout, P. Saha and T. Punniyamurthy, Tetrahedron Lett., 2008, 49, 1484–1487 CrossRef CAS; (c) Y. Zhang, K. C. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495–3498 CrossRef CAS.
- (a) Z. Tber, M. A. Hiebel, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Berteina-Raboin, RSC Adv., 2016, 6, 72030–72036 RSC; (b) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 1697–1700 CrossRef CAS; (c) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880–2883 CrossRef CAS; (d) Y. C. Wong, T. T. Jayanth and C. H. Cheng, Org. Lett., 2006, 8, 5613–5616 CrossRef CAS.
- (a) F. Sandfort, T. Knecht, T. Pinkert, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2020, 142, 6913–6919 CrossRef CAS; (b) M. Jiang, H. Li, H. Yang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 874–879 CrossRef CAS; (c) M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760–1763 CrossRef CAS.
- (a) Y. Wang, L. Deng, X. Wang, Z. Wu, Y. Wang and Y. Pan, ACS Catal., 2019, 9, 1630–1634 CrossRef CAS; (b) D. Liu, H. X. Ma, P. Fang and T. S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033–5037 CrossRef CAS.
- G. M. F. Batista, P. P. De Castro, J. A. Dos Santos, T. Skrydstrup and G. W. Amarante, Org. Chem. Front., 2020 10.1039/D0QO01226B.
- (a) S. Kim, M. J. Goldfogel, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 9902–9907 CrossRef CAS; (b) S. Y. Ni, C. X. Li, Y. Mao, J. L. Han, Y. Wang, H. Yan and Y. Pan, Sci. Adv., 2019, 5, eaaw9516 CrossRef CAS; (c) F. Chen, K. Chen, Y. Zhang, Y. He, Y. M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929–13935 CrossRef CAS; (d) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS; (e) X. Wang, S. Wang, W. Xue and H. G. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS; (f) C. Zhao, X. Jia, X. Wang and H. G. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS; (g) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS; (h) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS.
- (a) J. Li, W. Rao, S. Y. Wang and S. J. Ji, J. Org. Chem., 2019, 84, 11542–11552 CrossRef CAS; (b) Y. Fang, T. Rogge, L. Ackermann, S. Y. Wang and S. J. Ji, Nat. Commun., 2018, 9, 2240 CrossRef; (c) V. Gómez-Benítez, O. Baldovino-Pantaleón, C. Herrera-Álvarez, R. A. Toscano and D. Morales-Morales, Tetrahedron Lett., 2006, 47, 5059–5062 CrossRef; (d) S.-I. Fukuzawa, D. Tanihara and S. Kikuchi, Synlett, 2006, 2006, 2145–2147 CrossRef; (e) N. Taniguchi and T. Onami, J. Org. Chem., 2004, 69, 915–920 CrossRef CAS; (f) N. Taniguchi, J. Org. Chem., 2004, 69, 6904–6906 CrossRef CAS; (g) S. Chowdhury and S. Roy, Tetrahedron Lett., 1997, 38, 2149–2152 CrossRef CAS.
- (a) R. H. Wu, K. K. Huang, G. Y. S. Qiu and J.-B. Liu, Synthesis, 2019, 51, 3567–3587 CrossRef CAS; (b) G. W. Kabalka, M. S. Reddy and M.-L. Yao, Tetrahedron Lett., 2009, 50, 7340–7342 CrossRef CAS.
- (a) J. W. Lee, W. J. Zheng, C. A. Morales-Rivera, P. Liu and M.-Y. Ngai, Chem. Sci., 2019, 10, 3217–3222 RSC; (b) W. Zheng, J. W. Lee, C. A. Morales-Rivera, P. Liu and M.-Y. Ngai, Angew. Chem., Int. Ed., 2018, 57, 13795–13799 CrossRef CAS; (c) W. G. Zhang, Z. L. Zou, W. X. Zhao, S. Lu, Z. G. Wu, M. J. Huang, X. C. Wang, Y. Wang, Y. Liang, Y. Zhu, Y. X. Zheng and Y. Pan, Nat. Commun., 2020, 11, 2572 CrossRef CAS.
- E. Kraka, D. Setiawan and D. Cremer, J. Comput. Chem., 2016, 37, 130–142 CrossRef CAS.
- B. Bang-Andersen, T. Ruhland, M. Jorgensen, G. Smith, K. Frederiksen, K. G. Jensen, H. Zhong, S. M. Nielsen, S. Hogg, A. Mork and T. B. Stensbol, J. Med. Chem., 2011, 54, 3206–3221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06446g |
| This journal is © The Royal Society of Chemistry 2021 |